
Abstract
Helicobacter pylori neutrophil-activating protein (HP-NAP) activates several innate leukocytes including neutrophils, monocytes, and mast cells. It has been reported that HP-NAP induces degranulation and interleukin-6 (IL-6) secretion of rat peritoneal mast cells. However, the molecular mechanism is not very clear. Here, we show that HP-NAP activates human mast cell line-1 (HMC-1) cells to secrete histamine and IL-6. The secretion depends on pertussis toxin (PTX)-sensitive heterotrimeric G proteins but not on Toll-like receptor 2. Moreover, HP-NAP induces PTX-sensitive G protein-mediated activation of extracellular signal-regulated kinase 1/2 (ERK1/2), p38-mitogen-activated protein kinase (p38 MAPK), and Akt in HMC-1 cells. Inhibition of ERK1/2, p38 MAPK, or phosphatidylinositol 3-kinase (PI3K) suppresses HP-NAP-induced release of histamine and IL-6 from HMC-1 cells. Thus, the activation of HMC-1 cells by HP-NAP is through Gi-linked G protein-coupled receptor-mediated MAPKs and PI3K/Akt pathways.
Abbreviations
| BSA | = | bovine serum albumin |
| CFU | = | colony-forming units |
| DMEM | = | Dulbecco's modified Eagle's medium |
| D-PBS | = | Dulbecco's phosphate-buffered saline |
| E. coli | = | Escherichia coli |
| ERK1/2 | = | extracellular signal-regulated kinase 1/2 |
| FBS | = | fetal bovine serum |
| GADPH | = | glyceraldehyde 3-phosphate dehydrogenase; GPCR, G protein-coupled receptor |
| HMC-1 | = | human mast cell line-1 |
| H. pylori | = | Helicobacter pylori |
| HP-NAP | = | Helicobacter pylori neutrophil-activating protein |
| IL-6 | = | interleukin-6 |
| IMDM | = | Iscove's modified Dulbecco's medium |
| LPS | = | lipopolysaccharide |
| MEK1/2 | = | MAPK/ERK kinase 1/2 |
| MFI | = | mean fluorescent intensity; MMLV, Moloney murine leukemia virus |
| MOI | = | multiplicity of infection |
| NADPH | = | nicotinamide adenine dinucleotide phosphate |
| NF-κB | = | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NSAID | = | nonsteroidal anti-inflammatory drugs |
| p38 MAPK | = | p38 mitogen-activated protein kinase |
| PCR | = | polymerase chain reaction |
| PI3K | = | phosphatidylinositol 3-kinase |
| PMSF | = | phenylmethanesulfonyl fluoride |
| PIPES | = | 1,4-piperazinediethanesulfonic acid |
| PKC | = | protein kinase C |
| PTX | = | pertussis toxin |
| ROS | = | reactive oxygen species |
| RT | = | reverse-transcription |
| SDS-PAGE | = | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| shRNA | = | short hairpin RNA |
| TGF-β1 | = | transforming growth factor-β1 |
| TLR2 | = | Toll-like receptor 2 |
| TLR4 | = | Toll-like receptor 4 |
| TNF-α | = | tumor necrosis factor-α |
| TSA | = | trypticase soy agar |
| VSV-G | = | vesicular stomatitis virus G protein. |
Introduction
Helicobacter pylori (H. pylori) is well recognized as a causative agent of acute and chronic gastric inflammation. Infection with H. pylori is associated with chronic gastritis, peptic ulcer disease, and gastric cancer.Citation1 H. pylori neutrophil-activating protein (HP-NAP), a virulence factor of H. pylori, may play a crucial role in H. pylori-induced gastric inflammation due to its ability to attract and activate neutrophils.Citation2 HP-NAP stimulates neutrophils to produce reactive oxygen species (ROS) and secrete myeloperoxidase, chemokines, and pro-inflammatory cytokines.Citation3-5 It also induces neutrophil chemotaxis and neutrophil-endothelial adhesion.Citation3,6 Moreover, HP-NAP is capable of activating monocytes and mast cells.Citation6,7 The inflammatory mediators produced by these innate immune cells upon HP-NAP stimulation could contribute to the gastric mucosal damage induced by H. pylori infection.
The molecular mechanism by which HP-NAP stimulates neutrophils to produce ROS has been well characterized. The signaling is initiated by the activation of a pertussis toxin (PTX)-sensitive G protein-coupled receptor (GPCR).Citation6 Subsequent signaling events include the activation of phosphatidylinositol 3-kinase (PI3K), Src family tyrosine kinase, extracellular signal-regulated kinase 1/2 (ERK1/2) and p38-mitogen-activated protein kinase (p38 MAPK), and an elevation of cytosolic calcium level.Citation6,8 ROS is produced by the final step of nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase activation on the plasma membrane of neutrophils.Citation6 Among these signaling molecules, ERK1/2 and p38 MAPK are also involved in the HP-NAP-induced chemotaxis and adhesion of neutrophils.Citation8 In human blood mononuclear cells, production of tissue factor and plasminogen activator inhibitor-2 induced by HP-NAP requires protein tyrosine kinase, protein kinase C (PKC), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), but not NADPH-oxidase.Citation9 It is not clear if the PTX-sensitive GPCR is also involved in this event. HP-NAP is not only an agonist of GPCR but also a ligand of Toll-like receptor 2 (TLR2).Citation10 TLR2 has been shown to be involved in HP-NAP-stimulated release of interleukin-6 (IL-6) in splenocytes.Citation11 However, the signaling events downstream of TLR2 induced by HP-NAP are not clear. In human leukocytes, such as neutrophils and monocytes, it is possible that HP-NAP induces cytokine release through TLR2 activation and ROS production through GPCR signaling pathways.
Mast cells have been reported to participate in the pathogenesis of H. pylori-infected gastritis.Citation12 The mast cell density in the gastric antrum was shown to be higher in H. pylori-infected peptic ulcer patients than that in patients with other types of gastritis.Citation13 Additional studies have shown that the mast cell density in the epithelium of the antral mucosa in H. pylori-infected patients with chronic active gastritis was higher than that in those with chronic nonactive gastritis or in patients with nonsteroidal anti-inflammatory drugs (NSAID)-induced gastritis.Citation14,15 In these H. pylori-infected patients with either peptic ulcer or chronic active gastritis, mast cell degranulation in gastric mucosa was prominent.Citation13,15 Thus, H. pylori should be involved in mucosal mast cell activation and its subsequent secretion of chemical mediators.
The water extract of H. pylori has been reported to induce perivenular mast cell degranulation in ratsCitation16 and to stimulate histamine release from canine gastric mucosal mast cells.Citation17 These findings indicate that factors from H. pylori could activate mast cells. Indeed, HP-NAP, identified from water extracts of H. pylori, is able to induce β-hexosaminidase release and IL-6 production in rat peritoneal mast cells.Citation7 However, the molecular mechanism by which HP-NAP activates human mast cells is not fully understood. In addition, there is no report using established cell lines to study the function of HP-NAP. In the present study, whether HP-NAP could activate human mast cell line-1 (HMC-1) cells was examined for its ability to stimulate the release of histamine and IL-6. The signaling pathways involved in HP-NAP-induced release of these mediators were also investigated. We herein report the molecular mechanism by which HP-NAP induces release of histamine and IL-6 through PTX-sensitive heterotrimeric G protein-mediated MAPKs and PI3K/Akt pathways in HMC-1 cells.
