
ABSTRACT
The Gram-negative bacterium Francisella tularensis is the etiological agent of the zoonotic disease tularemia. Its life cycle is characterized by an ability to survive within phagocytic cells through phagosomal escape and replication in the cytosol, ultimately causing inflammasome activation and host cell death. Required for these processes is the Francisella Pathogenicity Island (FPI), which encodes a Type VI secretion system (T6SS) that is active during intracellular infection. In this study, we analyzed the role of the FPI-component IglE, a lipoprotein which we previously have shown to be secreted in a T6SS-dependent manner. We demonstrate that in F. tularensis LVS, IglE is an outer membrane protein. Upon infection of J774 cells, an ΔiglE mutant failed to escape from phagosomes, and subsequently, to multiply and cause cytopathogenicity. Moreover, ΔiglE was unable to activate the inflammasome, to inhibit LPS-stimulated secretion of TNF-α, and showed marked attenuation in the mouse model. In F. novicida, IglE was required for in vitro secretion of IglC and VgrG. A mutagenesis-based approach involving frameshift mutations and alanine substitution mutations within the first ∼ 38 residues of IglE revealed that drastic changes in the sequence of the extreme N-terminus (residues 2–6) were well tolerated and, intriguingly, caused hyper-secretion of IglE during intracellular infection, while even subtle mutations further downstream lead to impaired protein function. Taken together, this study highlights the importance of IglE in F. tularensis pathogenicity, and the contribution of the N-terminus for all of the above mentioned processes.
Introduction
Bacteria use secretion systems for transporting a variety of protein substrates across their membranes and into target cells, thereby interfering in various ways with host cell processes. The Type VI Secretion System (T6SS) was first described just 10 y ago, but appears to be ubiquitously present in many clinically important Gram-negative pathogens.Citation1 Despite large heterogeneity, reflecting differences in their phylogenetic origin, certain T6SS components are highly conserved, including homologues of Vibrio cholerae IcmF, DotU, ClpV, VipA, VipB, VgrG, and Hcp proteins.Citation1,2 The T6SS performs key roles for the ability of the pathogen to infect eukaryotes and many of the effectors possess enzymatic functions associated with virulence, e.g. phospholipase activity, ADP ribosylation, actin cross-linking, and fusion of eukaryotic membranes.Citation3-6
A feature distinguishing the T6SS from all other bacterial secretion systems is, however, its central role in interbacterial competition.Citation7,8 Many T6SS-encoded effectors are enzymes directed against highly conserved and essential components of bacterial cells, such as the peptidoglycan layer, bacterial membranes or DNA, thus leading to growth arrest or lysis.Citation7-12 In many cases, antibacterial T6SS effectors occur in tandem with corresponding immunity proteins that inhibit the activity of the cognate toxin in the host bacterium, preventing self-damage.Citation7,9-12 Notably, almost all secreted substrates of T6SSs lack classical N-terminal signal peptides,Citation13 and only recently, motifs distinguishing such effectors from other T6S components have been identified in a wide variety of Gram-negative bacteria.Citation14 The motifs, denoted “marker for type 6 effectors (MIX),’ are located predominantly at the N-terminal end of proteins, with predicted effector activity domains at the C-terminus. The latter include, e.g., peptidoglycan-binding domains, PyocinS and colicin DNase bacteriocidal domains, a bacterial ribosomal inactivating RNase domain, and a Rho-activating domain of cytotoxic necrotizing factor.Citation14 Still, many of the MIX-containing proteins have long C-termini lacking homology to other proteins, suggesting additional functions. Thus, T6SS effectors appear to perform very broad functions, presumably targeting both prokaryotic and eukaryotic organisms. Another recently identified T6SS-associated motif is that of T6SS effector chaperone (TEC) proteins.Citation15 These proteins are not secreted but required for effector delivery through binding to VgrG and effector proteins and are encoded upstream of their cognate effector genes.
