
ABSTRACT
FR-900098 is an inhibitor of 1-deoxy-d-xylulose-5-phosphate (DXP) reductoisomerase, the second enzyme in the non-mevalonate isoprenoid biosynthesis pathway. In previous studies, FR-900098 was shown to possess potent antimalarial activity in vitro and in a murine malaria model. In order to provide a basis for further preclinical and clinical development, we studied the acute toxicity and genotoxicity of FR-900098. We observed no acute toxicity in rats, i.e. there were no clinical signs of toxicity and no substance-related deaths after the administration of a single dose of 3000 mg/kg body weight orally or 400 mg/kg body weight intravenously. No mutagenic potential was detected in the Salmonella typhimurium reverse mutation assay (Ames test) or an in vitro mammalian cell gene mutation test using mouse lymphoma L5178Y/TK+/− cells (clone 3.7.2C), both with and without metabolic activation. In addition, FR-900098 demonstrated no clastogenic or aneugenic capability or significant adverse effects on blood formation in an in vivo micronucleus test with bone marrow erythrocytes from NMRI mice. We conclude that FR-900098 lacks acute toxicity and genotoxicity, supporting its further development as an antimalarial drug.
Introduction
Isoprenoid biosynthesis in Plasmodium falciparum, the causative agent of malignant tertian malaria, solely depends on the 1-deoxy-d-xylulose-5-phosphate (DXP) pathway, also known as the 2-C-methyl-d-erythritol-4-phosphate (MEP) pathway, whereas isoprenoids in humans are derived from the unrelated mevalonate pathway. The DXP pathway is used by most bacteria and is also found in the plastids of algae and higher plants. Likewise, DXP pathway enzymes in P. falciparum are located in a plastid-like organelle that is present in most parasites of the phylum Apicomplexa, and is therefore called the apicoplast.Citation1 DXP reductoisomerase, the second enzyme in the DXP pathway, is inhibited by the natural antimicrobial compound fosmidomycin and its close derivative FR-900098.Citation2,3 Both compounds display potent in vitro antimalarial activity, but FR-900098, which differs from fosmidomycin by the presence of a single additional methyl group (), inhibits the growth of cultured P. falciparum parasites with approximately twice the efficacy of fosmidomycin.Citation4 The activity of fosmidomycin and FR-900098 against 34 fresh clinical Cameroonian P. falciparum isolates was compared by Tahar and Basco.Citation5 The geometric mean IC50 values (95% confidence interval) were 301 nM (245–370 nM) for fosmidomycin and 118 nM (93.3–149 nM) for FR-900098. Furthermore, FR-900098 also displayed twice the activity of fosmidomycin in the P. vinckei mouse model, following intraperitoneal and oral administration. Citation4,6 The IC50 value of FR900098 for recombinant P. falciparum DXP reductoisomerase was 18 nM compared to 32 nM for fosmidomycin,Citation7 suggesting that the more potent activity of FR-900098 against malaria parasites results mainly from its higher affinity for the target enzyme. Indeed, the structural analysis of P. falciparum DXP reductoisomerase bound to FR-900098 revealed that the additional methyl group of FR-900098 forms a van der Waals contact with the side chain of a tryptophan residue, which could explain why FR-900098 is more active than fosmidomycin.Citation8
Figure 1. Chemical structures of fosmidomycin and FR-900098.
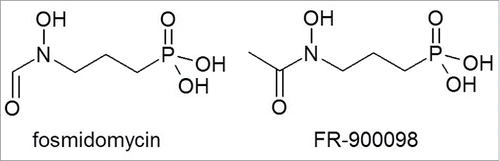
Before the discovery of the DXP pathway,Citation9,10 fosmidomycin and FR-900098 were isolated as natural antibacterial compounds from the culture broth of Streptomyces lavendulae and S. rubellomurinus, respectively.Citation11,12 Both compounds were found to be active against a number of clinically important Gram-negative bacteria,Citation13 but only fosmidomycin was developed further due to its superior antibacterial activity. In a phase I study of 127 healthy male volunteers, fosmidomycin was administered intravenously (i.v.) at a dose of 2 g every 6 h for 7 days, intramuscularly (i.m.) at a dose of 1 g every 6 h for 5 days, or orally (p.o.) at a dose of 1 g every 6 h for 7 d.Citation14,15 No adverse events were reported except for mild to moderate irritation at the site of injection in the i.v. and i.m. treatment groups. The strong antibacterial efficacy of fosmidomycin was confirmed in a pilot phase II trial of 70 patients with acute urinary tract infections although no details were published, and only minor adverse effects were reported including cases of nausea, vomiting and loose stools.Citation15 It is unclear why the clinical development of fosmidomycin as an antibacterial agent was discontinued at that time, but probable reasons include lower efficacy compared to other antibiotics in development, the lack of activity against streptococci and staphylococci, and the development of resistance.
Interest in fosmidomycin was renewed following the discovery of its molecular target and its potential use as an antimalarial drug. In clinical phase II studies, oral treatment with fosmidomycin led to the rapid reduction of parasitemia in patients with acute, uncomplicated P. falciparum malaria.Citation16,17 However, a high rate of recrudescent infections precludes the use of fosmidomycin as a monotherapy. Nevertheless, the combination of fosmidomycin with clindamycin emerged as a new potential antimalarial treatment, wherein the antimalarial activity of clindamycin is probably mediated by inhibiting the prokaryotic-like protein synthesis of the apicoplast. Clindamycin, if administered as a single agent, results in a peculiar delayed onset of parasite growth inhibition (sometimes referred to as delayed kill effect), making it unsuitable for monotherapy.Citation18 Three-day regimens with 2 doses per day of fosmidomycin (30 mg/kg body weight) and clindamycin (10 mg/kg body weight) resulted in 28-day cure rates of approximately 90% in Gabon and Thailand.Citation19-22 Cure rates of 100% were achieved with longer treatment durations (4 and 7 d in Gabon and Thailand, respectively).Citation19,23,24 Lower cure rates (62% and 45.9% after a 3-day regimen in 2 different studies) were observed in children younger than 3 y.Citation20,25 Because the relatively low efficacy in this group of patients probably reflects inadequate formulation, the authors of a recent meta-analysis advocate the further clinical development of fosmidomycin.Citation26 Currently, a combination of fosmidomycin with piperaquine is under investigation in a phase IIa proof-of-concept study in Lambaréné, Gabon (ClinicalTrials. gov Identifier: NCT02198807). In the case of FR-900098, the paucity of toxicological data currently hinders its clinical evaluation, despite demonstrably superior antimalarial activity in vitro and in mice. Here, we present preliminary studies concerning the toxicology of FR-900098 to promote its further development as an antimalarial drug.
