
ABSTRACT
Macrophages are a permissive niche for E. coli K1 multiplication for which the interaction of the bacterial outer membrane protein A and its cognate receptor CD64 are critical. Using in vitro immunofluorescence and live microscopy with ex vivo macrophage cultures from RFP-Lifeact mice, we show that cytotoxic necrotizing factor 1 (CNF1) secreted by E. coli K1 sequesters cellular actin toward microspike formation, thereby limiting actin availability for OmpA-mediated bacterial invasion. Surprisingly, the observed effects of CNF1 occur despite the absence of 67-kDa laminin receptor in macrophages. Concomitantly, the CNF1 deletion mutant of E. coli K1 (Δcnf1) invades macrophages and the brains of newborn mice in greater numbers compared to wild-type. However, the Δcnf1 strain induces less severe pathology in the brain. These results suggest a novel role for CNF1 in limiting E. coli K1 entry into macrophages while exacerbating disease severity in the brains of newborn mice.
Introduction
Neonatal bacterial meningitis still causes significant morbidity and mortality despite the use of effective antibiotics and supportive care. E. coli K1 is the second most common bacteria that cause meningitis in newborns (0 to 1 month of age).Citation1-3 Half of the survivors suffer from neurological deficits such as learning disabilities, mental retardation and hearing impairment.Citation4 If left untreated, the mortality rates due to E. coli K1 meningitis reach 100% in these infected neonates.Citation5,6 This poor outcome is due to the incomplete understanding of the pathophysiology of the disease, emphasizing the need to develop novel preventative strategies. Several virulence factors responsible for the onset of meningitis have been reported, including the outer membrane protein A (OmpA) and cytotoxic necrotizing factor 1 (CNF1).Citation7-9 Lack of OmpA expression or mutation of 3 or 4 amino acids at a time in the extracellular loops effectively prevents meningitis despite the expression of other virulence factors.Citation10-12 E. coli K1 also survives in both neutrophils and macrophages in an OmpA-dependent manner for multiplication and to reach a threshold level of bacteremia required to cross the blood-brain barrier.Citation13,14 Importantly, lack of either neutrophils or macrophages in newborn mice prevents the onset of meningitis, suggesting that these 2 immune cells are critical for initial E. coli K1 replication.Citation15-17 Our studies have shown that OmpA interacts with distinct N-glycosylation sites on the extracellular domains of CD64 in macrophages, and not with the IgG-binding domain for internalization.Citation11 Concurrently, adoptive transfer of bone marrow-derived macrophages (BMDMs) expressing N-glycosylation mutant CD64 into CD64−/− mouse pups prevents E. coli K1 from causing meningitis in these animals; the introduction of full-length CD64 into CD64−/− BMDMs followed by adoptive transfer into CD64−/− mouse pups renders them susceptible to E. coli K1-induced meningitis.Citation12 Therefore, E. coli K1 entry into macrophages requires the interaction of OmpA with CD64, and is independent of the phagocytosis mechanism in macrophages, although dependent on microfilament and microtubule dynamics.Citation13,18 Hence, we have addressed this bacterial-mediated entry process as “invasion.”
Pathogenic E. coli strains expressing CNF1 are known to cause meningitis and urinary tract infections.Citation7,19 CNF1 acts on several eukaryotic cell types by activating small GTP-binding proteins, such as RhoA, Rac1, and Cdc42, which eventually modulates actin-myosin cytoskeleton.Citation20-22 Previous studies have demonstrated that human brain microvascular endothelial cells (HBMEC), an in vitro blood-brain barrier model, treated with CNF1 are highly susceptible to E. coli K1 invasion in a RhoA-dependent mechanism. Lack of CNF1 in E. coli K1 significantly inhibits its invasion in HBMEC and the onset of meningitis in a newborn rat model.Citation23 The CNF1-mediated invasion of E. coli in HBMEC is due to the ability of CNF1 to interact with 67-kDa laminin receptor (67LR). The interaction of E. coli with HBMEC increases the expression of 67LR in HBMEC, but not by E. coli K1 that lacks the cnf1 gene.Citation24 Notably, polymorphonuclear leukocytes pre-treated with CNF1 shows a significant reduction in the phagocytosis of E. coli.Citation25 However, the precise role of CNF1 in the modulation of actin dynamics and the entry of E. coli K1 in macrophages is not currently known. Therefore, in the present study, we sought to examine the effects of the lack of CNF1 expression in E. coli K1 invasion of macrophages and the onset of meningitis in a newborn mouse model of meningitis.
Results
CNF1 expression inhibits E. coli K1 invasion of macrophages
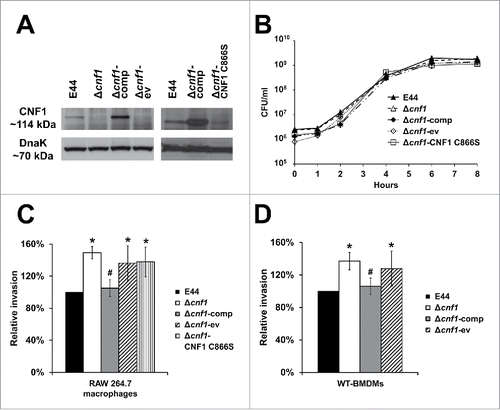
To determine the role played by CNF1 in E. coli K1 internalization of macrophages, we have used bacterial strains with or without the cnf1 gene in macrophage invasion assays. Prior to infection studies, the expression of CNF1 in these E. coli K1 strains was examined by Western blotting of the bacterial lysates with the anti-CNF1 antibody. Lysates from the E. coli K1 Δcnf1 mutant (Δcnf1) and the empty vector control (Δcnf1-ev) showed no expression of the CNF1 protein (). CNF1 expression was observed in lysates from the wild-type E. coli K1 strain E44, the complemented strain (Δcnf1-comp), and the Δcnf1 mutant transformed with a plasmid containing the cnf1 gene with a point mutation which abrogates the deamidase activity of CNF1 (Δcnf1-CNF1 C866S), thereby producing a functionally inactive CNF1 protein. Bacterial DnaK protein showed equal loading in all lanes. Furthermore, deletion of the cnf1 gene or complementation of the full-length CNF1 or CNF1 C866S did not affect the growth patterns compared to E44 (). These strains were used for invasion assays on RAW 264.7 macrophages and bone marrow-derived macrophages from C57BL/6 mice (WT-BMDMs). The graphs for invasion represent the data as a relative percentage compared to E44 taken as 100%. The observed CFU counts for invasion assays ranged between 4.2 × 104 and 1.52 × 105 CFU/ml. The results showed that Δcnf1 exhibited significantly increased entry into RAW 267.4 macrophages compared to E44 (). The Δcnf1-comp strain partially restored the wild-type phenotype, which was significantly less than the invasion levels of Δcnf1-ev. Moreover, the Δcnf1-CNF1 C866S strain invades similarly to Δcnf1-ev. This pattern of increased internalization of bacterial strains lacking CNF1 expression was also observed in WT-BMDMs, in which the Δcnf1 invaded to a higher degree than E44 (). Correspondingly, the invasion of Δcnf1-comp was similar to E44, and the Δcnf1-ev was similar to the Δcnf1. Since the invasion of the Δcnf1-ev and Δcnf1-CNF1 C866S strains was comparable to the Δcnf1 strain, we proceeded only with the Δcnf1 and Δcnf1-comp strains for subsequent experiments. Taken together, these results indicate that the presence of CNF1 in E. coli K1 exerts an inhibitory role in the invasion of both RAW 264.7 macrophages and WT-BMDMs.