While some of the mechanisms underlying Type VI Secretion (T6S) of enterobacteria in general and, in particular, V. cholerae, have been identified, T6S of Francisella tularensis is poorly understood at the molecular level. This highly virulent and facultative intracellular bacterium causes tularemia in humans and animals.Citation16 Its primary replication site in humans appears to be macrophages and involves escape from the phagosome prior to lysosomal fusion, followed by extensive cytosolic replication.Citation16-18 This eventually leads to host cell-death and the subsequent release of intracellular bacteria through mechanisms that involve activation of both caspase-3- and caspase-1-dependent pathways and the release of proinflammatory cytokines.Citation19-21 This Francisella-induced cell death has also been proposed to be an innate immune macrophage response to cytosolic bacteria aimed at restricting bacterial multiplication.Citation20 Francisella actively interferes with host cell intracellular signaling, and suppresses the ability of both dendritic cells and macrophages to secrete cytokines in response to secondary stimuli.Citation22-27 Thus, F. tularensis is clearly able to modulate many levels of the host immune response to facilitate its intracellular survival, although the factors employed by the bacterium to coordinate these mechanisms are, so far, poorly understood.
Genes necessary for phagosomal escape, intracellular survival and virulence are found within the Francisella Pathogenicity Island (FPI), a 34 kb gene cluster consisting of ∼ 16–19 open reading frames that is duplicated in the highly virulent F. tularensis subsp. tularensis and F. tularensis subsp holarctica (reviewed inCitation28). The FPI encodes a phylogenetically aberrant T6SS variantCitation1 that we have previously shown to be active during intracellular infection. Thus, when fused to a TEM β-lactamase reporter, IglCEFIJ, PdpAE, as well as VgrG, most of which are unique to Francisella, were found to be secreted by LVS into the macrophage cytosol.Citation29 Importantly, secretion was dependent on the T6SS core components DotU, VgrG and IglC (Hcp), as well as IglG. The latter was recently demonstrated to be a member of the DUF4280 protein family, which like PAAR proteins, caps the T6SS and thereby modulates T6S.Citation30 Moreover, secretion of VgrG and IglC by F. novicida was recently shown to be triggered in vitro by the presence of KCl in the growth medium. Interestingly, although most FPI components are essential for the phagosomal escape, a recent study demonstrated that FPI proteins like IglA and IglC, are not essential for the subsequent intracytosolic replication, indicating that the T6S machinery is required only for the initial escape.Citation31 Still, many important questions remain unanswered, such as the identity of the signals that trigger recognition and secretion of substrates in vivo, as well as the underlying regulatory mechanisms.
The present study was focused on improving our understanding of the role of IglE in T6SS. Our interest for the protein was the result of our previous finding that out of all the 17 FPI proteins of LVS, it is the most highly secreted FPI component by F. tularensis LVS during intracellular infection and is also efficiently secreted by intracellular F. novicida.Citation29,32 The efficient secretion seems paradoxical in view of publications that demonstrated that it is an outer membrane lipoprotein.Citation33,34 In the present study, we analyzed the role of the IglE protein of F. tularensis LVS as it is a conserved T6S component present in all species of F. tularensis and therefore, as previously suggested, is likely to perform similar and essential functions. In agreement, IglE was found to be essential for F. tularensis phagosomal escape, cytopathogenicity, inflammasome activation, and virulence in mice. In F. novicida, IglE was required for in vitro secretion of the T6SS substrates IglC and VgrG. Moreover, a functional mapping of the protein identified key residues within the N-terminus as critical for the IglE function.
Results
Construction and characterization of an ΔiglE mutant of LVS
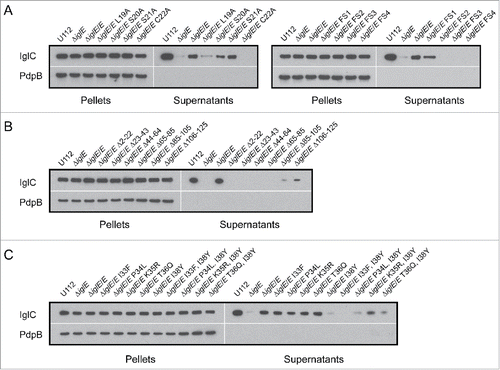
To better characterize the role of the F. tularensis FPI protein IglE, we constructed an in-frame ΔiglE deletion mutant in the live vaccine strain, LVS, removing most of the gene (Δ6–119 of 125 amino acids). To confirm that IglE was missing in the mutant, we used Western blot analysis with antibodies directed against IglE as well as RT-PCR, to confirm that no IglE protein or transcript was being produced. To complement the mutant, we expressed GSK-tagged IglE in trans from the GroEL promoter of pMOL52.Citation35 Compared to LVS, the complemented mutant ΔiglE/E expressed elevated levels of IglE, while no IglE was produced by the deletion mutant (Fig. S1). Importantly, no visible effect on the production of PdpB/IcmF or VgrG was observed in the mutant (Fig. S1), demonstrating the absence of polar effects on the pdpB and vgrG genes encoded immediately upstream and downstream of iglE. This was also confirmed using RT-PCR (data not shown).