Results and discussion
Acute oral and intravenous toxicity in rats
FR-900098 acute toxicity testing in Wistar (WU) rats was carried out as a limit test with single doses of 3000 mg/kg body weight p.o. and 400 mg/kg body weight i.v. No clinical signs of toxicity were observed in either group. Furthermore, FR-900098 did not affect body weight gain in either group during the 14-day observation period. Therefore, the LD50 (approximate lethal range) value of FR-900098 under the described conditions was > 3000 mg/kg body weight following oral administration and > 400 mg/kg body weight following intravenous administration. In a previous preliminary FR-900098 toxicity study in ICR mice, 5 animals received a single i.v. dose of 100 mg (4000–5000 mg/kg body weight). All mice survived without toxic symptoms during 14 d of observation after injection.Citation11 These data indicate that FR-900098 may also lack significant toxicity in humans at therapeutically relevant doses of 15–30 mg/kg body weight.
Salmonella typhimurium reverse mutation assay (Ames test)
The mutagenic potential of FR-900098 in bacteria was determined using the Ames test, which is part of the standard battery of genotoxicity tests for pharmaceuticals [ICH S2(R1)] and is therefore a regulatory requirement before novel drugs can be registered. The test is performed in different Salmonella typhimurium strains with mutations in genes involved in histidine synthesis.Citation27 Reverse mutation (his− → his+) in the presence of mutagens restores the ability of bacteria to grow on histidine-free substrates. The use of tailor-made tester strains that specifically test for frameshifts (TA 98, TA 1537) and substitutions (TA 100, TA 102, TA 1535) in the genes required for histidine synthesis allows the detection of mutagens with different modes of action.
A preliminary test with the S. typhimurium strain TA 100 was performed in order to examine the direct antibacterial activity of FR-900098 under the relevant assay conditions, employing a plate incorporation test without metabolic activation. The pretest was performed in duplicate with 10 concentrations ranging from 0.0316–1000 µg per plate. No revertant colonies appeared at 1000 and 316 µg per plate. At 100 µg per plate, the number of revertants was reduced (45 and 48 colonies on the 2 plates, respectively) compared to the solvent control (118 and 110 colonies, respectively). Therefore, we selected 100 µg per plate as the maximum concentration in the full series of tests. For the main study, 2 independent test designs were used (the plate incorporation and pre-incubation methods) and the experiments were each carried out with and without metabolic activation using rat liver S9-mix. In contrast to the preliminary test, there was no reduction of the number of revertant colonies at 100 µg per plate, which might reflect slight differences in the actual experimental conditions. Nevertheless, a scarce background lawn was indicative for the direct antibacterial activity of FR-900098 at this dose. The background lawn typically appearing in the Ames test results from limited growth of the non-revertant bacteria due to the trace of histidine added to the top agar.
At all dose levels, FR-900098 did not increase the number of revertant colonies in any of the 5 test strains when compared with the negative control plates treated solely with the solvent dimethylsulfoxide (DMSO) regardless of the test design and the presence or absence of S9-mix (; ). The positive controls with and without S9-mix resulted in the induction of revertant colonies in all 5 strains, indicating that the test worked correctly and that the S9-mix had sufficient activity. The inability of FR-900098 to induce the formation of revertant colonies therefore confirmed the lack of mutagenic activity of FR-900098 in the S. typhimurium test strains up to the concentration causing direct antibacterial activity.
Table 1. Salmonella typhimurium reverse mutation assay without metabolic activation.a
Table 2. Salmonella typhimurium reverse mutation assay with metabolic activation.a
In vitro mammalian cell gene mutation test in mouse lymphoma L5178Y/TK+/− cells
The genotoxicity of compounds can differ substantially depending on whether they are tested against bacteria or mammalian cells, so a bacterial mutagenicity assay (Ames test) must be complemented by an in vitro genotoxicity test using mammalian cells prior to the approval of new pharmaceuticals. The Ames test was thus complemented by an in vitro mouse lymphoma assay (MLA) using L5178Y/TK+/− cells, representing the second (mammalian-based) genotoxicity test in option 1 of the updated ICH S2(R1) guidance. This MLA is based on the quantification of forward mutations in the thymidine kinase (TK) locus induced by test substances.Citation28 L5178Y/TK+/− cells possess TK activity and are therefore sensitive to the cytotoxic effects of the nucleoside analog trifluorothymidine (TFT), which substitutes for thymidine in the salvage pathway. However, TK-deficient cells generated by the forward mutation TK+/− → TK−/− are resistant to TFT and continue to grow, because thymidine can also be synthesized de novo.