Figure 1. CNF1 expression inhibits E. coli K1 invasion of macrophages. (A) Total bacterial lysates (60 µg) were subjected to Western blotting with the anti-CNF1 antibody. DnaK was used as a loading control. The panel contains 2 blots performed to verify CNF1 expression in various strains in 2 different experiments. (B) E. coli K1 strains were grown in LB at 37°C with shaking for 8 hours to log phase. Data are representative of 3 independent experiments, and error bars indicate standard deviation. (C) RAW 264.7 macrophages and (D) wild-type bone marrow-derived macrophages from C57BL/6 mice (WT-BMDMs) were used for gentamicin protection assays. Data represent relative invasion percentages compared to E44 taken as 100% and are averages of 3 independent experiments. Error bars indicate standard deviation. *p < 0.05 by Student's t-test compared to the wild-type strain E44; # p < 0.05 by Student's t-test for transformed strains compared to Δcnf1-ev strain.
E. coliK1 internalization induces a greater number of microspikes in macrophages compared to Δcnf1
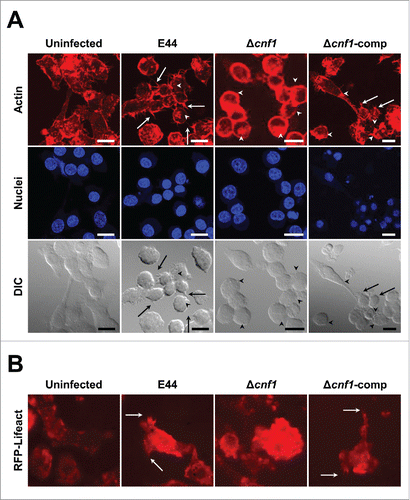
Since Δcnf1 entered macrophages with higher efficiency, it is possible that it induces prominent actin remodeling in these cells. We have previously demonstrated that E. coli K1 recruits actin filaments underneath the bacterial attachment sites.Citation13,26-29 Therefore, we treated RAW 264.7 macrophages with various E. coli K1 strains listed above and then stained for actin filaments with phalloidin. E44, as expected, induced actin filament remodeling in these cells beneath the bacterial attachment sites; however, E44 also triggered more microspikes that protruded from RAW 264.7 macrophages compared to uninfected cells (). Notably, Δcnf1 entered macrophages in higher numbers, and thus, showed more actin remodeling underneath the bacteria, but exhibited microspikes to a lesser degree than E44-infected macrophages. Infection with Δcnf1-comp revealed actin microspikes similar to those induced by E44. To analyze whether a similar phenotype is observed in primary macrophage cultures, we also infected BMDMs isolated from RFP-Lifeact mice (transgenic mice that express the Lifeact peptide fused to mRFPruby, which can specifically label F-actin without adversely affecting actin dynamicsCitation30) and analyzed the actin dynamics in real time with confocal live imaging. The microspike pattern or lack thereof in E44, Δcnf1, and Δcnf1-comp-infected RFP-Lifeact-BMDMs was similar to that seen in RAW 264.7 macrophages (). Videos of the actin dynamics are presented in Supplementary Figure S1. These results along with invasion data suggest that the CNF1 secreted by E44 into macrophages exert excessive actin redistribution, which partially results in the formation of microspikes, thereby inducing effective sequestration of actin and resulting in the unavailability of the actin required for the optimal invasion of E44.Citation13,26,28 In contrast, Δcnf1 could not induce microspikes due to lack of CNF1, and thus, all actin in the cells are available for efficient bacterial invasion compared to E44. This is a novel observation that CNF1, a bacterial virulence factor, plays a role in limiting the internalization of the bacteria into the host cell by modulating actin availability.
Figure 2. E. coli K1 expressing CNF1 induces microspikes in macrophages. (A) RAW 264.7 macrophages were infected with bacteria and stained for actin. The nucleus was stained with DAPI. Arrows indicate microspikes and arrowheads indicate bacterial attachment sites or actin accumulation. The brightness was adjusted to the same degree in all images to clearly show the microspikes. The number of cells with microspikes as a fraction of total cells in these particular fields of view: Uninfected (2/12); E44 (10/15); Δcnf1 (4/14), Δcnf1-comp (8/13). Scale bars = 15 µm. (B) BMDMs isolated from RFP-Lifeact mice were infected as above and videos obtained. A screenshot of a single representative field of view is shown to demonstrate actin dynamics. Arrows indicate microspikes. The corresponding videos are provided in Supplementary Figure S1.