IglE is an outer membrane protein
To determine the subcellular localization of IglE, we fractionated LVS bacteria into soluble, inner- and outer membrane fractions and determined the amount of IglE in each fraction by immunoblot analysis. The data from this experiment verified previous studies demonstrating that IglE is predominantly localized to the outer membrane fraction, with only a small portion localized to the inner membrane (Fig. S2).Citation33,34 As anticipated, no IglE was detected in samples from ΔiglE, while the localization of the control proteins IglC, PdpB/IcmF and Tul4 remained the same as for LVS (Fig. S2). Expression of IglE in trans from the strong GroEL promoter of pMOL52 resulted in equal proportions of IglE in the inner membrane and outer membrane fractions, likely reflecting the incomplete translocation of the protein from the former to the latter location due to its overexpression (data not shown). Similar results were also obtained by Nguyen et al also when they expressed IglE in trans.Citation33
An ΔiglE mutant of LVS is deficient for phagosomal escape and cytopathogenicity, but not for intracellular replication
We investigated the location of F. tularensis ΔiglE during infection using confocal immuno-fluorescence microscopy of bacterial colocalization with LAMP-1 positive membranes (reviewed in Citation36). Unlike LVS and ΔiglE/E that had escaped from their original phagosomes at 6 h, 3.3 ± 1.8% and 7.5 ± 1.9% colocalization with LAMP-1, respectively, ΔiglE and the control strain ΔiglC remained enclosed within vacuoles, 94.9 ± 3.4% and 95.6 ± 3.5% colocalization with LAMP-1, respectively; P < 0.001 vs. LVS. We also confirmed the LAMP-1 data by transmission EM. At 6 h, 90% of LVS and 86% of ΔiglE/E were found to be free in the cytoplasm, while 10% and 8%, respectively, were in highly damaged phagosomes (). In contrast, a majority of the ΔiglC and ΔiglE mutant bacteria was found within intact phagosomes, 83% vs. 85%, respectively, or within fairly intact phagosomes, 14% vs. 13%, respectively (). Thus, IglE is essential for phagosomal escape by LVS, similar to what was previously shown for an iglE mutant of SCHU S4.Citation34
Figure 1. Phagosomal escape of F. tularensis. J774 cells were infected with F. tularensis at an MOI of 1,000 for 2 h and, after washing, incubated for another 6 h before they were fixed and analyzed by transmission electron microscopy (TEM). (A) Electron micrographs of infected J774 cells were acquired with a JEOL JEM 1230 Transmission Electron Microscope (JEOL Ltd., Tokyo, Japan). Black arrows indicate vacuolar membranes surrounding intracellular bacteria. (B) Bacteria were divided into one of 4 categories based on the membrane integrity of the surrounding vacuolar membrane. Micrographs in (A) illustrate the categories “Cytoplasm” (LVS and ΔiglE/E) or “Intact phagosome” (ΔiglE and ΔiglC).
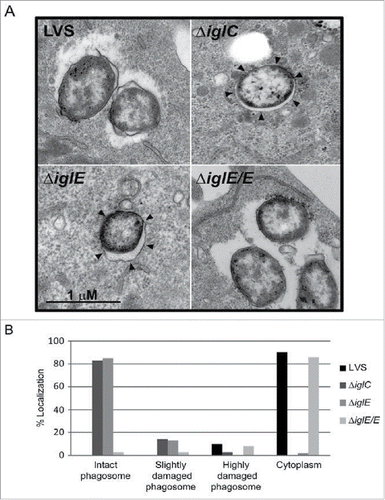
There is a strong correlation between phagosomal escape, cytopathogenicity and intracellular growth (reviewed in Citation28). A previous study employing an iglE mutant of SCHU S4 demonstrated a critical role of IglE for intracellular replication in bone marrow-derived macrophages (BMDM).Citation34 In contrast, an F. novicida iglE insertion mutant was found to be able to grow within J774 cells, albeit with impaired efficiency.Citation33 To determine the role of IglE for the cytopathogenic response and intracellular growth of LVS, we infected J774 cells or BMDM with LVS, ΔiglE and ΔiglE/E. We collected cell culture supernatants over a 48-h time course and sampled them for LDH release, a direct consequence of the cytopathogenic response resulting from an F. tularensis infection.Citation19 While LVS and ΔiglE/E both induced high levels of LDH release, ΔiglE and ΔiglC were both unable to induce a cytopathogenic response ( and data not shown).