The cytotoxicity of FR-900098 toward L5178Y/TK+/− cells was evaluated in a pre-test in the presence and absence of S9-mix, using a broad range of concentrations (0, 3.2, 8, 20, 50, 125, 250, 500, 1000 and 2200 µg/ml) up to the limit concentration of 10 mM for nontoxic compounds set out in OECD Guideline No. 476 and ICH guidance S2(R1). After exposure to FR-900098 for 4 h, there were only minor indications of direct cytotoxicity both in the presence and absence of S9-mix, without evidence of a concentration-dependent effect. The maximum reduction of relative cell counts was observed at 20 µg/ml FR-900098 (to 63% of the vehicle control) in the absence of S9-mix, and at 50 µg/ml FR-900098 (to 76% of the vehicle control) in the presence of S9-mix. The relative cell counts at the top dose of 2200 µg/ml were 100% and 116% in the absence and presence of S9-mix, respectively. These results indicated that FR-900098 was relatively non-toxic toward mouse lymphoma cells and that the requirement for the MLA to test up to 80–90% reduction in relative total growth (RTG) could not be achieved at concentrations up to the defined limit concentration. Therefore, a broad range of concentrations was also used for the main series of tests, covering both the OECD-defined limit concentration of 10 mM (approximately 2200 µg/ml) and low concentrations to detect hormesis-like phenomena.
The main MLA experiments were carried out using FR-900098 concentrations of 1, 10, 25, 125, 250 and 2200 µg/ml (without S9-mix), and 1, 10, 50, 250 and 2200 µg/ml (with S9-mix). Similar to the pre-test, there was no evidence for direct cytotoxicity of FR-900098 as judged by suspension growth (SG) and RTG (). Without S9-mix, FR-900098 induced a marginal and not relevant increase in the mean mutant frequency (MF) after exposure for 4 h, i.e., 97.6 (= 128%) per 106 viable cells at 25 µg/ml and 98.7 (= 130%) at 125 µg/ml, compared to 76.0 (= 100%) for the negative control (). This remained within our historical range for negative controls (66.7–169.7; 13 independent experiments) and the corresponding negative control range of 50–170 resistant mutants per 106 viable cells, as proposed by Moore et al.Citation29 As expected, the positive control methyl methanesulfonate (MMS) caused a relevant increase in the mean MF, based on the concept of the Global Evaluation Factor,Citation29,30 which amounted to 475.5 mutants per 106 viable cells (= 626% compared to the negative controls). After exposure to FR-900098 for 4 h in the presence of S9-mix, there were no signs of relevant increases in the number of TK−/− mutants. There was a marginal and non-relevant increase in mean MF at the limit concentration of 2200 µg/ml, which amounted to 96.7 (= 112%) mutants per 106 viable cells, compared to 86.1 (= 100%) for the negative controls (), falling well within the corresponding historical negative control range of 63.9–160.5 TFT-resistant mutants per 106 viable cells. The positive control again demonstrated a significant increase in mean MF with 415.7 mutants per 106 viable cells (= 483% compared to the negative controls). Based on the Global Evaluation Factor concept,Citation29,30 FR-900098 therefore did not induce any relevant increase in MF either in the presence or absence of S9-mix. All cultures exposed to FR-900098 exhibited MFs within the normal historical ranges for negative controls in this assay. Therefore, under the restrictions of this assay, FR-900098 is considered to lack potency for the induction of mutations in L5178Y/TK+/− cells, a mammalian somatic cell model that carries a dysfunctional p53 tumor-suppressor protein and is thus hypersensitive to genotoxic stress.Citation31
Table 3. In vitro mammalian cell gene mutation test in mouse lymphoma L5178Y/TK+/− cells.
Mammalian erythrocyte micronucleus test
The micronucleus test in mice is a cytogenetic in vivo assay with bone marrow erythrocytes.Citation32 Like the Ames test and the MLA, the micronucleus test is a prerequisite for the registration of new drugs and represents the in vivo part of the standard battery of genotoxicity tests for pharmaceuticals [ICH S2(R1)]. Micronuclei arise from chromosomal fragments or chromosomes that are not included in the daughter nuclei at cell division. They are easy to detect in young erythrocytes because the main nucleus is expelled a few hours after the final mitosis is completed, but micronuclei persist in the cytoplasm. For acute treatment regimens, micronuclei are analyzed in young erythrocytes to be sure they were induced by the test substance. Immature polychromatic erythrocytes (PCE) are less than 1 day old and contain fragments of nuclear material in the cytoplasm. This material consists mainly of RNA, which gradually disappears with maturation and stains blue with Giemsa. In contrast, mature normochromatic erythrocytes (NCE) stain pink with Giemsa due to the absence of RNA in the cytoplasm. The spontaneous incidence of micronucleated PCE in NMRI mice is ∼0.2%. Chemicals with chromosome-breaking (clastogenic) or spindle-disrupting (aneugenic) activity can increase the frequency of micronucleated PCE.
Before the main micronucleus test in NMRI mice, a preliminary test with 5 male and 5 female animals was carried out using an FR-900098 dose of 2000 mg/kg body weight, representing the defined limit dose for non-toxic substances (OECD Guideline No. 474). The total dose was administered as 2 consecutive oral gavages of 1000 mg/kg body weight at intervals of 6 h in order to achieve prolonged exposure. FR-900098 showed no signs of toxicity, and there was no reduction in body weight. For the main test, a 3-dose study design was chosen: 2 × 1000 mg/kg body weight (total 2000 mg/kg body weight), 2 × 200 mg/kg body weight (total 400 mg/kg body weight) and 2 × 40 mg/kg body weight (total 80 mg/kg body weight). This decision was taken for safety reasons, even though a limit test with the limit concentration alone may have been sufficient. The additional low and mid doses were included to exclude any potential confounding anti-proliferative effects of FR-900098 on bone marrow at the high limit concentration of 2000 mg/kg body weight.