Results
HP-NAP-induced release of histamine and IL-6 from HMC-1 cells
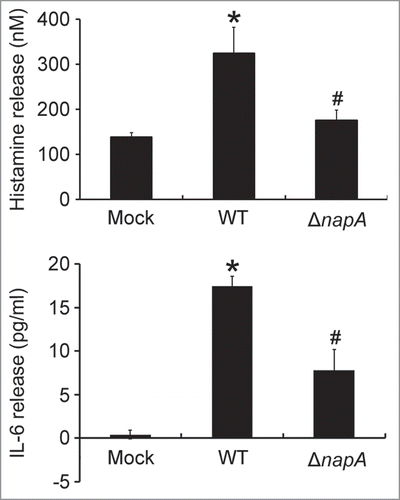
The HMC-1 is a well-established and widely used cell line for studying the function of mast cells.Citation18 To investigate whether HMC-1 cells can be activated by HP-NAP, we measured histamine release from cells stimulated with increasing concentration of HP-NAP ranging from 0.25 to 1 μM. As shown in , HP-NAP induced histamine release in a dose-dependent manner and up to 2.2-fold at the concentration of 1 μM. Another human mast cell line, LAD2 cells, also releases histamine in response to HP-NAP stimulation (Fig. S1). A time course study was performed to determine whether HP-NAP is able to induce IL-6 release from HMC-1 cells. We found that HP-NAP induced IL-6 release in a time-dependent manner (). The amount of IL-6 released from HMC-1 cells was increased to a maximum of 4.5-fold over basal level at 4 h of HP-NAP stimulation but eventually declined to 1.8-fold at 16 h of stimulation due to an elevated basal IL-6 release (). Heat-inactivated HP-NAP was not able to induce IL-6 release from HMC-1 cells (), indicating that induction of IL-6 release by HP-NAP was not due to the presence of lipopolysaccharide (LPS) contamination in the purified HP-NAP. Thus, HP-NAP is able to activate HMC-1 cells by inducing the release of histamine and IL-6. To further determine whether HP-NAP is involved in H. pylori-induced mast cell activation, we examined the release of histamine and IL-6 from HMC-1 cells infected with H. pylori wild-type strain NCTC 11637 and its isogenic mutant strain lacking HP-NAP. As shown in , the release of histamine and IL-6 induced by H. pylori infection was significantly inhibited by 80% and 57%, respectively, in cells infected by the isogenic napA H. pylori mutant strain as compared with the wild type H. pylori strain. These results indicate that HP-NAP plays a major role in H. pylori-induced release histamine and IL-6 from HMC-1 cells. Even though the H. pylori strain NCTC 11637 used for this infection experiment is different from the H. pylori strain 26695, whose napA gene was used as the source for production of recombinant HP-NAP, the highly conserved amino acid sequences of HP-NAP between these 2 strains (Fig. S2) suggested that the effect of HP-NAP on H. pylori-induced release histamine and IL-6 from HMC-1 cells for these 2 strains could be similar.
Figure 1. Induction of histamine release from HMC-1 cells by HP-NAP. HMC-1 cells were left unstimulated or stimulated with indicated concentrations of HP-NAP or 10 μM Ca2+ ionophore A23187 as a positive control at 37°C for 30 min. Histamine release from HMC-1 cells was determined by measuring the concentration of histamine in the cell-free supernatants as described in Materials and Methods. Data were represented as the mean ± SD of 4 independent experiments. *P < 0.05 as compared with unstimulated cells.
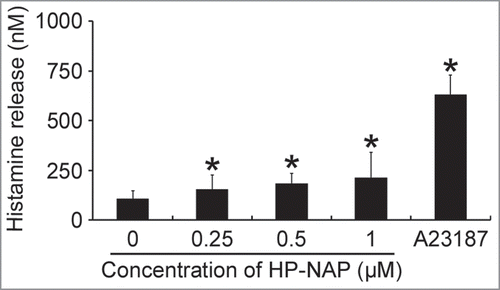
Figure 2. Induction of IL-6 release from HMC-1 cells by HP-NAP. (A) HMC-1 cells were left unstimulated or stimulated with 1 μM HP-NAP or 10 μg/mL E. coli LPS as a positive control at 37°C for the indicated time. IL-6 release from HMC-1 cells was determined by measuring the concentration of IL-6 in the cell-free supernatants as described in Materials and Methods. Data were represented as the mean ± SD of 3 independent experiments. *P < 0.05 as compared with unstimulated control cells at the same time point. (B) HMC-1 cells were left unstimulated or stimulated with 1 μM HP-NAP, 1 μM heat-inactivated (HI) HP-NAP, or 10 μg/mL E. coli LPS at 37°C for 16 h for measurement of IL-6 release as described in (A). Data were represented as the mean ± SD of 4 independent experiments. *P < 0.05 as compared with unstimulated control cells; #P < 0.05 as compared with HP-NAP-stimulated cells.
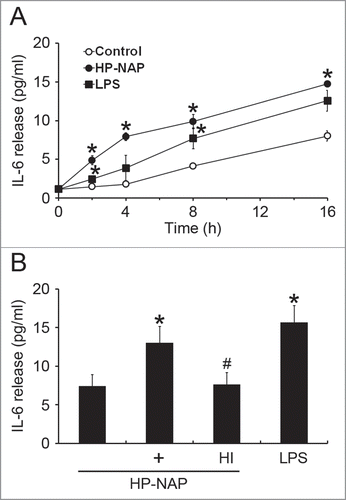
Figure 3. Induction of histamine and IL-6 release from HMC-1 cells infected with the wild-type H. pylori and isogenic napA knockout H. pylori mutant strains. HMC-1 cells were left uninfected (mock) or infected with wild-type (WT) H. pylori NCTC 11637 strain and its isogenic napA knockout mutant strain (ΔnapA) at 37°C for 30 min or 4 h for measurement of the release of histamine or IL-6, respectively. Release of histamine and IL-6 from HMC-1 cells was determined as described in , respectively. Data were represented as the mean ± SD of 3 independent experiments *P < 0.05 as compared with uninfected mock cells; #P < 0.05 as compared with wild-type H. pylori-infected cells.