Figure 2. Cytopathogenicity of F. tularensis strains. Culture supernatants of infected J774 cells were assayed for LDH activity at 0, 24 and 48 h and the activity was expressed as a percentage of the level of non-infected lysed cells (positive lysis control). Means and SD of triplicate wells from one representative experiment of 2 are shown. The asterisks indicate that the cytopathogenicity levels were significantly higher than those of LVS-infected cells at a given time point as determined by a 2-sided t-test with equal variance, including the Bonferroni correction for multiple pair-wise comparisons (**, P ≤ 0.01; ***, P ≤ 0.001).
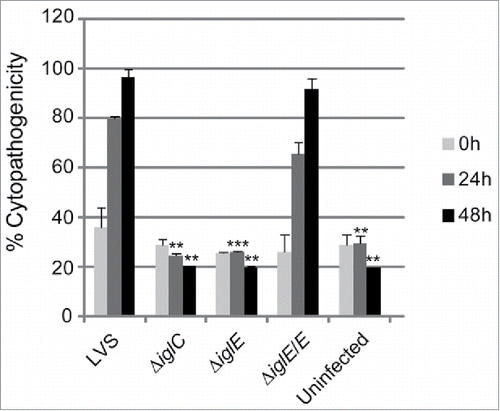
Similar to the control strain, ΔiglC, ΔiglE was unable to grow within macrophages over the same time period, while expression of iglE in trans restored growth to wild-type levels (Fig. S3 and data not shown). Importantly, since FPI mutants defective for phagosomal escape never reach the growth-permissive cytosol to which their metabolism is adapted,Citation37 it is very difficult to determine the requirement of a given FPI protein for intracellular replication. To circumvent the need for phagosomal escape, we therefore injected the ΔiglE mutant directly into the cytoplasm of J774 cells and compared its replication to that of ΔiglC and LVS. The injected ΔiglC mutant, in contrast to that of LVS, replicated efficiently in a high number of cells (mean of 139.33 vs. 53.57; ), confirming our recent findings.Citation31 Strikingly, the microinjected ΔiglE mutant also exhibited efficient replication (mean 122.33; ), suggesting that IglE, similar to IglC, is essential for the initial phagosomal escape step only, and not for the subsequent intracellular replication.
Table 1. Growth of F. tularensis in J774 cells upon microinjection.
IglE is required for modulation of the macrophage cytokine response
Phagosomal escape of F. tularensis is a prerequisite for the inflammasome-dependent induction of IL-1β secretion during a macrophage infection.Citation20,25,35,38-40 As a result, mutants with no or delayed phagosomal escape, e.g., ΔiglC, ΔdotU, ΔvgrG, ΔiglI, and ΔiglG, exhibit no or very diminished IL-1β release.Citation35,38,40,41 We measured the levels of this cytokine in cell culture supernatants of mouse peritoneal exudate cells (PECs) infected with LVS, ΔiglE, ΔiglE/E, or the control strain ΔiglC at 24 h. In supernatants from LVS- and ΔiglE/E-infected cell cultures, significant levels were detected at 24 h, whereas levels were below or just above the detection level of the assay for ΔiglE- and ΔiglC-infected cultures or uninfected cells ().
Figure 3. Cytokine secretion of F. tularensis-infected macrophages. Uninfected J774 cells or cells infected with F. tularensis at an MOI of 500 for 2 h were washed and subsequently incubated in the presence of E. coli-derived LPS (50 ng/ml) for an additional 2 h. The average TNF-α secretion in pg/ml and SEM of quadruple samples (n = 4) from one out of 2 representative experiments is shown. In the absence of LPS, the cytokine levels were below the limit of detection for the assay (< 15 pg/ml) (data not shown). Francisella-infected or non-infected PEC cells were incubated for 24 h after gentamicin treatment and the average IL-1β secretion in pg/ml and SEM of 6 samples (n = 6) from one out of 2 representative experiments is presented. A Student´s 2-sided t-test, including the Bonferroni correction for multiple pair-wise comparisons, was used to determine whether the cytokine release induced by each of the strains or the uninfected control were significantly different to the parental strain (*, P ≤ 0.05; ***, P ≤ 0.001).