The p.o. treatment of 5 male and 5 female animals in each treatment group indicated no significant inhibition of blood formation in the bone marrow, 24 and 48 h after application, compared to the control animals (). In male animals at the intermediate dose, there was a slight but statistically significant induction of blood formation, amounting to 123 ± 10.1 PCE per 200 red blood cells (RBC) compared to 110 ± 8.1 PCE per 200 RBC for the corresponding negative control animals. However, absence of concentration-dependency means that this outcome is unlikely to be biologically relevant. In the present study, FR-900098 did not significantly increase the number of micronucleated PCE in male and female mice at any of the doses tested, either 24 or 48 h after oral treatment. A very slight trend toward a higher frequency of micronucleated PCE was observed, particularly in FR-900098 treated female mice after 24 h (2 × 200 mg/kg body weight), but was also evident in female and male mice after 48 h (2 × 1000 mg/kg body weight). Micronucleus frequencies amounted to 0.25 ± 0.061, 0.24 ± 0.119, and 0.25 ± 0.122%, respectively, as compared to 0.12 ± 0.097, 0.18 ± 164, and 0.20 ± 0.141% for the concurrent negative controls. These slightly higher mean values were not considered relevant, because e.g. the micronucleus frequency of the FR-900098 treated females, 24 h after administration, was still in the range of the respective historical negative control data (0.12–0.55% micronucleated PCE), with a very low group mean value of the concurrent negative controls of 0.12% ± 0.097, compared to 0.24 ± 0.14% for the respective historical mean value. In contrast, the positive control animals showed a statistically significant increase in the number of micronucleated PCE in the bone marrow, with group mean values of 5.57 ± 1.717% for males and 3.72 ± 0.690% for females, confirming that the test worked as expected. As sufficient systemic availability of FR-900098 after oral administration can be derived from former in vivo studies in the P. vinckei mouse model,Citation4,6 our results strongly suggest that FR-900098 does not have clastogenic and/or aneugenic effects in NMRI mice at the dose levels tested, agreeing with the in vitro mutagenicity tests.
Table 4. Bone marrow erythrocyte micronucleus test in NMRI mice.a
Conclusions
In conclusion, the present study indicates that FR-900098 does not cause acute toxicity in rats, and there was no evidence for genotoxicity or mutagenicity in 2 different in vitro assays and one in vivo assay in mice. The exceptionally low toxicity of FR-900098 may not only reflect the absence of the molecular target DXP reductoisomerase in mammalian cells, but may also indicate a specific uptake mechanism in erythrocytes infected with P. falciparum, which appears to be absent in mammalian cells. Radiolabeled FR-900098 does not penetrate human fibroblasts or uninfected erythrocytes, but enters infected erythrocytes, because the parasite can influence cell membrane permeability.Citation33 FR-900098 has a novel and highly specific mode of action, low toxicity in rats, and no significant genotoxicity according to the comprehensive assays required by both the former ICH S2A and S2B guidelines and the current ICH S2(R1) guidance on genotoxicity testing of pharmaceuticals intended for human use, thus strongly encouraging the further development of FR-900098 as an antimalarial drug.
Materials and methods
FR-900098 administration
FR-900098 monosodium salt (systematic chemical name [3-(acetyl-hydroxy-amino)-propyl]-phosphonic acid monosodium; molar mass 219.11 g/mol; CAS No. 73226-73-0) was synthesized using a combination of previously described methods.Citation34,35 Infrared spectrometry and 1H-, 13C- and 31P-nuclear magnetic resonance spectrometry were used to confirm the identity of the substance. The purity of batch AR62 used in these experiments was ≥ 97.6% as determined by ion chromatography and photometry. For the in vivo tests, FR-900098 was dissolved in water and homogenized by ultrasonication for 15 min in a water bath directly before application. For the Ames test, FR-900098 was dissolved in DMSO for sterilization and was further diluted to the desired concentration using sterile water. For the mouse lymphoma assay, FR-900098 was accurately weighed and initially dissolved in pure ethanol in a sterile tube to avoid bacterial contamination. The ethanol was then allowed to evaporate under a sterile hood. Directly before use, FR-900098 was dissolved in treatment medium (culture medium with 5% rather than 10% heat-inactivated horse serum) by stirring, and then diluted to the desired concentrations. The stability of FR-900098 was confirmed both in ethanol and aqueous solution.
Relevant guidelines and regulations at the time of testing
All animal experiments complied with the regulations of the German Animal Protection Law (Tierschutzgesetz, May 18, 2006). The acute oral and intravenous toxicity study with rats and the genotoxicity modules were both conducted in compliance with the Principles of Good Laboratory Practice (GLP, German Chemical Law § 19a, Appendix 1, July 02, 2008) and with the appropriate Organization for Economic Cooperation and Development (OECD) Guidelines for the Testing of Chemicals No. 423 (Acute Oral Toxicity – Acute Toxic Class Method, March 22, 1996), No. 471 (Bacterial Reverse Mutation Test, July 21, 1997), No. 476 (In Vitro Mammalian Cell Gene Mutation Test, July 21, 1997), and No. 474 (Genetic Toxicology: Mammalian Erythrocyte Micronucleus Test, July 21, 1997), which were in part recently updated. The genotoxicity studies followed, in addition, the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) Harmonised Tripartite Guidelines S2A (Guidance on Specific Aspects of Regulatory Genotoxicity Tests for Pharmaceuticals, July 19, 1995) and S2B (Genotoxicity: A Standard Battery for Genotoxicity Testing of Pharmaceuticals, July 16, 1997). These ICH guidelines were combined and updated in June 2012 to form the ICH S2(R1) Guidance on Genotoxicity Testing and Data Interpretation for Pharmaceuticals Intended for Human Use.
Acute oral and intravenous toxicity in rats
To determine the acute toxicity of FR-900098 in vivo, an acute toxicity study was carried out with 3 male and 3 female Wistar rats per treatment group (strain Crl:WU, 8 weeks old, obtained from Charles River, Germany). Water was used as the vehicle for both p.o. and i.v. administration. For the p.o. route, 3000 mg FR-900098 was dissolved in 10 ml of water and administered at a dose of 10 ml/kg body weight using a stomach tube. For the i.v. route, 1000 mg FR-900098 was dissolved in 5 ml water and administered at a dose of 2 ml/kg body weight via the tail vein. After treatment, the animals were monitored for clinical signs of toxicity and morbidity for 14 d. At the end of the observation period, necropsy was carried out, including macroscopic inspection of major organs.