Involvement of PTX-sensitive heterotrimeric G protein but not TLR2 in HP-NAP-induced release of histamine and IL-6 in HMC-1 cells
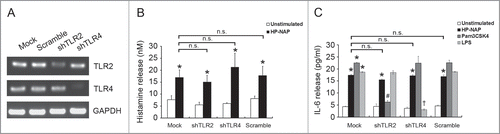
To investigate whether GPCR and TLR2 contribute to HP-NAP-induced activation of HMC-1 cells, we first used PTX, an inhibitor of Gi, to examine the involvement of GPCR in HP-NAP-induced release of histamine and IL-6 from HMC-1 cells. As shown in , the release of both histamine and IL-6 induced by HP-NAP was significantly inhibited in cells pretreated with PTX. We next examined whether TLR2 is involved in HP-NAP-induced release of histamine and IL-6 from HMC-1 cells by pretreatment of TLR2-neutralizing antibody or by knockdown of TLR2. In these experiments, pretreatment of Toll-like receptor 4 (TLR4)-neutralizing antibody or knockdown of TLR4 were used to further rule out a possible role of contaminating LPS in the purified HP-NAP. Flow cytometry analysis showed that both TLR2 and TLR4 were expressed in HMC-1 cells (). When cells were pretreated with TLR2-neutralizing antibody, HP-NAP-induced histamine release was not inhibited (). As expected, pretreatment of cells with TLR4-neutralizing antibody did not affect HP-NAP-induced histamine release (). Also, pretreatment of HMC-1 cells with either TLR2- or TLR4-neutralizing antibodies did not inhibit HP-NAP-induced IL-6 release (). However, IL-6 release induced by Pam3Csk4, a TLR2 agonist, and LPS, a TLR4 agonist, from HMC-1 cells was significantly inhibited by the pretreatment of TLR2- and TLR4-neutralizing antibodies, respectively, as compared with those in untreated cells or cells pretreated with the isotype control antibody (). These results indicate that TLR2 is not involved in HP-NAP-induced release of histamine and IL-6 from HMC-1 cells. In the experiment to knock down the expression of TLR2 and TLR4 in HMC-1 cells, lentiviral-transduced shRNA targeting these 2 TLRs was employed. The levels of mRNA expression of TLR2 and TLR4 were suppressed in cells transduced with shRNA targeting TLR2 and TLR4, respectively, as compared with those in cells transduced with scramble control shRNA. (). The reduced expression of either TLR2 or TLR4 in HMC-1 cells did not affect HP-NAP-induced release of histamine and IL-6 (). However, knockdown of the expression of TLR2 and TLR4 in HMC-1 cells significantly inhibited IL-6 release induced by Pam3Csk4 and LPS, respectively (). These results further support the finding that TLR2 is not involved in HP-NAP-induced release of histamine and IL-6 from HMC-1 cells. Thus, HP-NAP-induced release of histamine and IL-6 from HMC-1 cells depends on PTX-sensitive heterotrimeric G proteins but not on TLR2.
Figure 4. Inhibition of HP-NAP-induced release of histamine and IL-6 from HMC-1 cells by the treatment with PTX. HMC-1 cells were pretreated with 100 ng/mL PTX at 37°C for 16 h and then stimulated with 1 μM HP-NAP at 37°C for 30 min or 16 h for measurement of the release of histamine or IL-6, respectively. Release of histamine and IL-6 from HMC-1 cells was determined as described in . Data were represented as the mean ± SD of 6 independent experiments for histamine release and 5 independent experiments for IL-6 release. *P < 0.05 as compared with unstimulated cells in each group; #P < 0.05 as compared with HP-NAP-stimulated cells in the control group.
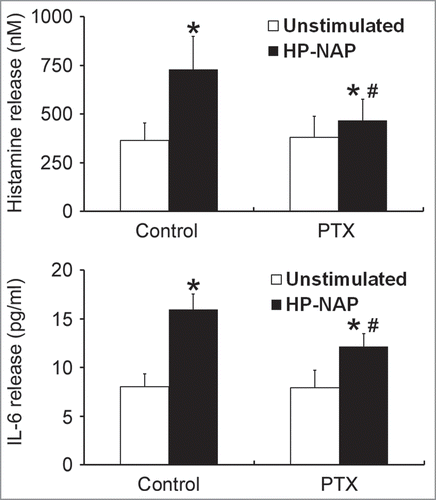
Figure 5. Surface expression of TLR2 and TLR4 on HMC-1 cells. The surface expression of TLR2 or TLR4 on HMC-1 cells was determined by flow cytometry using anti-TLR2, anti-TLR4, or mouse IgG2a isotype control antibodies. Data in the left panel show the representative histograms. Filled histograms represent isotype control and open histograms represent TLR expression. Data in the right panel show the fold differences of mean fluorescent intensity (MFI) with respect to that of the isotype control and are represented as the mean ± SD of 3 independent experiments. *P < 0.05 as compared with the isotype control.
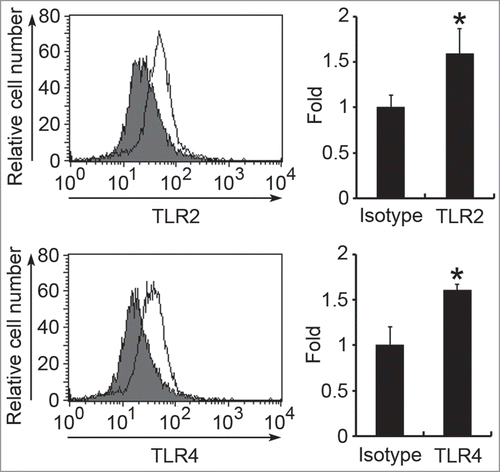
Figure 6. Effect of TLR2 and TLR4 neutralizing antibodies on HP-NAP-induced histamine and IL-6 release from HMC-1. HMC-1 cells were pretreated with 10 μg/mL of either anti-TLR2, anti-TLR4, or mouse IgG2a isotype antibodies at 37°C for 1 h. Then, the cells were stimulated with 1 μM HP-NAP at 37°C for 30 min or 16 h for measurement of histamine release (A) or IL-6 release (B), respectively, or stimulated with 10 μg/mL Pam3CSK4 or 10 μg/mL E. coli LPS at 37°C for 16 h for measurement of IL-6 release (C). Release of histamine and IL-6 from HMC-1 cells was determined as described in . Data were represented as the mean ± SD of 3 independent experiments. *P < 0.05 as compared with unstimulated cells in each group; #P < 0.05 as compared with Pam3CSK4-stimulated cells in the isotype control group; †P < 0.05 as compared with LPS-stimulated cells in the isotype control group; n.s., non-significant.
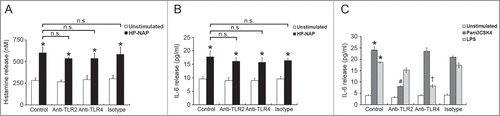
Figure 7. Effect of knockdown of TLR2 and TLR4 on HP-NAP-induced histamine and IL-6 release from HMC-1. HMC-1 cells were left untransduced (mock) or transduced with lentiviral particles bearing either scramble control shRNA or shRNA specific to TLR2 (shTLR2) or TLR4 (shTLR4) for 2 d. Lentivirally transduced HMC-1 cells were then under puromycin selection for 2 d The mRNA expressions of TLR2, TLR4, and GAPDH, as a loading control, in these cells were analyzed by using RT-PCR (A) as described in Materials and Methods. Untransduced or TLR-specific lentivirally transduced HMC-1 cells were stimulated with 1 μM HP-NAP at 37°C for 30 min for measurement of histamine release (B) or stimulated with 1 μM HP-NAP, 10 μg/mL Pam3CSK4, or 10 μg/mL E. coli LPS at 37°C for 16 h for measurement of IL-6 release (C). Release of histamine and IL-6 from HMC-1 cells were determined as described in . Data were represented as the mean ± SD of 3 independent experiments. *P < 0.05 as compared with unstimulated cells in each group; #P < 0.05 as compared with Pam3CSK4-stimulated cells in the scramble control group; †P < 0.05 as compared with LPS-stimulated cells in the scramble control group; n.s., non-significant.