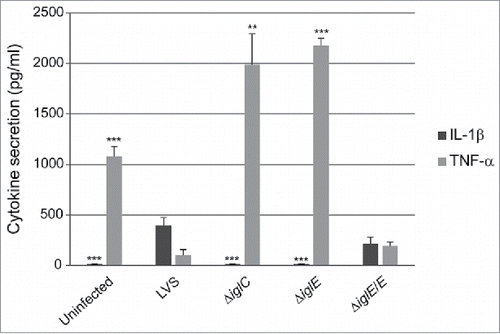
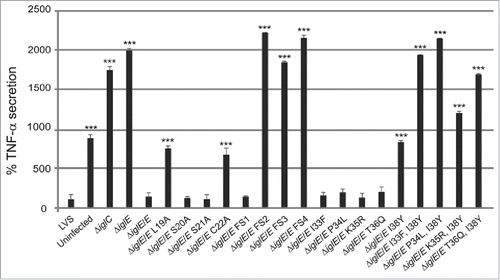
F. tularensis actively suppresses the ability of host cells to secrete the inflammasome-independent cytokine TNF-α in response to E. coli LPS.Citation24,26 In contrast, mutants confined to intact phagosomes lack this suppressive property. Citation26,35,41,42 To characterize the effects of the ΔiglE mutant, J774 cells were infected and cell culture supernatants analyzed for the presence of TNF-α after 120 min of LPS-stimulation. Efficient inhibition of TNF-α release was observed after infection with LVS and ΔiglE/E, but not after infection with ΔiglE, the control strain ΔiglC, or from uninfected cells (). As observed before, uninfected cells release less TNF-α than the ΔiglE and ΔiglC mutants, approximately 2-fold less.Citation37
The ΔiglE mutant is attenuated in vivo
Intracellular growth, cytopathogenicity and cytokine modulation are important factors for Francisella pathogenicity in vivo (reviewed in Citation28). To determine whether the ΔiglE mutant was defective for virulence, C57BL/6 mice were infected via the intradermal route with LVS, ΔiglE, or the complemented mutant. With an infection dose of 5.0 × 107 CFU (approximately 2.5 × LD50),Citation43 LVS caused 100% mortality (mean time to death 4.4 ± 0.5 days). In contrast, no mice died after infection with ∼7.0 × 108 CFU of ΔiglE. Complementation of ΔiglE resulted in 60% mortality with a dose of 3.0 × 107 CFU (mean time to death 5.0 ± 0.0 days) and 100% with a dose of 7.0 × 108 CFU (mean time to death 3.6 ± 1.5 days) (). This demonstrates the critical role of IglE for virulence of F. tularensis LVS, similar to what was previously shown for IglE of F. novicida and SCHU S4.Citation33,34
Figure 4. IglE is required for lethality in C57BL/6 mice. Mice were infected intradermally with 5 × 107 CFU of LVS (diamond), 7 × 108 CFU of ΔiglE (square), 3 × 107 CFU (triangle) or 7 × 108 CFU (circle) of the complemented mutant. Mice were monitored for signs of morbidity for up to 22 d post infection. The data represents one representative experiment out of 3 where groups of 5 (n = 5) mice were used.