Salmonella typhimurium reverse mutation assay (Ames test)
The Ames test to determine the mutagenicity of FR-900098 in bacteria was carried out by the LPT Laboratory of Pharmacology and Toxicology, Germany, using S. typhimurium strains TA 98, TA 100, TA 102, TA 1535 and TA 1537 obtained directly from Dr. Bruce N. Ames. Two experimental designs were used, i.e. the standard plate incorporation method and the pre-incubation method. Based on a preliminary test using strain TA 100, we chose 1.0, 3.16, 10, 31.6 and 100 µg/plate as the test concentrations for the main experiments, with 3 plates analyzed per concentration and bacterial tester strain. Both experimental designs were carried out with and without metabolic activation using S9-mix, consisting of a post-mitochondrial fraction (S9 fraction) and the corresponding co-factors.Citation27 The S9 fraction was prepared from rats treated with Aroclor 1254 (Analabs, USA), as described by Maron and Ames.Citation36 Reference items included DMSO as the solvent/negative control and strain-specific positive controls. The concurrent positive controls without metabolic activation comprised sodium azide in water (10 µg/plate) for TA 1535 and TA 100, 2-nitrofluorene in DMSO (10 µg/plate) for TA 98, 9-aminoacridine in ethanol (100 µg/plate) for TA 1537, and methyl methanesulfonate in DMSO (1300 µg/plate) for TA 102. The concurrent positive controls with metabolic activation comprised 2-aminoanthracene in DMSO (2 µg/plate) for TA 98, TA 102 and TA 1537, and cyclophosphamide in water (1500 µg/plate) for TA 100 and TA 1535. A test item was considered to show a positive response if the number of revertants was significantly increased (p ≤ 0.05, U-test) compared with the solvent control to at least 2-fold for TA 98, TA 100 and TA 102, and 3-fold for TA 1535 and TA 1537.
In vitro mammalian cell gene mutation test with mouse lymphoma L5178Y/TK+/− cells
The mutagenic potential of FR-900098 in mammalian cells was determined in vitro using the microwell method of the mouse lymphoma TK mutation assay (MLA), according to Honma et al.Citation28 Heterozygous L5178Y/TK+/− mouse lymphoma cells (clone 3.7.2C; the model system) were provided by Dr. Heike Schramke (Philip Morris Research Laboratories GmbH, Germany). The cells were cultured in RPMI-1640 medium, containing 2 mM glutamine supplemented with 100 U/ml penicillin G, 100 µg/ml streptomycin sulfate, and 10% heat-inactivated horse serum (all components were purchased from GIBCO/Invitrogen, Germany) at 37°C and 5% CO2 in a humidified atmosphere. Prior to use, spontaneous TK−/− mutants were removed as described by Clive and Spector.Citation37 Cultures of proliferating L5178Y/TK+/− cells (1 × 107) in 20 ml treatment medium were exposed to FR-900098 at concentrations of 1, 10, 25, 125, 250 and 2200 µg/ml (without S9-mix) and 1, 10, 50, 250 and 2200 µg/ml (with S9-mix). These concentrations were based on a preliminary cytotoxicity test complying with the relevant guidelines. Negative controls (medium alone) and positive controls (methyl methanesulfonate for assays without metabolic activation and cyclophosphamide monohydrate for assays with metabolic activation; both substances from Sigma, Germany) were included. The cells were exposed for 4 h in the presence or absence of S9 fraction from phenobarbital and β-naphthoflavone treated rats (RCC Cytotest Cell Research GmbH, Germany) and the appropriate co-factors, as described by Maron and Ames.Citation36 The cells were subsequently washed and subcultured for 48 h to determine cytotoxicity (cell counts post-treatment and over the 2-day expression period as well as plating efficiency) and to allow for phenotypic expression (with daily cell population adjustment to 6 × 106 cells/30 ml of culture medium) prior to mutant selection. The cytotoxicity directly after the treatment period (survivor I) and after the expression period (survivor II) was determined by plating ∼1.6 cells per well in 200 µl non-selective growth medium (20% instead of 10% horse serum) on 2 96-well plates per treatment and time point. Cells were grown for additional 6 d until colonies were detected. The colonies were counted and the plating efficiencies (PE) calculated [PE = –ln (number of empty wells/number of total wells plated) / 1.6]. Mutant frequencies were determined by seeding ∼2 × 103 cells per well in 4 96-well plates per treatment group, using 200 µl restrictive medium per well, i.e., growth medium with 20% instead of 10% horse serum, supplemented with 3 µg/ml TFT (Sigma). After selection for 16–17 days, mutant colonies were counted and qualified (small or large colonies) under an inverted microscope and the plating efficiencies were calculated [PE mutant cells = PE of the TFT selection plates = –ln (number of empty wells/number of total wells plated) / 2000].