PTX-sensitive heterotrimeric G protein-dependent activation of ERK1/2, p38 MAPK, and Akt in HMC-1 cells induced by HP-NAP
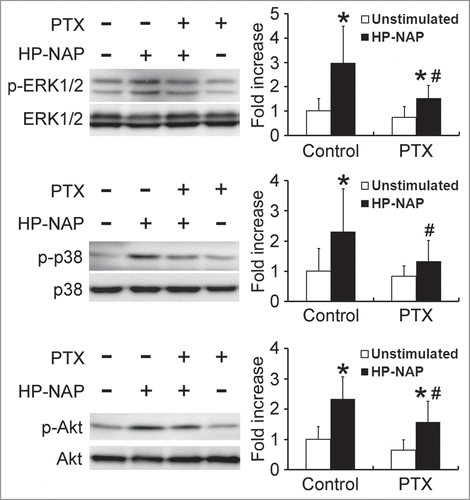
Both ERK1/2 and p38 MAPK are involved in HP-NAP-induced activation of neutrophils.Citation8 Also, our finding that PTX-sensitive G protein participates in HP-NAP-induced activation of HMC-1 cells indicates that Gβγ-mediated activation of PI3Kγ may be involved. A time course experiment was performed to determine whether HP-NAP activates ERK1/2, p38 MAPK, and Akt, a downstream effector of PI3K, in HMC-1 cells. The phosphorylation of ERK1/2 and p38 MAPK was significantly increased at 10 and 20 min of HP-NAP stimulation, respectively, while the phosphorylation of Akt increased at 1.5 min of stimulation (). The phosphorylation of all 3 molcules continued to increase up to 30 min (). HP-NAP also induced the phosphorylation of these 3 molcules in human LAD2 mast cells (Fig. S3). We next examined whether G proteins act upstream of ERK1/2, p38 MAPK, and Akt in HMC-1 cells in response to HP-NAP stimulation by pretreating cells with PTX. HP-NAP-induced phosphorylation of ERK1/2, p38 MAPK, and Akt was significantly inhibited in cells pretreated with PTX (). Thus, HP-NAP activates ERK1/2, p38 MAPK, and Akt in HMC-1 cells through a PTX-sensitive heterotrimeric G protein-dependent signaling pathway.
Figure 8. Activation of ERK1/2, p38 MAPK, and Akt in HMC-1 cells induced by HP-NAP. Serum-starved HMC-1 cells were left unstimulated or stimulated with 1 μM HP-NAP at 37°C for the indicated time and then lysed. Whole cell lysates were subjected to immunoblotting for phospho-ERK1/2, ERK, phospho-p38, p38, phospho-Akt, and Akt. The quantitative results were expressed in fold increase by defining the amounts of the phosphorylated proteins in unstimulated cells as 1 and represented as the mean ± SD of at least 4 independent experiments. *P < 0.05 as compared with unstimulated cells.
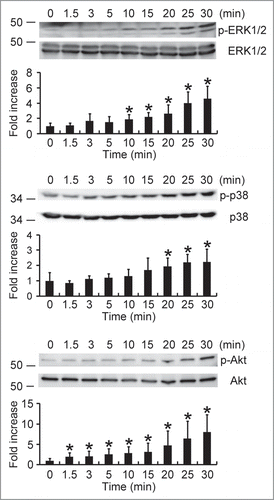
Figure 9. Inhibition of HP-NAP-induced activation of ERK1/2, p38 MAPK, and Akt in HMC-1 cells by the treatment with PTX. Serum-starved HMC-1 cells were not pretreated (control) or pretreated with 100 ng/mL PTX at 37°C for 16 h and then left unstimulated or stimulated with 1 μM HP-NAP at 37°C for 30 min. Cells were lysed and whole cell lysates were subjected to immunoblotting for phospho-ERK1/2, ERK, phospho-p38, p38, phospho-Akt, and Akt. The quantitative results were expressed in fold increase by defining the amounts of the phosphorylated proteins in cells without any treatment as 1 and represented as the mean ± SD of at least 5 independent experiments. *P < 0.05 as compared with unstimulated cells in each group; #P < 0.05 as compared with HP-NAP-stimulated control cells.
Involvement of ERK1/2, p38 MAPK, and PI3K/Akt in HP-NAP-induced histamine and IL-6 release from HMC-1 cells
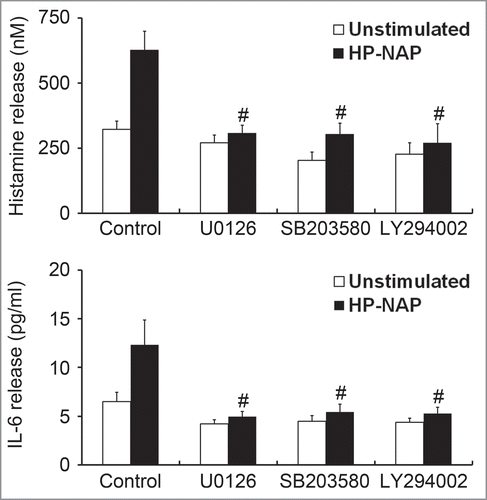
To further determine whether MAPKs and PI3K/Akt are involved in HP-NAP-induced release of histamine and IL-6 from HMC-1 cells, we performed experiments with cells pretreated with U0126, a MAPK/ERK kinase 1/2 (MEK1/2) inhibitor to block ERK1/2 activation; SB203580, a p38 MAPK inhibitor; or LY294002, a PI3K-specific inhibitor. Pretreatment of HMC-1 cells with U0126, SB203580, and LY294002 suppressed HP-NAP-induced phosphorylation of ERK1/2, p38 MAPK, and Akt, respectively (). Unexpectedly, the phosphorylation of Akt induced by HP-NAP was also slightly inhibited in cells pretreated with U0126 (Fig. S4), suggesting that HP-NAP-induced AKT phosphorylation in HMC-1 cells partially relies on MEK1/2. This same pathway has also been shown to mediate the transforming growth factor-β1 (TGF-β1) signaling in primary human monocytes.Citation19 Nevertheless, the release of histamine and IL-6 from HMC-1 induced by HP-NAP was blocked by any one of these 3 inhibitors (). These results indicate that concomitant activation of ERK1/2, p38 MAPK, and PI3K is required for HP-NAP-induced release of histamine and IL-6 in HMC-1 cells. Thus, HP-NAP-induced activation of HMC-1 cells is through PTX-sensitive heterotrimeric G protein-dependent ERK1/2, p38 MAPK, and PI3K/Akt signaling pathways.
Figure 10. Inhibition of HP-NAP-induced activation of ERK1/2, p38 MAPK, and Akt in HMC-1 cells by the treatment with U0126, SB203580, and LY294002. Serum-starved HMC-1 cells were not pretreated (control) or pretreated with 5 μM U0126, a MEK1/2 inhibitor, 10 μM SB203580, a p38 MAPK inhibitor, or 10 μM LY294002, a PI3K inhibitor, at 37°C for 1 h and then left unstimulated or stimulated with 1 μM HP-NAP at 37°C for 30 min. Cells were lysed and whole cell lysates were subjected to immunoblotting for phospho-ERK1/2, ERK, phospho-p38, p38, phospho-Akt, and Akt. The quantitative results were expressed in fold increase by defining the amounts of the phosphorylated proteins in cells without any treatment as 1 and represented as the mean ± SD of 3 independent experiments. *P < 0.05 as compared with unstimulated cells in each group; #P < 0.05 as compared with HP-NAP-stimulated control cells.