IglE of F. novicida is required for T6S in vitro
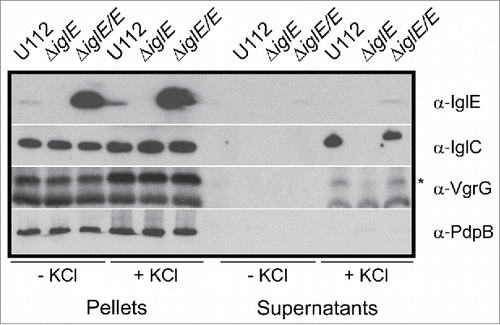
Recently, IglA-dependent secretion of IglC and VgrG was demonstrated for the F. novicida strain U112 when grown in TSB supplemented with 5% KCl.Citation44 With regard to the essential role of IglE in Francisella pathogenesis, we wanted to determine whether IglE is required also for in vitro secretion. No secretion could, however, be detected when LVS was grown in KCl-supplemented medium, in fact, even low concentrations (2% KCl) resulted in marked growth inhibition of LVS (data not shown). This finding supports previous observations that KCl is toxic for other Francisella subspecies than F. novicida.Citation30,45 For this reason, we generated an ΔiglE mutant of F. novicida U112. Similar to the LVS ΔiglE mutant, it was unable to replicate in J774 macrophages after phagocytosis, while growth was readily restored upon expression of IglE in trans (data not shown). We then tested the mutant, together with parental U112, and the complemented strain in the secretion assay. When grown in the presence of 5% KCl, we observed a significant increase in IglC levels in the bacterial lysates from all 3 strains, suggesting that KCl induces the expression of this protein (). This was not specific for IglC, in fact, levels of most of the FPI proteins, including PdpABC, VgrG and IglAH, tested were found to be induced in the presence of 5% KCl with similar levels in all 3 strains ( and data not shown). While the secretion of the inner membrane protein PdpB is not affected by the presence of KCl, IglC and VgrG are translocated into the culture supernantant of U112 and the complemented strain, whereas the ΔiglE mutant secreted no or very little of these proteins (). Since we have previously observed that IglE is secreted during macrophage infection,Citation29 we wanted to test whether IglE was also secreted in vitro. We found that IglE expression was induced in strain U112, however, IglE secretion was not found in the supernatant fraction (). This may be due to the relatively low IglE levels detected in U112 under the present conditions, however, the complemented ΔiglE mutant expressed high levels of IglE from the GroEL promoter and still IglE was secreted at only very low levels in a KCl-independent fashion (). Thus, in F. novicida, IglE is required for efficient secretion of IglC and VgrG in the presence of KCl, but is only weakly secreted under the same in vitro conditions.
Figure 5. IglC and VgrG are secreted in response to KCl. Indicated F. novicida strains were grown in TSB with or without 5 % KCl and FPI protein synthesis (pellet fractions) and secretion (cleared culture supernatants) were analyzed using SDS-PAGE and immunoblotting with specific antiserum. The band highlighted with an asterisk corresponds to VgrG, while the lower band corresponds to an unspecific band recognized by the antiserum. The inner membrane protein PdpB was included as a lysis control. The experiment was repeated 3 times and a representative example is shown.
Mapping of functional domains within IglE using deletion mutagenesis
Limited data exists on how IglE might exert its critical functions. A recent study by Robertson et al., in which in-frame deletions were used to map functional domains, suggested that regions within the C-terminus of IglE are required for intracellular growth and pathogenesis, while removing a region incorporating residues 2–23, and thus parts of the lipobox, led to protein instability.Citation34 To identify additional regions of importance for IglE function, we generated a series of deletion mutants, removing ∼20 residues at a time across the entire protein, resulting in mutants Δ2–22, Δ23–43, Δ44–64, Δ65–85, Δ86–105 and Δ106–125. All mutant proteins, similar to wild-type IglE, were expressed in trans as C-terminal GSK-fusions from the Francisella GroEL promoter of pMOL52, enabling their detection using anti-GSK antiserum.
We screened each mutant for its ability to restore the defects exhibited by LVS ΔiglE with regard to (i) intracellular growth after phagocytosis, (ii) induction of LDH release and (iii) inhibition of TNF-α secretion. Surprisingly, in contrast to wild-type IglE, none of the deletion mutants were able to complement LVS ΔiglE in any of the assays (data not shown). To determine whether this was due to a loss of protein expression/protein instability, we used immunoblot analysis to detect the different IglE mutant forms in bacterial lysates. Indeed, IglE mutants within the extreme N-terminus, i.e. Δ2–22 and Δ23–43, were found to be completely absent, suggesting that these variants most likely are unstable (Fig. S4A). Similar results were previously obtained upon deleting IglE residues 2–23.Citation34 In contrast, efficient synthesis of mutant proteins Δ44–64, Δ65–85 and Δ86–105 was observed, while levels of mutant Δ106–125 were low in comparison to the wild-type protein (Fig. S4A). To exclude that a general inherent instability of the aforementioned deletion variants was responsible for their inability to rescue the ΔiglE null mutant strain, we also performed a protein stability assay by which protein levels were determined by immunoblotting up to 240 min after stopping protein de novo synthesis with chloramphenicol. IglB levels were monitored at the same time as a loading control. We predicted that an IglE mutant form would be subject to constitutive proteolysis if unstable, resulting in a reduction of protein levels over time, however, similar to wild-type IglE, the cellular abundance of all of the mutant proteins were more or less unchanged up to 240 min post chloramphenicol addition (Fig. S4B). Thus, protein instability does not explain why the mutants fail to complement LVS ΔiglE in the cell assays. These results suggest that a region involving residues 44 to 125 is clearly critical for IglE function. A high-resolution crystal structure of IglE has unfortunately not yet been resolved,Citation46 however, according to Psipred (http://bioinf.cs.ucl.ac.uk/psipred/), this region predominantly contains β-strands and coils, with the exception of an α-helix in positions 54 to 61. Previously, Robertson et al identified a region encompassing residues 96 to 120 as important for IglE function,Citation34 and this overlaps with our identified region.