Mammalian erythrocyte micronucleus test
The genotoxic potential of FR-900098 in vivo was determined using the bone marrow micronucleus test in NMRI mice as described by Hayashi et al.Citation32 Young adult male and female NMRI mice (8–12 weeks at delivery) were obtained from Harlan Winkelmann (Germany) and were randomized by weight into the different treatment groups of 5 male and 5 female animals. Body weights were recorded at arrival, prior to treatment and before bone marrow preparation. Before administration of the test and reference substances, the animals were starved overnight. FR-900098 was dissolved in water and administered at a dose of 10 ml/kg body weight by oral gavage using a stomach tube. Three dose groups were used. Because the limit dose of 2000 mg/kg body weight (administered at 2 doses of 1000 mg/kg body weight) did not induce clinical signs of toxicity in a preliminary toxicity test, the limit dose was chosen as maximum dose, 2 × 200 mg/kg body weight was chosen as the mid-range dose, and 2 × 40 mg/kg body weight was chosen as the low dose. In each case, the 2 doses were administered at intervals of 6 h. The positive control cyclophosphamide monohydrate was administered once orally 24 h before sacrifice at a dose of 60 mg/kg body weight. After administration, animals were observed at defined intervals of 0.5, 2.5, 5 and 24 h after the first dose, and 0.5, 2.5, and 24 h following the second dose to promptly detect toxic effects and treatment-related suffering. Bone marrow was sampled 24 and 48 h after the first dose of FR-900098. At the first sampling interval, animals in all 5 treatment groups (negative control, positive control and 3 FR-900098 dose levels) were prepared for necropsy. At the second sampling interval, additional animals in the highest dose group as well as additional negative control animals were prepared for necropsy. Two femurs were isolated from each mouse, the ends of the femurs were removed, and the bone marrow was transferred to a tube by washing out with fetal calf serum. The bone marrow suspension was gently pulled up and down in the tube to achieve a fine cell suspension. The bone marrow was then centrifuged for 5 min and most of the supernatant was discarded. The cell pellet was carefully re-suspended in a small volume of fetal calf serum, yielding about 2 drops of bone marrow cell suspension per animal. From this suspension 2 smears (A and B) were prepared on defatted clean slides. The smears were air-dried for 24 h and stained with May-Grünwald and Giemsa solutions. The slides were coded prior to analysis. For each animal, the number of micronucleated cells per 2000 PCE was determined and the number of PCE and NCE per 200 erythrocytes was scored to determine the toxic effects of FR-90098 on bone marrow cells and thus blood formation.Citation38-40 The slides were decoded post-analysis.
Abbreviations
| CP | = | cyclophosphamide monohydrate |
| DMSO | = | dimethyl sulfoxide |
| DXP | = | 1-deoxy-d-xylulose-5-phosphate |
| GLP | = | good laboratory practice |
| ICH | = | International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use |
| i.v. | = | intravenously |
| MF | = | mutant frequency |
| MLA | = | mouse lymphoma assay |
| MMS | = | methyl methanesulfonate |
| NCE | = | normochromatic erythrocytes |
| OECD | = | Organization for Economic Cooperation and Development |
| PCE | = | polychromatic erythrocytes |
| PE | = | plating efficiency |
| p.o. | = | orally |
| RBC | = | red blood cells |
| RTG | = | relative total growth |
| SG | = | suspension growth |
| TFT | = | trifluorothymidine |
| TK | = | thymidine kinase |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank Dr. Richard M. Twyman and Dr. David Hutchinson for editing the manuscript.
Additional information
Funding
References
- Wiesner J, Reichenberg A, Heinrich S, Schlitzer M, Jomaa H. The plastid-like organelle of apicomplexan parasites as drug target. Curr Pharm Des 2008; 14:855-71; PMID:18473835; http://dx.doi.org/10.2174/138161208784041105
- Kuzuyama T, Shizimu T, Takahashi S, Seto H. Fosmidomycin, a specific inhibitor of 1-deoxy-d-xylulose 5-phosphate reductoisomerase in the nonmevalonate pathway of isoprenoidbiosynthesis. Tetrahedron Lett 1998; 39:7913-6; http://dx.doi.org/10.1016/S0040-4039(98)01755-9
- Zeidler J, Schwender J, Müller C, Wiesner J, Weidemeyer C, Beck E, Jomaa H, Lichtenthaler HK. Inhibition of the non-mevalonate 1-deoxy-D-xylulose-5-phosphate pathway of plant isoprenoid biosynthesis by fosmidomycin. Z Naturforsch C 1998; 53:980-6
- Jomaa H, Wiesner J, Sanderbrand S, Altincicek B, Weidemeyer C, Hintz M, Türbachova I, Eberl E, Zeidler J, Lichtenthaler HK, Soldati D, Beck E. Inhibitors of the nonmevalonate pathway of isoprenoid biosynthesis as antimalarial drugs. Science 1999; 285:1573-6; PMID:10477522; http://dx.doi.org/10.1126/science.285.5433.1573