E. coli K1 requires CD64, and not 67LR, for the invasion of macrophages
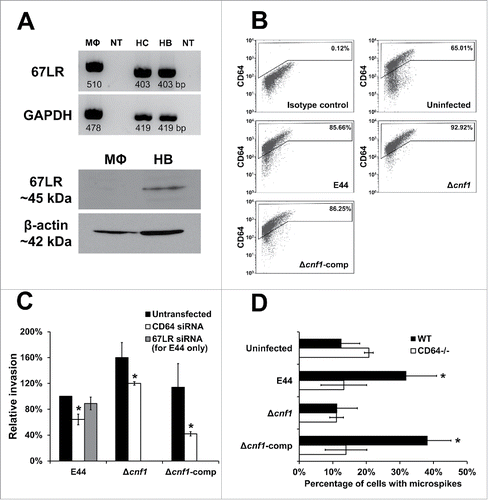
Previous reports indicate that CNF1 recognizes the 67-kDa laminin receptor (67LR) in human brain microvascular endothelial cells (HBMEC) and aids in the invasion of E. coli K1.Citation24 To determine if E44 also requires 67LR in RAW 264.7 macrophages, we examined the presence of 67LR at the mRNA level by RT-PCR. The transcript for 67LR is present in RAW 264.7 macrophages, as well as in the human cerebral endothelial cell/D3 line (HCMEC/D3) and primary HBMEC, which were used as positive controls (). However, Western blot analysis also showed monomeric subunits of the 67LR protein (∼45 kDa) only in HBMEC, but not in RAW 264.7 macrophages, indicating that RAW 264.7 macrophages do not translate the 67LR mRNA for corresponding protein expression. Previous studies have demonstrated that E. coli K1 interaction with RAW 264.7 macrophages increases the expression of CD64, which is used by the bacteria as a receptor to invade the cells.Citation15 Therefore, we also examined whether Δcnf1 and Δcnf1-comp infection also increase CD64 expression in RAW 264.7 macrophages by flow cytometry. Infection with these E. coli K1 strains increased the number of macrophages expressing CD64 compared to uninfected cells (). In addition, invasion of the E. coli K1 strains in RAW 264.7 macrophages transfected with siRNA to CD64 showed a significant decrease in intracellular bacteria compared to untransfected cells (). In contrast, E44 invasion of 67LR siRNA-transfected RAW 264.7 macrophages was not affected. This supports the previous observation that the 67LR protein is not expressed in RAW 264.7 macrophages, thereby precluding further analysis of the role of 67LR in the invasion of the other bacterial strains. Invasion assays performed in BMDMs isolated from CD64−/− mice also yielded results similar to those observed with RAW 264.7 macrophages in the presence of CD64 siRNA (data not shown). To determine whether the CD64 expression is critical for E. coli K1-induced microspikes in macrophages, WT-BMDMs and CD64−/− BMDMs were infected with various E. coli K1 strains and stained for actin filaments. Blinded counting of the samples without designations was performed, and the number of microspikes was expressed as a percentage of cells with microspikes in a given field. The number of microspikes in CD64−/− BMDMs was similar to the number observed in WT-BMDMs, while E44 infection increased the number of microspikes in WT-BMDMs by 20% (). In contrast, Δcnf1 did not induce microspikes in WT-BMDMs or CD64−/− BMDMs. WT-BMDMs infected with Δcnf1-comp induced microspikes similar to that of E44, which were absent in CD64−/− BMDMs. These results exemplify the role of CD64 as the predominant receptor in the invasion of E. coli K1 in macrophages, even in the absence of CNF1.
Figure 3. E. coli K1 requires CD64, and not 67LR, for the invasion of macrophages. (A) Total RNA extracted from murine RAW 264.7 macrophages (MΦ), human cerebral microvascular endothelial cells/D3 (HC), and primary human brain microvascular endothelial cells (HB) were used for RT-PCR using primers specific to murine or human templates. NT indicates no template control. Total cell lysates (25 µg) were subjected to Western blotting with the anti-67LR antibody. β-actin was used as a loading control. (B) RAW 264.7 macrophages were infected with bacteria and subjected to flow cytometry for CD64 expression. Staining with secondary antibody alone was used as an isotype control. Similar patterns of CD64 expression was observed in 3 independent experiments. (C) RAW 264.7 macrophages were transfected with siRNA and then used for invasion assays. Data represent relative invasion percentages compared to untransfected E44 taken as 100% and are averages of 3 independent experiments. Error bars indicate standard deviation. * p < 0.05 by Student's t-test compared to the untransfected controls. (D) BMDMs isolated from wild-type or CD64−/− mice were infected with bacteria and stained for actin. The number of microspikes were counted in a blinded fashion by analyzing Z-stacks and expressed as a percentage of cells with microspikes. Data are averages of 3 independent fields of view, and error bars indicate standard deviation. * p < 0.05 by Student's t-test compared to uninfected cells.
Microspike formation mediated by Rho GTPases modulates bacterial invasion in RAW 264.7 macrophages
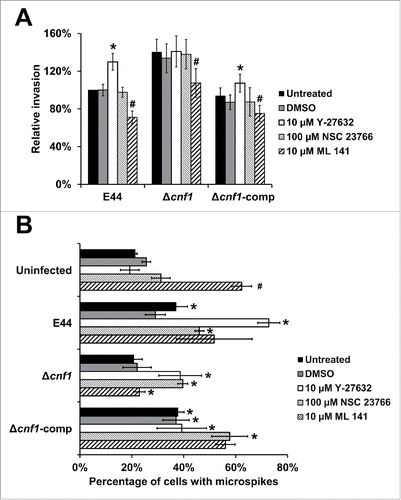
CNF1 constitutively activates Rho GTPases, such as RhoA, Rac1, and Cdc42, by the deamidation of glutamine residues and inhibition of the GTP-hydrolyzing activity, promoting actin polymerization.Citation20-22 To examine the specific role of Rho GTPases in macrophages infected with E. coli K1 strains, we pre-treated the cells with Y-27632 (inhibitor of Rho-associated protein kinase p160ROCK), NSC 23766 (Rac1 inhibitor), and ML 141 (Cdc42 inhibitor).Citation23,31-33 Pre-treatment with Y-27632 increased the invasion of E44 and Δcnf1-comp compared to untreated macrophages (). However, Y-27632 pre-treatment of macrophages did not affect the invasiveness of the Δcnf1 strain. NSC 23766 produced no discernible differences in bacterial invasion, whereas ML 141 in DMSO resulted in a decrease of invasion, regardless of CNF1 expression. DMSO alone did not affect the observed patterns. These results show that Cdc42 activation may play a role in bacterial invasion, while Rac1 does not participate in the invasion process. Furthermore, RhoA activation in the presence of CNF1 expression may affect the ability of CNF1 to limit bacterial invasion. Therefore, it is possible that CNF1 secretion by E44 in the presence of Y-27632 (absence of RhoA activation) increases activation of both Rac1 and Cdc42, of which excess activation of Cdc42 is responsible for invasion. In the case of Δcnf1, the activation of Cdc42 is mediated by the aforementioned OmpA-CD64 interaction, thereby demonstrating similar invasion patterns between untreated and Y-27632-treated cells. Rac1 and Cdc42 have been previously shown to facilitate microspike formation.Citation34 Therefore, we speculated that the reduced bacterial invasion in the presence of Cdc42 inhibitor may correspond with decreased microspike formation in RAW 264.7 macrophages. To verify this, RAW 264.7 macrophages were pre-treated with inhibitors, infected with bacteria, fixed, and stained for actin filaments. All bacterial strains induced microspikes in RAW 264.7 macrophages similar to their respective levels in . Infection of E44, Δcnf1, and Δcnf1-comp in macrophages pre-treated with Y-27632 or NSC 23766 resulted in increased microspike formation compared to untreated cells. Interestingly, pre-treatment of RAW 264.7 macrophages with ML 141 in DMSO in the absence of bacteria resulted in a significant increase in microspikes compared to the DMSO control, suggesting that Rac1 could be responsible for microspikes in these cells.Citation34 Most strikingly, infection with the highly invasive Δcnf1 strain yielded a significant decrease in microspikes (). This strongly suggests that while ML 141 induced microspike formation, the Δcnf1 strain was able to repossesses actin for its invasion, resulting in the commensurate decrease in microspikes. This shows that microspike formation in the presence of an inhibitor to a given Rho GTPase may be influenced by the overcompensation of another Rho GTPase. In addition, the degree of Rho GTPase activation may vary in the presence or absence of CNF1 independent of the activation mediated by bacterial invasion. This complex interplay between Rho GTPase-driven microspike formation in the presence of different stimuli warrants further study.