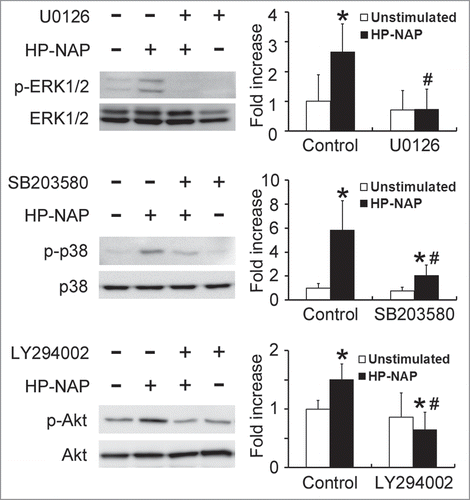
Figure 11. Inhibition of HP-NAP-induced release of histamine and IL-6 from HMC-1 cells by the treatment with U0126, SB203580, and LY294002. Serum-starved HMC-1 cells were not pretreated (control) or pretreated with 5 μM U0126, a MEK1/2 inhibitor; 10 μM SB203580, a p38-MAPK inhibitor; or 10 μM LY294002, a PI3K inhibitor at 37°C for 1 h and then left unstimulated or stimulated with 1 μM HP-NAP at 37°C for 30 min or 16 h for measurement of the release of histamine or IL-6, respectively. Release of histamine and IL-6 from HMC-1 cells was determined as described in . Data were represented as the mean ± SD of 3 independent experiments. *P < 0.05 as compared with unstimulated control cells; #P < 0.05 as compared with HP-NAP-stimulated control cells.
Discussion
In this study, we show that HP-NAP can activate HMC-1 cells by stimulating the release of histamine and IL-6 though the concomitant activation of ERK1/2, p38 MAPK, and PI3K/Akt signaling pathways. Upon HP-NAP stimulation, initiation of the signaling to these protein kinase cascades in HMC-1 cells requires PTX-sensitive heterotrimeric G proteins but not TLR2. These results indicate that GPCRs may play a critical role in HP-NAP-induced degranulation and cytokine release in human mast cells. Most importantly, the HMC-1 is the first cell line model being used to study the function of HP-NAP.
The finding that HP-NAP-induced release of histamine and IL-6 in HMC-1 cells is PTX-sensitive () indicates that the GPCR activated by HP-NAP is Gi-linked. However, the partial inhibition of HP-NAP-induced release of histamine and IL-6 by PTX points to the involvement of both PTX-sensitive and PTX-insensitive pathways. Since TLR2, a receptor of HP-NAP, is not involved in HP-NAP-induced release of histamine and IL-6 from HMC-1 cells (), a PTX-insensitive heterotrimeric Gq pathway leading to activation of phospholipase CβCitation20,21 may contribute to HP-NAP-induced activation of HMC-1 cells. In the other study, TLR2 has been reported to mediate the release of histamine from HMC-1 cells induced by bacterial peptidoglycan,Citation22 which is in contrast to our finding with HP-NAP. Since TLR2 is a receptor of HP-NAP and is expressed on HMC-1 cells (), the finding that the release of histamine and IL-6 from HMC-1 cells induced by HP-NAP did not depend on TLR2 suggested that TLR2 may mediate other mast cell responses induced by HP-NAP.
Histamine not only stimulates gastric acid secretion but may also be involved in the pathogenesis of H. pylori-associated gastroduodenal disease. It has been reported that H. pylori-positive patients have lower histamine concentration in gastric mucosa than H. pylori-negative subjects.Citation23,24 This finding might be due to an increased release and subsequent depletion of histamine from mast cells in human gastric mucosa of H. pylori-positive patients. Histamine released from mast cells has been reported to regulate gastric mucosal injury-repair in humans.Citation25 In a study investigating the effect of H. pylori on rat gastric mucosal microcirculation, macromolecular leakage induced by H. pylori was found to depend on the release of histamine from mast cells.Citation26 Moreover, in histamine-deficient mice, H. pylori induced lower secretion of tumor necrosis factor-α (TNF-α) and IL-6 in their gastric mucosa than in wild-type mice.Citation27 In our study, we have shown that recombinant HP-NAP stimulates histamine release from HMC-1 cells. Also, a marked decrease in histamine release was found in cells infected with H. pylori isogenic mutant strain lacking HP-NAP as compared with the wild-type strain (). These results, together with the previous findings just mentioned, support the idea that HP-NAP plays a role in H. pylori-induced gastric mucosal injury through activating human mast cells.
In summary, HP-NAP induces release of histamine and IL-6 from HMC-1 through PTX-sensitive heterotrimeric G protein-mediated activation of ERK1/2, p38 MAPK, and PI3K/Akt. The secretion of granule-associated histamine and pleiotropic cytokine IL-6 in human mast cells induced by HP-NAP might be essential in the initiation and modulation of gastric inflammatory responses during H. pylori infection. Also, the HP-NAP-induced signal pathways identified in HMC-1 cells should provide an understanding of how HP-NAP activates human mast cells.
Materials and Methods
Cell culture
The human mast cell line HMC-1.2Citation18 was maintained in Iscove's modified Dulbecco's medium (IMDM) (Invitrogen, Carlsbad, CA, USA) supplemented with 1.2 mM monothioglycerol (Sigma, St. Louis, MO, USA), 10% heat-inactivated fetal bovine serum (FBS) (Biological Industries, Kibbutz Beit Haemek, Israel), 100 units/mL penicillin, and 50 μg/mL streptomycin at 37°C under 5% CO2 and saturated humidity. The human mast cell line LAD2Citation28 was cultured in the Stem-Pro-34 serum-free medium (Invitrogen) in the presence of 2 mM glutamine, 100 ng/mL recombinant human stem cell factor, purchased from Peprotech (Rocky Hill, NJ, USA), 100 units/mL penicillin, and 50 μg/mL streptomycin at 37°C under 5% CO2 and saturated humidity. The human embryonic kidney cell line HEK293T (ATCC CRL-3216) was maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen) supplemented with 10% FBS, 100 units/mL penicillin, and 50 μg/mL streptomycin at 37°C under 5% CO2 and saturated humidity.
H. pylori strains and culture conditions
H. pylori strain NCTC 11637 (ATCC 43504) was obtained from the Bioresource Collection and Research Center (BCRC, Taiwan) and its isogenic napA knockout mutant strain was a generous gift from Dr. Jin-Town Wang at the College of Medicine at National Taiwan University, Taiwan.Citation29 Both strains were grown for 3 d under microaerophilic condition using GasPak EZ Container System Sachets (BD BBL; Sparks, MD, USA) and 100% humidity at 37°C on trypticase soy agar (TSA) with 5% sheep blood (BD BBL). For expanding the culture, the H. pylori colonies from TSA plate were transferred into Brucella broth (BD BBL) with 10% horse serum (JRH Biosciences, Lenexa, KS, USA) and then grown for 2 d. For the culture suspension, 1 OD unit at 600 nm is roughly equivalent to 3.78×108 colony-forming units (CFU)/mL. Before infecting HMC-1 cells with H. pylori, the bacteria were harvested and then resuspended in IMDM supplemented with 10% or 1% heat-inactivated FBS for the experiments measuring the release of histamine or IL-6, respectively. HMC-1 cells were infected with H. pylori at a multiplicity of infection (MOI) of 200.