- Tahar R, Basco LK. 2007. Molecular epidemiology of malaria in Cameroon. XXV. In vitro activity of fosmidomycin and its derivatives against fresh clinical isolates of Plasmodium falciparum and sequence analysis of 1-deoxy-d-xylulose 5-phosphate reductoisomerase. Am J Trop Med Hyg 2007; 77:214-20; PMID:17690389
- Reichenberg A, Wiesner J, Weidemeyer C, Dreiseidler E, Sanderbrand S, Altincicek B, Beck E, Schlitzer M, Jomaa H. Diaryl ester prodrugs of FR900098 with improved in vivo antimalarial activity. Bioorg Med Chem Lett 2001; 11:833-5; PMID:11277531; http://dx.doi.org/10.1016/S0960-894X(01)00075-0
- Giessmann D, Heidler P, Haemers T, Van Calenbergh S, Reichenberg A, Jomaa H, Weidemeye C, Sanderbrand S, Wiesner J, Link A. Towards new antimalarial drugs: synthesis of non-hydrolyzable phosphate mimics as feed for a predictive QSAR study on 1-deoxy-d-xylulose-5-phosphate reductoisomerase inhibitors. Chem Biodivers 2008; 5:643-56; PMID:18421757; http://dx.doi.org/10.1002/cbdv.200890060
- Umeda T, Tanaka N, Kusakabe Y, Nakanishi M, Kitade Y, Nakamura KT. Molecular basis of fosmidomycin's action on the human malaria parasite Plasmodium falciparum. Sci Rep 2011; 1:9; PMID:22355528; http://dx.doi.org/10.1038/srep00009
- Horbach S, Sahm H, Welle R. Isoprenoid biosynthesis in bacteria: two different pathways? FEMS Microbiol Lett 1993; 111:135-40; PMID:8405922; http://dx.doi.org/10.1111/j.1574-6968.1993.tb06375.x
- Sprenger GA, Schorken U, Wiegert T, Grolle S, de Graaf AA, Taylor SV, Begley TP, Bringer-Meyer S, Sahm H. Identification of a thiamin-dependent synthase in Escherichia coli required for the formation of the 1-deoxy-d-xylulose 5-phosphate precursor to isoprenoids, thiamin, and pyridoxol. Proc Natl Acad Sci USA 1997; 94:12857-62; PMID:9371765; http://dx.doi.org/10.1073/pnas.94.24.12857
- Okuhara M, Kuroda Y, Goto T, Okamoto M, Terano H, Kohsaka M, Aoki H, Imanaka H. Studies on new phosphonic acid antibiotics. I. FR-900098, isolation and characterization. J Antibiot (Tokyo) 1980; 33:13-7; PMID:6768704; http://dx.doi.org/10.7164/antibiotics.33.13
- Okuhara M, Kuroda Y, Goto T, Okamoto M, Terano H, Kohsaka M, Aoki H, Imanaka H. Studies on new phosphonic acid antibiotics. III. Isolation and characterization of FR-31564, FR-32863 and FR-33289. J Antibiot (Tokyo) 1980; 33:24-8; PMID:6768705; http://dx.doi.org/10.7164/antibiotics.33.24
- Mine Y, Kamimura T, Nonoyama S, Nishida M, Goto S, Kuwahara S. In vitro and in vivo antibacterial activities of FR-31564, a new phosphonic acid antibiotic. J Antibiot (Tokyo) 1980; 33:36-43; PMID:7372548; http://dx.doi.org/10.7164/antibiotics.33.36
- Kuemmerle HP, Murakawa T, Soneoka K, Konishi T. Fosmidomycin: a new phosphonic acid antibiotic. Part I: Phase I tolerance studies. Int J Clin Pharmacol Ther Toxicol 1985; 23:515-20; PMID:4066075
- Kuemmerle HP, Murakawa T, Sakamoto H, Sato N, Konishi T, De Santis F. Fosmidomycin, a new phosphonic acid antibiotic. Part II: 1. Human pharmacokinetics. 2. Preliminary early phase IIa clinical studies. Int J Clin Pharmacol Ther Toxicol 1985; 23:521-528; PMID:4066076
- Missinou MA, Borrmann S, Schindler A, Issifou S, Adegnika AA, Matsiegui PB, Binder R, Lell B, Wiesner J, Baranek T, Jomaa H, Kremsner PG. Fosmidomycin for malaria. Lancet 2002; 360:1941-2; PMID:12493263; http://dx.doi.org/10.1016/S0140-6736(02)11860-5
- Lell B, Ruangweerayut R, Wiesner J, Missinou MA, Schindler A, Baranek T, Hintz M, Hutchinson D, Jomaa H, Kremsner PG. Fosmidomycin, a novel chemotherapeutic agent for malaria. Antimicrob Agents Chemother 2003; 47:735-8; PMID:12543685; http://dx.doi.org/10.1128/AAC.47.2.735-738.2003
- Lell B, Kremsner PG. Clindamycin as an antimalarial drug: review of clinical trials. Antimicrob Agents Chemother 20002; 46:2315-20; PMID:12121898; http://dx.doi.org/10.1128/AAC.46.8.2315-2320.2002
- Borrmann S, Issifou S, Esser G, Adegnik AA, Ramharter M, Matsiegui PB, Oyakhirome S, Mawili-Mboumba PD, Missinou MA, Kun JF, et al. Fosmidomycin-clindamycin for the treatment of Plasmodium falciparum malaria. J Infect Dis 2004; 190:1534-40; PMID:15478056; http://dx.doi.org/10.1086/424603
- Borrmann S, Lundgren I, Oyakhirome S, Impouma B, Matsiegui PB, Adegnika A, Issifou S, Kun JF, Hutchinson D, Wiesner J, et al. Fosmidomycin plus clindamycin for treatment of pediatric patients aged 1 to 14 years with Plasmodium falciparum malaria. Antimicrob Agents Chemother 2006; 50:2713-18; PMID:16870763; http://dx.doi.org/10.1128/AAC.00392-06
- Oyakhirome S, Issifou S, Pongratz P, Barondi F, Ramharter M, Kun JF, Missinou, Lell B, Kremsner PG. Randomized controlled trial of fosmidomycin-clindamycin versus sulfadoxine-pyrimethamine in the treatment of Plasmodium falciparum malaria. Antimicrob Agents Chemother 2007; 51:1869-1871; PMID:17325227; http://dx.doi.org/10.1128/AAC.01448-06
- Ruangweerayut R, Looareesuwan S, Hutchinson D, Chauemung A, Banmairuroi V, Na-Bangchang K. Assessment of the pharmacokinetics and dynamics of two combination regimens of fosmidomycin-clindamycin in patients with acute uncomplicated falciparum malaria. Malar J 2008; 7:225; PMID:18973702; http://dx.doi.org/10.1186/1475-2875-7-225