Figure 4. Rho GTPases modulate E. coli K1 entry of macrophages. (A) RAW 264.7 macrophages were pre-treated with 10 µM Y-27632 in water, 100 µM NSC 23766 in water, or 10 µM ML 141 in DMSO and used for invasion assays. Data represent relative invasion percentages compared to untreated E44 taken as 100% and are averages of 3 independent experiments. Error bars indicate standard deviation. *p < 0.05 by Student's t-test compared to the untreated cells; # p < 0.05 by Student's t-test compared to the DMSO-treated cells. (B) RAW 264.7 macrophages pre-treated with inhibitors and stained for actin. The number of microspikes was counted in a blinded fashion by analyzing Z-stacks and expressed as a percentage of cells with microspikes. Data are averages of 3 independent fields of view. Error bars indicate standard deviation. *p < 0.05 by Student's t-test compared to uninfected cells; # p < 0.05 by Student's t-test compared to the uninfected, DMSO-treated cells.
CNF1 expression is required by E. coli K1 to induce gross pathology in the newborn mouse model of meningitis
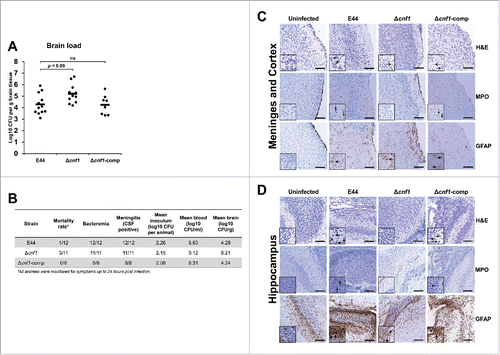
Using a well-established mouse model of meningitis, we sought to determine the virulence traits of Δcnf1.Citation18 The E. coli K1 strains were injected into 3-day-old pups via intraperitoneal route, and the bacterial load and cytokines levels in the blood, cerebrospinal fluid (CSF), and brain were evaluated up to 24 hours post-infection. All infected animals displayed similar levels of bacteremia; no statistically significant differences by ANOVA (). Furthermore, serum cytokine analysis revealed that Δcnf1 induced greater amounts of pro-inflammatory cytokine IL-1β compared to E44 (). There was no difference in IL-6 levels (). There was a slight, but not statistically significant increase of TNFα between Δcnf1 and E44 (), and no detectable levels of pro-inflammatory cytokines IFNγ or IL-12p70 or anti-inflammatory cytokines IL-4, IL-5, or IL-10 were observed (Supplementary Fig. S2). Furthermore, consistent with the in vitro and ex vivo invasion data, the Δcnf1 was present at significantly higher levels in the brain when compared to E44 by ANOVA (). All animals were positive for bacteria in the CSF, indicative of meningitis (). Histological analyses of sagittal brain sections of E44-infected or Δcnf1-comp-infected mice showed increased neuronal cell death and elevated neutrophil and glial cell infiltration in the hippocampus compared to uninfected brains. Interestingly, the Δcnf1-infected brain, despite the presence of high bacterial load, showed less pathological features (). These results suggest that lack of CNF1 expression might have reduced the pathological state in the brain. Nonetheless, the animals exhibited significant bacterial load in the blood and brain, and greater amounts of IL-1β.
Figure 5. Bacterial load and cytokine profile in the blood of neonatal mice infected with E. coli K1 strains. (A) Blood samples from infected mouse pups were collected 24 hours post-infection and examined for the presence of bacteria. Statistical analyses were performed by Student's t-test and ANOVA; ns = not significant. Serum samples were used for ELISA analysis for (B) IL-1β, (C) IL-6, and (D) TNFα. Statistical analyses were performed by Student's t-test and ANOVA; p-values indicated; ns = not significant.
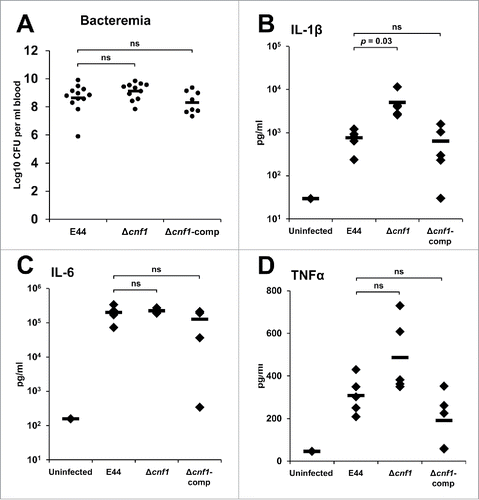
Figure 6. Lack of CNF1 expression in E. coli K1 causes meningitis in newborn mice with mild pathological features in the brain. (A) Brains from infected mouse pups were collected 24 hours post-infection and examined for the presence of bacteria. Statistical analyses were performed by Student's t-test and ANOVA; p-values indicated; ns = not significant. (B) CSF samples were collected and examined for the presence of bacteria. Positive bacterial growth in CSF was indicative of meningitis. Brain sections of the (C) meninges and cortex or (D) hippocampus were stained with H&E (open arrows indicate neuronal cell death), MPO (closed arrows indicate neutrophil infiltration), and GFAP (spear arrows indicate glial cell infiltration). Scale bars = 100 µm.