Lentivirus generation and short hairpin RNA transduction
HEK293T cells were seeded at a density of 2.5 × 106 cells per 100-mm culture dish 24 h before transfection. Cells were cotransfected with 10 μg of the pLKO.1-based lentiviral vectors expressing short hairpin RNA (shRNA) targeting TLR2 (Clone ID: TRCN0000057019) or TLR4 (Clone ID: TRCN0000056894) or scrambled control shRNA, obtained from the National RNAi Core Facility, Academia Sinica, Taiwan, together with 9 μg of the Δ8.9 plasmid and 2.5 μg of the vesicular stomatitis virus G protein (VSV-G) envelope vector, by using calcium phosphate precipitation. After 12 h of transfection, HEK293T cells were washed with Dulbecco's phosphate-buffered saline (D-PBS), pH 7.2, and then cultured in fresh DMEM complete medium. After 24 h of transfection, the medium was replaced with 6 ml of IMDM complete medium. After 48 h post-transfection, the cell supernatants containing lentiviral particles were collected by centrifugation at 800 × g for 5 min at room temperature and then filtered through a 0.45-mm filter (Pall, Ann Arbor, MI, USA). Lentivirus-mediated shRNA transduction of HMC-1 cells was then conducted by the addition of 1 mL of the filtered cell supernatant to a T75 culture flask containing 6×106 HMC-1 cells in 19 mL of IMDM complete medium containing 8 μg/mL polybrene (Sigma, St Louis, MO). After 2 d of transduction, the virus-containing medium was removed and fresh IMDM complete medium containing 2 μg/mL puromycin (invitrogen) was added. Two days after puromycin selection, the survival cells were subjected to the subsequent analysis.
Production of HP-NAP
HP-NAP, encoded by the napA gene from genomic DNA of H. pylori strain 26695 (accession no. AE000543, ATCC), was expressed in Escherichia coli (E. coli) and purified by 2 consecutive gel filtration steps using a XK 16/100 column packed with Sephacryl S-300 high resolution resin (Sephacryl S-300 HR) (GE Healthcare Bio-Sciences) and a HiLoad 16/60 Superdex 200 prep grade (Superdex 200 pg) gel filtration column (GE Healthcare Bio-Sciences) as previously described,Citation4 except that 1,4-piperazinediethanesulfonic acid (PIPES)-glucose-calcium-magnesium (PGCM) buffer was used in the second gel filtration step. PGCM buffer was 25 mM PIPES, pH 7.4, containing 110 mM NaCl, 5 mM KCl, 0.1% glucose, 1 mM CaCl2, and 1 mM MgCl2. Recombinant HP-NAP was eluted as a single peak, and the peak fractions containing HP-NAP were pooled. The pooled fraction of HP-NAP was added with bovine serum albumin (BSA) at a final concentration of 0.1% and then subjected to syringe filtration for endotoxin removal as described previously.Citation4 The amount of endotoxin present in the purified HP-NAP was less than 8.13 endotoxin units (EU)/mg of HP-NAP as examined by SUPER LABORATORY (Taipei, Taiwan). The purity of HP-NAP was higher than 95% as confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) stained with PhastGel Blue R (Fig. S5). Heat inactivated HP-NAP was prepared by incubating the purified HP-NAP at 95°C for 5 min.
Treatment with inhibitors and antibodies
Pretreatment of HMC-1 cells with inhibitors and antibodies was performed at 37°C after resuspending cells in the PGCM buffer supplemented with 0.1% BSA (PAGCM buffer). PTX (Calbiochem, San Diego, CA, USA) at a concentration of 100 ng/mL was used for pretreating cells for 16 h to block the activation of Gi. U0126, a MEK1/2 inhibitor to block ERK1/2 activation, SB203580, a p38 MAPK inhibitor, and LY294002, a PI3K inhibitor, were purchased from Calbiochem and used for pretreating cells for 1 h at 5, 10, and 10 μM, respectively. Functional graded monoclonal mouse anti-human TLR2 (clone TL2.1), functional graded monoclonal mouse anti-human TLR4 (clone HTA125), and functional graded mouse IgG2a isotype control antibodies, purchased from eBioscience (San Diego, CA, USA), at a concentration of 10 μg/mL were used for pretreating cells for 1 h.
Histamine release assay
HMC-1 cells at a density of 5×105 were infected with H. pylori at 37°C for 30 min or resuspended in PAGCM buffer to a density of 5×105 cells/mL, followed by the treatment with indicated stimuli at 37°C for 30 min. LAD2 cells were resuspended in PAGCM buffer to a density of 1.65 × 105 cells/mL and then treated with indicated stimuli at 37°C for 30 min. Calcium ionophore A23187 (Sigma, St. Louis, MO) was used as a positive control for histamine release of HMC-1 cells. The cell suspension was centrifuged at 100 × g for 5 min, and the supernatant was collected. The amount of histamine in the supernatant was determined by using a histamine EIA kit (Cayman Chemical, Ann Arbor, MI, USA), according to the manufacturer's instructions.
IL-6 release assay
HMC-1 cells were serum starved with 1% heat-inactivated FBS for 6 h. The cells at a density of 1×106 cells/mL were infected with H. pylori at 37°C for 4 h. Alternatively, the cells were resuspended in PAGCM buffer to a density of 1×106 cells/mL and then treated with various stimuli at 37°C for 16 h unless specified. E. coli LPS-EB ultrapure (Invivogen, San Diego, CA, USA), a TLR4 agonist, was used as a positive control for IL-6 release of HMC-1 cells. Pam3CSK4 (Invivogen), a TLR2 agonist, was used as a positive control to induce IL-6 release through TLR2 activation. The cell suspension was centrifuged at 100 × g for 5 min, and the supernatant was collected. The amount of IL-6 in the supernatant was determined by using IL-6 ELISA kits (Bender MedSystems, Vienna, Austria and e-Biosence, San Diego, CA, USA), according to the manufacturers' instructions.
Flow cytometry
HMC-1 cells were centrifuged at 400 × g for 5 min, and the cell pellet was resuspended in FACS buffer containing D-PBS, pH 7.2, supplemented with 2.5% BSA. Cells were stained with 1 μg of anti-TLR2 (TL2.1), anti-TLR4 (HTA125), and mouse IgG2a isotype control antibodies per 5×105 cells at 25°C for 1 h, and then washed twice with FACS buffer. Subsequently, the cells were stained with 0.5 μg Alexa 488-conjugated goat anti-mouse secondary antibody (Invitrogen, Carlsbad, CA, USA) at 25°C for 1 h, and then washed twice with FACS buffer to remove unbound antibodies. At least 10000 cells were analyzed by flow cytometry on a FACScalibur equipped with CellQuest software (BD Bioscience; San Jose, CA, USA).