- Borrmann S, Adegnika AA, Matsiegui PB, Issifou S, Schindler A, Mawili-Mboumba DP, Baranek T, Wiesner J, Jomaa H, Kremsner PG. Fosmidomycin-clindamycin for Plasmodium falciparum Infections in African children. J Infect Dis 2004; 189:901-8; PMID:14976608; http://dx.doi.org/10.1086/381785
- Na-Bangchang K, Ruengweerayut R, Karbwang J, Chauemung A, Hutchinson D. Pharmacokinetics and pharmacodynamics of fosmidomycin monotherapy and combination therapy with clindamycin in the treatment of multidrug resistant falciparum malaria. Malar J 2007; 6:70; PMID:17531088; http://dx.doi.org/10.1186/1475-2875-6-70
- Lanaspa M, Moraleda C, Machevo S, González R, Serrano B, Macete E, Cisteró P, Mayor A, Hutchinson D, Kremsner PG, Alonso P, Menéndez C, Bassat Q. Inadequate efficacy of a new formulation of fosmidomycin-clindamycin combination in Mozambican children less than three years old with uncomplicated Plasmodium falciparum malaria. Antimicrob Agents Chemother 2012; 56:2923-8; PMID:22430959; http://dx.doi.org/10.1128/AAC.00018-12
- Fernandes JF, Lell B, Agnandji ST, Obiang RM, Bassat Q, Kremsner PG, Mordmüller B, Grobusch MP. Fosmidomycin as an antimalarial drug: a meta-analysis of clinical trials. Future Microbiol 2015; 10:1375-90; PMID:26228767; http://dx.doi.org/10.2217/FMB.15.60
- Ames BN, McCann J, Yamasaki E. Methods for detecting carcinogens and mutagens with the Salmonella/mammalian-microsome mutagenicity test. Mutat Res 1975; 31:347-64; PMID:768755; http://dx.doi.org/10.1016/0165-1161(75)90046-1
- Honma M, Hayashi M, Shimada H, Tanaka N, Wakuri S, Awogi T, Yamamoto KI, Kodani NU, Nishi Y, Nakadate M, Sofuni T. Evaluation of the mouse lymphoma tk assay (microwell method) as an alternative to the in vitro chromosomal aberration test. Mutagenesis 1999; 14:5-22; PMID:10474816; http://dx.doi.org/10.1093/mutage/14.1.5
- Moore MM, Honma M, Clements J, Bolcsfoldi G, Burlinson B, Cifone M, Clarke J, Delongchamp R, Durward R, Fellows M, et al. Mouse lymphoma thymidine kinase gene mutation assay: follow-up meeting of the International Workshop on Genotoxicity Testing–Aberdeen, Scotland, 2003–Assay acceptance criteria, positive controls, and data evaluation. Environ Mol Mutagen 2006; 47(1):1-5; PMID:15991242; http://dx.doi.org/10.1002/em.20159
- Moore MM, Honma M, Clements J, Bolcsfoldi G, Cifone M, Delongchamp R, Fellows M, Gollapudi B, Jenkinson P, Kirby P, et al. Mouse lymphoma assay workgroup. 2003. mouse lymphoma thymidine kinase gene mutation assay: international workshop on genotoxicity tests workgroup report–plymouth, UK 2002. Mutat Res 2003; 540:127-40; PMID:14550497; http://dx.doi.org/10.1016/j.mrgentox.2003.07.003
- Storer RD, Kraynak AR, McKelvey TW, Elia MC, Goodrow TL, DeLuca JG. The mouse lymphoma L5178Y Tk+/− cell line is heterozygous for a codon 170 mutation in the p53 tumor suppressor gene. Mutat Res 1997; 373(2):157-65; PMID:9042396; http://dx.doi.org/10.1016/S0027-5107(96)00227-8
- Hayashi M, Tice RR, MacGregor JT, Anderson D, Blakey DH, Kirsch-Volders M, Oleson FB Jr, Pacchierotti F, Romagna F, Shimada H, et al. In vivo rodent erythrocyte micronucleus assay. Mutat Res 1994; 312:293-304; PMID:7514741; http://dx.doi.org/10.1016/0165-1161(94)90039-6
- Baumeister S, Wiesner J, Reichenberg A, Hintz M, Bietz S, Harb OS, Roos DS, Kordes M, Friesen J, Matuschewski K, et al. Fosmidomycin uptake into Plasmodium and Babesia-infected erythrocytes is facilitated by parasite-induced new permeability pathways. PLoS One 2011; 6:e19334; PMID:21573242; http://dx.doi.org/10.1371/journal.pone.0019334
- Kamiya T, Hashimoto M, Hemmi K, Takeno H. Hydroxyaminohydrocarbon-phosphonic acids. Fujisawa Pharmaceutical Co. Ltd. U.S. Patent Application No. 4,206,156, 3 June 1980.
- Öhler E, Kanzler S. Regioselective palladium(0) catalyzed amination of carbonates of allylic a-hydroxyphosphonates with hydroxylamine derivatives: A convenient route to phosphonic acids related to the antibiotic fosmidomycin. Synthesis 1995;539-43
- Maron DM, Ames BN. Revised methods for the Salmonella mutagenicity test. Mutat Res 1983; 113:173-215; PMID:6341825; http://dx.doi.org/10.1016/0165-1161(83)90010-9
- Clive D, Spector JF. Laboratory procedure for assessing specific locus mutations at the TK locus in cultured L5178Y mouse lymphoma cells. Mutat Res 1975; 31:17-29; PMID:1093012; http://dx.doi.org/10.1016/0165-1161(75)90059-X
- Schmid W. The micronucleus test for cytogenetic analysis. In Chemical mutagens, principle and methods for their detection A. Hollaender (ed.), vol. 1. Plenum Press: New York, NY. 1976; pp. 31-53.
- Heddle JA, Stuart E, Salamone MF. 1984. The bone marrow micronucleus test, In G. J. Kilbey, et al. (ed.) Handbook of mutagenicity test procedures, 2nd ed. Elsevier: Amsterdam. p. 441-457.
- Mavournin KH, Blakey DH, Cimino MC, Salamone MF, Heddle JA. The in vivo micronucleus assay in mammalian bone marrow and peripheral blood. A report of the U.S. environmental protection agency gene-tox program. Mutat Res 1990; 239:29-80; PMID:2195332; http://dx.doi.org/10.1016/0165-1110(90)90030-F