Discussion
Several bacterial virulence factors have been shown to be responsible for the pathogenesis of meningitis induced by E. coli K1, which include OmpA and CNF1.Citation7,9 Except OmpA, the influence of other factors on evasion of immune cell defense is not known. In this study, we show that CNF1 expression in E. coli K1 exhibits a negative effect on the entry of the bacterium in macrophages. The Δcnf1 strain invaded macrophages in greater numbers compared to E44 or Δcnf1-comp by inducing effective actin remodeling. These results are quite contrary to the invasion pattern of E. coli K1 in HBMEC in which lack of CNF1 expression prevented the bacterial entry.Citation23 CNF1 treatment of HBMEC induced RhoA activation for stress fibers formation, which enhanced the invasion of E. coli K1 into the cells.Citation20 E44-infected cells also showed a greater number of microspikes (protrusions) compared to uninfected cells. Macrophages infected with Δcnf1 displayed a lower amount of microspikes, which may be due to minimal Cdc42 activation.Citation35 Furthermore, pre-treatment of E44-infected macrophages with ROCK inhibitor Y-27632, which blocks RhoA activation, increased invasion as well as microspike formation. This suggests that in the absence of RhoA activation or CNF1 expression, the invasion process may be dependent on Cdc42-mediated actin rearrangement.
OmpA of E. coli K1 interacts with CD64 for binding to and invasion of macrophages, whereas CNF1 has been shown to bind 67LR to enter HBMEC.Citation24 However, RAW 264.7 macrophages showed no expression of 67LR at the protein level, although 67LR mRNA transcript was present. Thus, E. coli K1 utilizes CD64 for invasion in macrophages, since the knockdown of CD64 with siRNA significantly prevented bacterial invasion, despite the presence or absence of CNF1. In addition, our unpublished data revealed that the OmpA-CD64 interaction induces the expression of Filamin A, a protein that associates with and stabilizes the CD64 at the membrane. Filamin A is shown to utilize Rac1 for phagocytosis of apoptotic cells by macrophages.Citation36 Since E44 still invades in the absence of Cdc42, it is assumed that Rac1 activation mediated by FilaminA-CD64 interaction may be responsible for the invasion of macrophages. The exact roles of Rac1 and Cdc42 in the invasion and microspike formation need to be investigated further.
We have recently demonstrated that avian pathogenic and other neonatal meningitis causing E. coli strains cause extensive brain damage by inducing neutrophil infiltration and neuronal apoptosis, although they lack CNF1.Citation18 An interesting observation in the present study is that Δcnf1-infected mouse brains showed lower neuronal cell death, neutrophil infiltration, and glial cell migration, which are prominent in E44 and Δcnf1-comp-infected mice, despite infection with Δcnf1 resulting in higher brain loads. Therefore, it is interesting to speculate that the prototypical E. coli K1 strain E44 has a unique mechanism of using CNF1 to modulate its pathogenic process. These results are in contrast to the results obtained previously in E. coli K1 mutant strain lacking CNF1 expression (Δcnf1) did not cause meningitis.Citation23 These differences in the results could be due to the infection route (intraperitoneal versus subcutaneous), or the animal model used for the experiments (mice vs. rats). Based on our observations, though it was tempting to assume that CNF1 is not necessary for the onset of meningitis, E44 may secrete CNF1 to induce extensive inflammatory brain damage, which is a hallmark of E. coli K1 meningitis. Future studies are required to fully elucidate the role of CNF1 in the modulation of brain pathology induced by E. coli K1.
Materials and methods
Bacterial strains
Escherichia coli K1 strain E44, a spontaneous rifampicin-resistant mutant of strain RS218 (O18:K1:H7), was used as the parental strain for all experiments (Supplementary Table S1). A knockout deletion mutant of E. coli K1 was constructed by replacing the cnf1 gene with a kanamycin resistance cassette as described previously.Citation23 The cnf1 deletion mutant strain of E. coli K1 (referred as Δcnf1) was complemented with a plasmid expressing the cnf1 gene from E44 on a pACYC184 vector constructed using the In-Fusion HD Cloning Kit (Clontech Laboratories, Cat. No. 638909) as per the manufacturer's instructions and the primer sequences are shown in Supplementary Table S2. The Δcnf1 strain was also complemented with a plasmid expressing the catalytically inactive CNF1 C866S on a pQE-30 vector (Dr. Emilie Andrio, Dr. Patrick Munro, and Dr. Emmanuel Lemichez, INSERM U1065, C3M); this strain was induced with 200 µM IPTG for overnight incubation.Citation37
Western blots
For Western blots of bacterial protein, 60 µg of whole cell bacterial lysate was run on SDS-PAGE and subjected to immunoblotting with goat anti-CNF1 antibody (Dr. Alison O'Brien, Uniformed Services University of the Health Sciences). Mouse anti-DnaK monoclonal antibody (8E2/2) (Enzo Life Sciences, Cat. No. ADI-SPA-880-D) was used to probe for DnaK as a loading control. For Western blots of eukaryotic protein, 25 µg of whole cell lysates of RAW 264.7 macrophages (American Type Culture Collection, Cat. No. TIB-71) or primary HBMEC Citation11,26,38 were run on SDS-PAGE and subjected to immunoblotting with anti-67LR monoclonal IgM (MLuC5) (Thermo Fisher Scientific, Cat. No. MA5-12840). Rabbit polyclonal anti-β-actin antibody (GeneTex, Cat. No. GTX109639) was used to probe for β-actin as a loading control.