Previously, IglE lacking the signal sequence (aa 23–125) was demonstrated to interact with the C-terminus of PdpB (aa 590–1093) in a B2H assay,Citation33 raising the possibility that our deletion mutants might be defective for PdpB binding. To test this, we employed a B2H assay. In contrast to the results by Nguyen et al, no significant β-galactosidase activity was observed for the reporter strain expressing ω- and Zif fusions of IglE aa 23–125 and PdpB aa 590–1093 respectively, although the positive control MglA-SspA Citation47 induced strong activation of the β-galactosidase reporter in the same assay (data not shown). We also tested the full-length proteins and all possible combinations of full-length and truncated IglE and PdpB proteins against each other, but with the same negative outcome (data not shown). Possibly, the presence of unstable proteins may be an explanation for the lack of interaction in our assay, since protein degradation products were observed for most of the B2H constructs (Fig. S5).
Dissecting the N-terminus of IglE – key residues identified
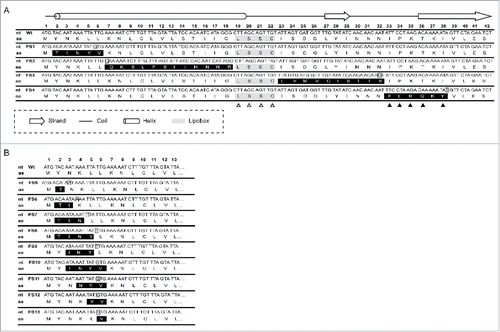
Our deletion mutagenesis strategy suggested that IglE is very sensitive for deletions within the extreme N-terminus, leading to instability. A different strategy was therefore necessary to further dissect this region. Thus, we generated a series of frameshift mutants, denoted FS1 to FS4 to identify key domains and residues within the first ∼38 N-terminal residues of IglE (for details see ). All four constructs carried significantly altered amino acid sequences although with only subtle changes to the mRNA sequence (). Using Psipred, the effect of the mutations on the expected secondary structure was estimated. The FS1 and FS2 mutations both overlapped with an expected N-terminal α-helix (), however, none of the mutations was predicted to alter this structure. The FS3 mutation overlapped with a predominantly coiled region containing a small β-strand formed by residues 27–29 (), which was predicted to include also residues 26 and 30 in the FS3 mutant protein. Finally, the FS4 mutation overlapped within a region of IglE expected to consist of a coil and a subsequent β-strand (), both of which were predicted to remain intact in the mutant according to Psipred. In addition to the frameshift mutants, we also generated single alanine substitution mutations within the lipobox motif, LSSC (residues 19 to 22), to determine the impact on IglE function (). Previously, a mutation of the invariant cysteine at position 22 resulted in an IglE variant that failed to be lipidated, nor did it support intracellular growth or virulence.Citation35,36 The importance of the remaining lipobox residues had so far not been investigated. The Psipred analysis suggested that for the WT (wild-type) and L19A proteins, the N-terminal α-helix ended with residue 19, while it also included residue 20 for mutants S20A, S21A and C22A. Again, all mutants were expressed as C-terminal GSK-fusions from pMOL52, enabling their detection using anti-GSK antiserum and each mutant was screened for its ability to complement LVS ΔiglE with regard to intracellular growth upon phagocytosis, induction of LDH release and inhibition of TNF-α secretion.