RNA extraction and Reverse transcription–polymerase chain reaction
Total RNA was extracted from the HMC-1 cells by using TriPure isolation reagent (Roche Basel, Swiss) according to the manufacturer's instructions. To exclude genomic DNA contamination, 6 μg of the extracted RNA was treated with RQ1 RNase-free DNase I (Promega) at 37°C for 30 min. In the reverse-transcription (RT) reaction, 2 μg of DNA-free RNA from each sample was incubated with 1 μg of the random hexamer primer at 70°C for 5 min to open the secondary structure, and then incubated with 10 mM dNTPs, 20 U RNase inhibitor, and 200 U Moloney murine leukemia virus (MMLV) reverse transcriptase (Promega) at 37°C for 1 h to transcribe cDNA, followed by heat inactivation at 92°C for 10 min. The polymerase chain reaction (PCR) was carried out with 1/20 of the cDNA product, the primer pairs for the gene of interest, and AmpFlSTR PCR Amplification Kit (ABI, Vernon, CA, USA) in a Mastercycler Gradient 5331 (Eppendorf, Germany). An initial denaturing phase of 94°C for 5 min was followed by 35 cycles of 94°C for 45 sec, 60°C for 30 sec, and 72 °C for 45 sec. The primer pairs used in PCR were: human TLR2 (NM_003264): sense 5′-TGCTGCCATTCTCATTCTTCTG-3′ and antisense 5′- AGGTCTTGGTGTTCATTATCTTCC-3′; human TLR4 (NM_003266): sense 5′- CTTCCTCTCACCCTTTAGC-3′ and antisense 5′- CATCATCCTGGCATCATCC-3′; human glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (NM_002046): sense 5′- ACACCCACTCCTCCACCTTT-3′ and antisense 5′- TACTCCTTGGAGGCCATGTG-3′. The PCR products were separated on a 2% agarose gel with FluoroDye DNA fluorescent loading dye (SMOBIO, Hsinchu, Taiwan) and the bands were imaged by LAS-3000 imaging system (Fujifilm).
Immunoblotting
HMC-1 and LAD2 cells were serum starved with heat-inactivated 1% FBS for 24 h or 8 h (for cells with PTX-pretreatment) and then resuspended in PAGCM buffer to a density of 3×106 and 8×105 cells/mL, respectively. After treatment with stimuli, cells were harvested in the lysis buffer containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, with the addition of 1 mM phenylmethanesulfonyl fluoride (PMSF) (Roche), 2 μg/mL aprotinin, 2 μg/mL leupeptin, 1 mM Na3VO4, and 1 mM NaF, and then lysed by passaging through a 27.5G needle 10 times. The insoluble material was removed by centrifugation at 13200 × g at 4°C for 30 min. Protein concentrations were determined by using BCA Assay kit (Pierce, Carlsbad, CA, USA). Equal amounts of 120 μg of cell lysates were subjected to SDS-PAGE and immunoblotting as previously described,Citation30 except that the membranes were probed with monoclonal mouse anti-human phospho-ERK1/2 (clone E10), polyclonal rabbit anti-human phospho-p38 MAPK, monoclonal mouse anti-human phospho-Akt (Ser473) (clone 587F11), polyclonal rabbit anti-human p38 MAPK, and polyclonal rabbit anti-Akt antibodies, purchased from Cell Signaling (Beverly, MA, USA); and anti-human p44/42 MAPK (ERK1/2) rabbit antiserum, purchased from Sigma. Chemiluminescent detection was performed using ECL (Amersham, Piscataway, NJ, USA). The bands were imaged by LAS-3000 imaging system (Fujifilm, Japan) and quantified using densitometry by UN-SCAN-IT gel 6.1 software.
Statistical analysis
All analysis was performed using Excel 2007 software. Data are presented as mean ± standard deviation (SD). Statistical difference was determined by a paired 2-tailed Student's t-test, except that analysis of paired data in flow cytometry and immunoblotting was determined by a 1-tailed Student's t-test. p < 0.05 is considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
KVIR_A_SUppl_1043505.doc
Download MS Word (1.3 MB)Acknowledgments
We appreciate Dr. Butterfield JH at Mayo Clinic, Rochester, MN, USA for providing HMC-1 cells and Dr. Kirshenbaum A and Metcalfe D. at National Institutes of Health, Bethesda, MD, USA for providing LAD2 cells. We thank Dr. Jin-Town Wang at the College of Medicine at National Taiwan University, Taiwan for providing the isogenic napA knockout mutant of H. pylori strain NCTC 11637 and Dr. Shih-Hsiung Wu at the Academia Sinica, Taiwan for preparing the bacteria. We also thank Dr. Yu-Ting Chou at the Institute of Biotechnology at National Tsing Hua University, Taiwan for providing HEK293T cells, the lentiviral vector expressing scrambled shRNA, the Δ8.9 plasmid, and the VSV-G envelope vector.
Funding
This work was supported by grants from the National Science Council of Taiwan (NSC101–2311-B-007–007 and NSC98–2311-B-007–006-MY3), the National Research Program for Genomic Medicine, National Science Council of Taiwan (NSC96–3112-B-007–003), the Joint Research Program of National Tsing Hua University and Mackay Memorial Hospital (100N7727E1, 101N2727E1, and 103N2773E1) and the research program of National Tsing Hua University (104N2052E1).
References
- Dunn BE, Cohen H, Blaser MJ. Helicobacter pylori. Clin Microbiol Rev 1997; 10:720-41; PMID:9336670.
- Fu HW. Helicobacter pylori neutrophil-activating protein: from molecular pathogenesis to clinical applications. World J Gastroenterol 2014; 20:5294-301; PMID:24833859; http://dx.doi.org/10.3748/wjg.v20.i18.5294.
- Evans DJ, Jr., Evans DG, Takemura T, Nakano H, Lampert HC, Graham DY, Granger DN, Kvietys PR. Characterization of a Helicobacter pylori neutrophil-activating protein. Infect Immun 1995; 63:2213-20; PMID:7768601.
- Wang CA, Liu YC, Du SY, Lin CW, Fu HW. Helicobacter pylori neutrophil-activating protein promotes myeloperoxidase release from human neutrophils. Biochem Biophys Res Commun 2008; 377:52-6; PMID:18823946; http://dx.doi.org/10.1016/j.bbrc.2008.09.072.
- Polenghi A, Bossi F, Fischetti F, Durigutto P, Cabrelle A, Tamassia N, Cassatella MA, Montecucco C, Tedesco F, de Bernard M. The neutrophil-activating protein of Helicobacter pylori crosses endothelia to promote neutrophil adhesion in vivo. J Immunol 2007; 178:1312-20; http://dx.doi.org/10.4049/jimmunol.178.3.1312.
- Satin B, Del Giudice G, Della Bianca V, Dusi S, Laudanna C, Tonello F, Kelleher D, Rappuoli R, Montecucco C, Rossi F. The neutrophil-activating protein (HP-NAP) of Helicobacter pylori is a protective antigen and a major virulence factor. J Exp Med 2000; 191:1467-76; PMID:10790422; http://dx.doi.org/10.1084/jem.191.9.1467.
- Montemurro P, Nishioka H, Dundon WG, de Bernard M, Del Giudice G, Rappuoli R, Montecucco C. The neutrophil-activating protein (HP-NAP) of Helicobacter pylori is a potent stimulant of mast cells. Eur J Immunol 2002; 32:671-6; PMID:11857341; http://dx.doi.org/10.1002/1521-4141(200203)32:3%3c671::AID-IMMU671%3e3.3.CO;2-X.
- Nishioka H, Baesso I, Semenzato G, Trentin L, Rappuoli R, Del Giudice G, Montecucco C. The neutrophil-activating protein of Helicobacter pylori (HP-NAP) activates the MAPK pathway in human neutrophils. Eur J Immunol 2003; 33:840-9; PMID:12672049; http://dx.doi.org/10.1002/eji.200323726.
- Montemurro P, Barbuti G, Dundon WG, Del Giudice G, Rappuoli R, Colucci M, De Rinaldis P, Montecucco C, Semeraro N, Papini E. Helicobacter pylori neutrophil-activating protein stimulates tissue factor and plasminogen activator inhibitor-2 production by human blood mononuclear cells. J Infect Dis 2001; 183:1055-62; PMID:11237830; http://dx.doi.org/10.1086/319280.