Invasion assays
RAW 264.7 macrophages were cultured to a density of 105 cells per well in 24-well plates. Bacteria were added at a multiplicity of infection (MOI) between 5–10 for 1 hour at 37°C with 5% CO2. The cells were further incubated with medium containing 50 µg gentamicin for 1 hour. The cells were lysed with 0.5% Triton X-100, and then intracellular bacteria recovered were plated on blood agar. Percentage invasion was determined by comparing each strain's recovered bacteria to the input. BMDMs were isolated from femurs of adult C57BL/6 mice (The Jackson Laboratory and Harlan Laboratories), and seeded in 24-well plates with growth media supplemented with 20% fetal bovine serum, 10 ng/ml macrophage colony-stimulating factor (M-CSF) (Peprotech, Cat. No. 315–02), and 1:1,000 Bac-Off antibiotic solution (Cell Systems Corporation, Cat. No. 4Z0–643). Invasion assays with BMDMs were performed as described above except the infection time was 2 hours. For transfection experiments, RAW 264.7 macrophages were transfected with either CD64 siRNA (ID: s65915) or 67LR siRNA (ID: MSS243649) (Thermo Fisher Scientific) using the TransIT-X2 Dynamic Delivery System (Mirus, Cat. No. MIR 6003) for 24 hours prior to performing invasion assays or flow cytometry analysis. For inhibition studies, RAW 264.7 macrophages were pre-treated with 10 µM Y-27632 in water (BioVision, Cat. No. 1596–1), 100 µM NSC 23766 in water (Tocris, Cat. No. 2161), or 10 µM ML 141 in DMSO (Sigma, Cat. No. SML0407) for 1 hour prior to performing invasion assays or staining.
Confocal microscopy
RAW 264.7 macrophages or BMDMs were grown to a density of 4 × 103 cells per well in 8-well slides. Bacteria were added at an MOI of 250 for 15 minutes at 37°C with 5% CO2, washed, fixed, permeabilized, and stained for actin using Alexa Fluor 568 Phalloidin (Thermo Fisher Scientific, Cat. No. A12380). The nucleus was stained with DAPI. Images with Z-stacks were obtained on a Zeiss LSM 700 confocal microscope (Carl Zeiss Microscopy) controlled by Micro-Manager software at 40× with multiple fields of view.Citation39
RT-PCR
RNA was extracted from RAW 264.7 macrophages, human cerebral microvascular endothelial cells/D3 cell line (HCMEC/D3),Citation18 and primary human brain microvascular endothelial cells (HBMEC)Citation11,26,38 using the Quick-RNA MiniPrep Kit (Zymo Research Corporation, Cat. No. R1054) and quantified on a NanoDrop 1000 spectrophotometer. RT-PCR was performed with 20 ng total RNA using the OneStep RT-PCR Kit (Qiagen, Cat. No. 210212). Primer sequences are shown in Supplementary Table S2.
Flow cytometry
RAW 264.7 macrophages were infected with bacteria at an MOI of 5–10 for 1 hour and subjected to flow cytometry analyses with rabbit anti-CD64 polyclonal IgG (H-250) (Santa Cruz Biotechnology, Cat. No. sc-15364) and goat anti-rabbit IgG Alexa Fluor 647 conjugate (Thermo Fisher Scientific, Cat. No. A-21244). Staining with secondary antibody alone was used as an isotype control. At least 10,000 cells per sample were analyzed with a BD FACS Calibur flow cytometer.
Mouse infections, brain histology, and ELISA
The animal studies were approved by the Institutional Animal Care and Use Committee of The Saban Research Institute at Children's Hospital Los Angeles (CHLA) (Protocol #59–14) and followed National Institutes of Health guidelines for the performance of animal experiments. Three-day-old mouse pups received an intraperitoneal inoculation of 100 colony forming units (CFU) of E. coli K1. The pups were monitored up to 24 hours post-infection for signs of illness. Pups were sacrificed, cerebrospinal fluid (CSF) was collected aseptically by cisternal puncture, 5 µl of blood was collected by cardiac puncture, and the brains were removed. CSF was directly inoculated into LB broth supplemented with the appropriate antibiotics, and incubated overnight at 37°C. The blood and brain bacterial load and histology of brain sections was performed as previously described.Citation18 ELISA was performed according to the ProcartaPlex Mouse Immunoassay Manual (eBioscience) using Mouse Th1/Th2 Essential 6Plex (Cat. No. EPX060-20831-SA), Mouse IL-10 Simplex (Cat. No. EPX010-20614-901), and Mouse IL-1β Simplex (Cat. No. EPX010-26002-901). Samples were analyzed on a Luminex-200 analyzer.
RFP-Lifeact live microscopy
BMDMs were isolated from femurs of adult RFP-Lifeact mice, and cultured as previously described.Citation18 Cells were cultured to a density of 4 × 103 cells per well. Bacteria were added at an MOI of 250, and actin dynamics were recorded at 37°C with 5% CO2 using the Axiovert 200 M microscope (Carl Zeiss Microscopy) with Orca-AG camera (Hamamatsu Photonics) and temperature/CO2 incubation system (PeCon GmbH) controlled by Micro-Manager software at 40× with multiple fields of view and multiple Z-stacks every 5 minutes for 1 hour.Citation26,39
Statistical analysis
The unpaired Student's t-test was used for statistical analysis of all assays. In addition, one-way ANOVA was used for statistical analysis of blood and brain bacterial counts from newborn mice and serum cytokine ELISA. P-values < 0.05 were considered statistically significant.
Abbreviations
| 67LR | = | 67-kDa laminin receptor |
| BMDMs | = | bone marrow-derived macrophages |
| CNF1 | = | cytotoxic necrotizing factor 1 |
| HBMEC | = | human brain microvascular endothelial cells |
| HCMEC/D3 | = | human cerebral endothelial cell/D3 line |
| OmpA | = | outer membrane protein A |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KVIR_S_1192730.zip
Download Zip (6.8 MB)Acknowledgments
We sincerely thank G. Esteban Fernandez for assistance with confocal imaging. The plasmid pQE-30 CNF1C866S was a kind gift from Dr. Emilie Andrio, Dr. Patrick Munro, and Dr. Emmanuel Lemichez, INSERM U1065, C3M. The CNF1 antibody was a kind gift from Dr. Alison O'Brien, Uniformed Services University of the Health Sciences. The adult RFP-Lifeact mice were a kind gift from Dr. David Warburton, CHLA. We also thank Manisha Sajnani, Somtochukwu Uzoegwu, Amber Rock, and Rachel Trujillo for their valuable assistance.
Funding
This work was supported by funds from NICHD (NS73115) and NIAID (AI40567) to NVP.
References
- Mellata M. Human and avian extraintestinal pathogenic Escherichia coli: infections, zoonotic risks, and antibiotic resistance trends. Foodborne Pathog Dis 2013; 10:916-32; PMID:23962019; http://dx.doi.org/10.1089/fpd.2013.1533
- Bonacorsi S, Bingen E. Molecular epidemiology of Escherichia coli causing neonatal meningitis. Int J Med Microbiol 2005; 295:373-81; PMID:16238014; http://dx.doi.org/10.1016/j.ijmm.2005.07.011
- Barichello T, Fagundes GD, Generoso JS, Elias SG, Simoes LR, Teixeira AL. Pathophysiology of neonatal acute bacterial meningitis. J Med Microbiol 2013; 62:1781-9; PMID:23946474; http://dx.doi.org/10.1099/jmm.0.059840-0
- Chu SM, Hsu JF, Lee CW, Lien R, Huang HR, Chiang MC, Fu RH, Tsai MH. Neurological complications after neonatal bacteremia: the clinical characteristics, risk factors, and outcomes. PLoS One 2014; 9:e105294; PMID:25364821; http://dx.doi.org/10.1371/journal.pone.0105294
- Gaschignard J, Levy C, Romain O, Cohen R, Bingen E, Aujard Y, Boileau P. Neonatal Bacterial Meningitis: 444 Cases in 7 Years. Pediatr Infect Dis J 2011; 30:212-7; PMID:21416693; http://dx.doi.org/10.1097/INF.0b013e3181fab1e7
- Furyk JS, Swann O, Molyneux E. Systematic review: neonatal meningitis in the developing world. Trop Med Int Health 2011; 16:672-9; PMID:21395927; http://dx.doi.org/10.1111/j.1365-3156.2011.02750.x
- Kim KS. Current concepts on the pathogenesis of Escherichia coli meningitis: implications for therapy and prevention. Curr Opin Infect Dis 2012; 25:273-8; PMID:22395761; http://dx.doi.org/10.1097/QCO.0b013e3283521eb0
- Krishnan S, Prasadarao NV. Outer membrane protein A and OprF: versatile roles in Gram-negative bacterial infections. FEBS J 2012; 279:919-31; PMID:22240162; http://dx.doi.org/10.1111/j.1742-4658.2012.08482.x
- Xie Y, Kim KJ, Kim KS. Current concepts on Escherichia coli K1 translocation of the blood-brain barrier. FEMS Immunol Med Microbiol 2004; 42:271-9; PMID:15477040; http://dx.doi.org/10.1016/j.femsim.2004.09.001
- Prasadarao NV, Wass CA, Weiser JN, Stins MF, Huang SH, Kim KS. Outer membrane protein A of Escherichia coli contributes to invasion of brain microvascular endothelial cells. Infect Immun 1996; 64:146-53; PMID:8557332
- Krishnan S, Prasadarao NV. Identification of minimum carbohydrate moiety in N-glycosylation sites of brain endothelial cell glycoprotein 96 for interaction with Escherichia coli K1 outer membrane protein A. Microbes Infect 2014; 16:540-52; PMID:24932957; http://dx.doi.org/10.1016/j.micinf.2014.06.002
- Krishnan S, Liu F, Abrol R, Hodges J, Goddard WA, 3rd, Prasadarao NV. The interaction of N-glycans in Fcgamma receptor I α-chain with Escherichia coli K1 outer membrane protein A for entry into macrophages: experimental and computational analysis. J Biol Chem 2014; 289:30937-49; PMID:25231998; http://dx.doi.org/10.1074/jbc.M114.599407
- Sukumaran SK, Shimada H, Prasadarao NV. Entry and intracellular replication of Escherichia coli K1 in macrophages require expression of outer membrane protein A. Infect Immun 2003; 71:5951-61; PMID:14500515; http://dx.doi.org/10.1128/IAI.71.10.5951-5961.2003
- Sukumaran SK, Selvaraj SK, Prasadarao NV. Inhibition of apoptosis by Escherichia coli K1 is accompanied by increased expression of BclXL and blockade of mitochondrial cytochrome c release in macrophages. Infect Immun 2004; 72:6012-22; PMID:15385505; http://dx.doi.org/10.1128/IAI.72.10.6012-6022.2004
- Mittal R, Sukumaran SK, Selvaraj SK, Wooster DG, Babu MM, Schreiber AD, Verbeek JS, Prasadarao NV. Fcgamma receptor I α chain (CD64) expression in macrophages is critical for the onset of meningitis by Escherichia coli K1. PLoS Pathog 2010; 6:e1001203; PMID:21124939; http://dx.doi.org/10.1371/journal.ppat.1001203
- Mittal R, Prasadarao NV. gp96 expression in neutrophils is critical for the onset of Escherichia coli K1 (RS218) meningitis. Nat Commun 2011; 2:552; PMID:22109526; http://dx.doi.org/10.1038/ncomms1554
- Prasadarao NV. Identification of Escherichia coli outer membrane protein A receptor on human brain microvascular endothelial cells. Infect Immun 2002; 70:4556-63; PMID:12117968; http://dx.doi.org/10.1128/IAI.70.8.4556-4563.2002
- Krishnan S, Chang AC, Hodges J, Couraud PO, Romero IA, Weksler B, Nicholson BA, Nolan LK, Prasadarao NV. Serotype O18 avian pathogenic and neonatal meningitis Escherichia coli strains employ similar pathogenic strategies for the onset of meningitis. Virulence 2015; 6:1-10; PMID:26407066; http://dx.doi.org/10.1080/21505594.2015.1091914
- Garcia TA, Ventura CL, Smith MA, Merrell DS, O'Brien AD. Cytotoxic necrotizing factor 1 and hemolysin from uropathogenic Escherichia coli elicit different host responses in the murine bladder. Infect Immun 2013; 81:99-109; PMID:23090961; http://dx.doi.org/10.1128/IAI.00605-12
- Yu H, Kim KS. YgfZ contributes to secretion of cytotoxic necrotizing factor 1 into outer-membrane vesicles in Escherichia coli. Microbiology 2012; 158:612-21; PMID:22174383; http://dx.doi.org/10.1099/mic.0.054122-0