- Amedei A, Cappon A, Codolo G, Cabrelle A, Polenghi A, Benagiano M, Tasca E, Azzurri A, D'Elios MM, Del Prete G, et al. The neutrophil-activating protein of Helicobacter pylori promotes Th1 immune responses. J Clin Invest 2006; 116:1092-101; PMID:16543949; http://dx.doi.org/10.1172/JCI27177.
- Del Prete G, Chiumiento L, Amedei A, Piazza M, D'Elios MM, Codolo G, de Bernard M, Masetti M, Bruschi F. Immunosuppression of TH2 responses in Trichinella spiralis infection by Helicobacter pylori neutrophil-activating protein. J Allergy Clin Immunol 2008; 122:908-13.e5; PMID:18804852; http://dx.doi.org/10.1016/j.jaci.2008.08.016.
- Nakajima S, Bamba N, Hattori T. Histological aspects and role of mast cells in Helicobacter pylori-infected gastritis. Aliment Pharmacol Ther 2004; 20 Suppl 1:165-70; PMID:15298623; http://dx.doi.org/10.1111/j.1365-2036.2004.01974.x.
- Nakajima S, Krishnan B, Ota H, Segura AM, Hattori T, Graham DY, Genta RM. Mast cell involvement in gastritis with or without Helicobacter pylori infection. Gastroenterology 1997; 113:746-54; PMID:9287964; http://dx.doi.org/10.1016/S0016-5085(97)70167-7.
- Hofman V, Lassalle S, Selva E, Kalem K, Steff A, Hebuterne X, Sicard D, Auberger P, Hofman P. Involvement of mast cells in gastritis caused by Helicobacter pylori: a potential role in epithelial cell apoptosis. J Clin Pathol 2007; 60:600-7; PMID:17557865; http://dx.doi.org/10.1136/jcp.2006.040741.
- Caruso RA, Parisi A, Crisafulli C, Bonanno A, Lucian R, Branca G, Scardigno M, Fedele F. Intraepithelial infiltration by mast cells in human Helicobacter pylori active gastritis. Ultrastruct Pathol 2011; 35:251-5; PMID:21932987; http://dx.doi.org/10.3109/01913123.2011.606964.
- Kurose I, Granger DN, Evans DJ, Jr., Evans DG, Graham DY, Miyasaka M, Anderson DC, Wolf RE, Cepinskas G, Kvietys PR Helicobacter pylori-induced microvascular protein leakage in rats: role of neutrophils, mast cells, and platelets. Gastroenterology 1994; 107:70-9; PMID:8020691.
- Yakabi K, Arimura T, Koyanagi M, Uehigashi Y, Ro S, Minagawa Y, Nakamura T. Effects of interleukin-8 and Helicobacter pylori on histamine release from isolated canine gastric mucosal mast cells. J Gastroenterol 2002; 37:10-6; PMID:11824794; http://dx.doi.org/10.1007/s535-002-8126-y.
- Butterfield JH, Weiler D, Dewald G, Gleich GJ. Establishment of an immature mast cell line from a patient with mast cell leukemia. Leuk Res 1988; 12:345-55; PMID:3131594; http://dx.doi.org/10.1016/0145-2126(88)90050-1.
- Olieslagers S, Pardali E, Tchaikovski V, ten Dijke P, Waltenberger J. TGF-β1/ALK5-induced monocyte migration involves PI3K and p38 pathways and is not negatively affected by diabetes mellitus. Cardiovasc Res 2011; 91:510-8; PMID:21478266; http://dx.doi.org/10.1093/cvr/cvr100.
- Smrcka AV, Hepler JR, Brown KO, Sternweis PC. Regulation of polyphosphoinositide-specific phospholipase C activity by purified Gq. Science 1991; 251:804-7; PMID:1846707; http://dx.doi.org/10.1126/science.1846707.
- Taylor SJ, Chae HZ, Rhee SG, Exton JH. Activation of the β 1 isozyme of phospholipase C by α subunits of the Gq class of G proteins. Nature 1991; 350:516-8; PMID:1707501; http://dx.doi.org/10.1038/350516a0.
- Wu L, Feng BS, He SH, Zheng PY, Croitoru K, Yang PC. Bacterial peptidoglycan breaks down intestinal tolerance via mast cell activation: the role of TLR2 and NOD2. Immunol Cell Biol 2007; 85:538-45; PMID:17563761; http://dx.doi.org/10.1038/sj.icb.7100079.
- Queiroz DM, Mendes EN, Rocha GA, Barbosa AJ, Carvalho AS, Cunha-Melo JR. Histamine concentration of gastric mucosa in Helicobacter pylori positive and negative children. Gut 1991; 32:464-6; PMID:2040464; http://dx.doi.org/10.1136/gut.32.5.464.
- Queiroz DM, Mendes EN, Rocha GA, Cunha-Melo JR, Barbosa AJ, Lima GF, Jr., et al. Helicobacter pylori and gastric histamine concentrations. J Clin Pathol 1991; 44:612-3; PMID:1856297; http://dx.doi.org/10.1136/jcp.44.7.612.
- Bechi P, Romagnoli P, Panula P, Dei R, Bacci S, Amorosi A, Masini E. Gastric mucosal histamine storing cells. Evidence for different roles of mast cells and enterochromaffin-like cells in humans. Dig Dis Sci 1995; 40:2207-13; PMID:7587791; http://dx.doi.org/10.1007/BF02209008.
- Kalia N, Bardhan KD, Reed MW, Jacob S, Brown NJ. Mechanisms of Helicobacter pylori-induced rat gastric mucosal microcirculatory disturbances in vivo. Dig Dis Sci 2000; 45:763-72; PMID:10759248; http://dx.doi.org/10.1023/A:1005456029396.
- Klausz G, Buzas E, Scharek P, Tiszlavicz L, Gyulai Z, Fulop AK, Falus A, Mándi Y. Effects of Helicobacter pylori infection on gastric inflammation and local cytokine production in histamine-deficient (histidine decarboxylase knock-out) mice. Immunol Lett 2004; 94:223-8; PMID:15275970; http://dx.doi.org/10.1016/j.imlet.2004.05.005.
- Kirshenbaum AS, Akin C, Wu Y, Rottem M, Goff JP, Beaven MA, Rao VK, Metcalfe DD. Characterization of novel stem cell factor responsive human mast cell lines LAD 1 and 2 established from a patient with mast cell sarcoma/leukemia; activation following aggregation of FcepsilonRI or FcgammaRI. Leuk Res 2003; 27:677-82; PMID:12801524; http://dx.doi.org/10.1016/S0145-2126(02)00343-0.
- Yang FL, Hassanbhai AM, Chen HY, Huang ZY, Lin TL, Wu SH, Ho B. Proteomannans in biofilm of Helicobacter pylori ATCC 43504. Helicobacter 2011; 16:89-98; PMID:21435085; http://dx.doi.org/10.1111/j.1523-5378.2010.00815.x.
- Shih KS, Lin CC, Hung HF, Yang YC, Wang CA, Jeng KC, Fu HW. One-step chromatographic purification of Helicobacter pylori neutrophil-activating protein expressed in Bacillus subtilis. PloS One 2013; 8:e60786; PMID:23577158; http://dx.doi.org/10.1371/journal.pone.0060786.