- Chung JW, Hong SJ, Kim KJ, Goti D, Stins MF, Shin S, Dawson VL, Dawson TM, Kim KS. 37-kDa laminin receptor precursor modulates cytotoxic necrotizing factor 1-mediated RhoA activation and bacterial uptake. J Biol Chem 2003; 278:16857-62; PMID:12615923; http://dx.doi.org/10.1074/jbc.M301028200
- Grande KK, Meysick KC, Rasmussen SB, O'Brien AD. Cytotoxic necrotizing factor type 1-neutralizing monoclonal antibody NG8 recognizes three amino acids in a C-terminal region of the toxin and reduces toxin binding to HEp-2 cells. Infect Immun 2009; 77:170-9; PMID:18955470; http://dx.doi.org/10.1128/IAI.00943-08
- Khan NA, Wang Y, Kim KJ, Chung JW, Wass CA, Kim KS. Cytotoxic necrotizing factor-1 contributes to Escherichia coli K1 invasion of the central nervous system. J Biol Chem 2002; 277:15607-12; PMID:11877402; http://dx.doi.org/10.1074/jbc.M112224200
- Kim KJ, Chung JW, Kim KS. 67-kDa laminin receptor promotes internalization of cytotoxic necrotizing factor 1-expressing Escherichia coli K1 into human brain microvascular endothelial cells. J Biol Chem 2005; 280:1360-8; PMID:15516338; http://dx.doi.org/10.1074/jbc.M410176200
- Hofman P, Le Negrate G, Mograbi B, Hofman V, Brest P, Alliana-Schmid A, Flatau G, Boquet P, Rossi B. Escherichia coli cytotoxic necrotizing factor-1 (CNF-1) increases the adherence to epithelia and the oxidative burst of human polymorphonuclear leukocytes but decreases bacteria phagocytosis. J Leukoc Biol 2000; 68:522-8; PMID:11037974
- Krishnan S, Fernandez GE, Sacks DB, Prasadarao NV. IQGAP1 mediates the disruption of adherens junctions to promote Escherichia coli K1 invasion of brain endothelial cells. Cell Microbiol 2012; 14:1415-33; PMID:22519731; http://dx.doi.org/10.1111/j.1462-5822.2012.01805.x
- Maruvada R, Argon Y, Prasadarao NV. Escherichia coli interaction with human brain microvascular endothelial cells induces signal transducer and activator of transcription 3 association with the C-terminal domain of Ec-gp96, the outer membrane protein A receptor for invasion. Cell Microbiol 2008; 10:2326-38; PMID:18662321; http://dx.doi.org/10.1111/j.1462-5822.2008.01214.x
- Rudrabhatla RS, Selvaraj SK, Prasadarao NV. Role of Rac1 in Escherichia coli K1 invasion of human brain microvascular endothelial cells. Microbes Infect 2006; 8:460-9; PMID:16243562; http://dx.doi.org/10.1016/j.micinf.2005.07.012
- Prasadarao NV, Wass CA, Stins MF, Shimada H, Kim KS. Outer membrane protein A-promoted actin condensation of brain microvascular endothelial cells is required for Escherichia coli invasion. Infect Immun 1999; 67:5775-83; PMID:10531228
- Riedl J, Flynn KC, Raducanu A, Gartner F, Beck G, Bosl M, Bradke F, Massberg S, Aszodi A, Sixt M, et al. Lifeact mice for studying F-actin dynamics. Nat Methods 2010; 7:168-9; PMID:20195247; http://dx.doi.org/10.1038/nmeth0310-168
- Pfeiffer ZA, Aga M, Prabhu U, Watters JJ, Hall DJ, Bertics PJ. The nucleotide receptor P2´7 mediates actin reorganization and membrane blebbing in RAW 264.7 macrophages via p38 MAP kinase and Rho. J Leukoc Biol 2004; 75:1173-82; PMID:15075366; http://dx.doi.org/10.1189/jlb.1203648
- Levay M, Krobert KA, Wittig K, Voigt N, Bermudez M, Wolber G, Dobrev D, Levy FO, Wieland T. NSC23766, a widely used inhibitor of Rac1 activation, additionally acts as a competitive antagonist at muscarinic acetylcholine receptors. J Pharmacol Exp Ther 2013; 347:69-79; PMID:23887096; http://dx.doi.org/10.1124/jpet.113.207266
- Hsu CR, Pan YJ, Liu JY, Chen CT, Lin TL, Wang JT. Klebsiella pneumoniae translocates across the intestinal epithelium via Rho GTPase- and phosphatidylinositol 3-kinase/Akt-dependent cell invasion. Infect Immun 2015; 83:769-79; PMID:25452552; http://dx.doi.org/10.1128/IAI.02345-14
- Adams JC, Schwartz MA. Stimulation of fascin spikes by thrombospondin-1 is mediated by the GTPases Rac and Cdc42. J Cell Biol 2000; 150:807-22; PMID:10953005; http://dx.doi.org/10.1083/jcb.150.4.807
- Ikeda W, Nakanishi H, Tanaka Y, Tachibana K, Takai Y. Cooperation of Cdc42 small G protein-activating and actin filament-binding activities of frabin in microspike formation. Oncogene 2001; 20:3457-63; PMID:11429692; http://dx.doi.org/10.1038/sj.onc.1204463
- Shifrin Y, Arora PD, Ohta Y, Calderwood DA, McCulloch CA. The role of FilGAP-filamin A interactions in mechanoprotection. Mol Biol Cell 2009; 20:1269-79; PMID:19144823; http://dx.doi.org/10.1091/mbc.E08-08-0872
- Diabate M, Munro P, Garcia E, Jacquel A, Michel G, Obba S, Goncalves D, Luci C, Marchetti S, Demon D, et al. Escherichia coli α-hemolysin counteracts the anti-virulence innate immune response triggered by the Rho GTPase activating toxin CNF1 during bacteremia. PLoS Pathog 2015; 11:e1004732; PMID:25781937; http://dx.doi.org/10.1371/journal.ppat.1004732
- Krishnan S, Chen S, Turcatel G, Arditi M, Prasadarao NV. Regulation of Toll-like receptor 2 interaction with Ecgp96 controls Escherichia coli K1 invasion of brain endothelial cells. Cell Microbiol 2013; 15:63-81; PMID:22963587; http://dx.doi.org/10.1111/cmi.12026
- Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, Stuurman N. Advanced methods of microscope control using muManager software. J Biol Methods 2014; 1:1-10; PMID:25606571; http://dx.doi.org/10.14440/jbm.2014.36