Figure 6. Schematic representation of mutations generated within the IglE N–terminus. Numbers 1–42 indicate the nucleotide triplet positioned with respect to the start codon of IglE. (A) Frameshift mutants FS1 to 4 covering the first 38 residues of IglE are shown. Residues altered via frameshift mutations are shaded in black, inserted nucleotides are boxed. To generate FS1, the nucleotide ‘T’ at position 4 was removed, which was compensated by the insertion of a ‘G’ immediately after the sixteenth nucleotide of IglE. To generate FS2, the nucleotide ‘C’ was inserted immediately after the eighteenth nucleotide of codon 6. This insertion was compensated by the removal of a ‘G’ at position 54 to restore the reading frame after codon 1718. FS3 was generated by omission of nucleotide 67 (an ‘A’) and insertion of ‘G’ after nucleotide 95 of codon 32. FS4 was generated by removal of nucleotide 97 (an ‘A’) followed by the insertion of ‘T’ after nucleotide 114 of codon 38. None of the Frameshift mutations overlapped with the lipidation site, LSSC, which is shaded in gray. All lipobox residues were individually substituted by alanine (see open triangles), while residues within the FS4 region were replaced individually or in pairs with the corresponding residues of the FS4 frameshift mutation (see filled triangles) (B) Frameshift mutants FS5 to 13 covering the first 6 residues of IglE are shown. Residues altered via frameshift mutations are shaded in black, inserted nucleotides are boxed. To generate mutant FS5, the nucleotide ‘T’ at position 4 was removed, which was compensated by the insertion of an ‘A’ immediately after the sixth eight nucleotide of IglE. To generate FS6, the nucleotide ‘T’ at position 4 was removed, which was compensated by the insertion of an ‘A’ immediately after the ninth tenth nucleotide. To generate FS7, the nucleotide ‘T’ at position 4 was removed, which was compensated by the insertion of a ‘T’ immediately after the twelfth thirteenth nucleotide. FS8, was generated by removing ‘T’ at position 4 and inserting ‘T’ after the fifteenth sixteenth nucleotide. FS9 was generated by removing ‘A’ at position 7 and inserting ‘T’ after the fifteenth sixteenth nucleotide. To generate FS10, ‘A’ at position 7 was removed and compensated by insertion of ‘G’ after the sixteenth nucleotide. To generate FS11, ‘A’ at position 10 was removed and compensated by insertion of ‘G’ after the sixteenth nucleotide. To generate FS12, ‘T’ at position 13 was removed and compensated by insertion of ‘G’ after the sixteenth nucleotide. To generate FS13, ‘T’ at position 16 was removed and exchanged to ‘G’.“
We observed efficient complementation for ΔiglE expressing lipobox-mutants S20A, S21A or frameshift mutant FS1 in all assays tested (, ). In contrast, ΔiglE expressing frameshift mutants FS2, FS3 or FS4 were all defective in the aforementioned assays (, ). Strikingly, the numbers of FS4 mutant-expressing F. tularensis dropped significantly during the time course (, ). In contrast, lipobox mutants L19A and C22A were capable of promoting efficient intramacrophage growth, in particular at 48 h, the numbers of bacteria were higher than those seen for LVS or ΔiglE/E (, ). Intriguingly, while C22A was unable to induce LDH release from J774 cells, L19A again showed an intermediate phenotype with high levels of LDH released at 48 h (, ), both showed intermediate suppression of TNF-α secretion (, ). Interestingly, a C22G mutant was previously shown to be unable to complement ΔiglE with respect to intramacrophage replication and virulence.Citation33,34 We also generated C22G as well as C22S mutants and these showed identical phenotypes to the C22A mutant in all assays tested (data not shown).
Table 2. Summary of the IglE mutant phenotypes.
Figure 7. Intracellular growth of F. tularensis IglE mutant strains, including (A) lipobox mutants, (B) frameshift mutants, and (C) substitution mutants within the region overlapping with FS4. J774 cells were infected by various strains of F. tularensis at an MOI of 200 for 2 h. Upon gentamicin treatment, cells were allowed to recover for 30 min after which they were lysed immediately (corresponds to 0 h; light gray bars) or after 24 h (medium gray bars) or 48 h (black bars) with PBS-buffered 0.1 % sodium deoxycholate solution and plated to determine the number of viable bacteria (log10). All infections were repeated 2 times and a representative experiment is shown. Each bar represents the mean values and the error bar indicates the SD from triplicate data sets. The asterisks indicate that the log10 number of CFU was significantly different from the parental LVS strain as determined by a 2-sided t-test with equal variance, including the Bonferroni correction for multiple pair-wise comparisons (*, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001).
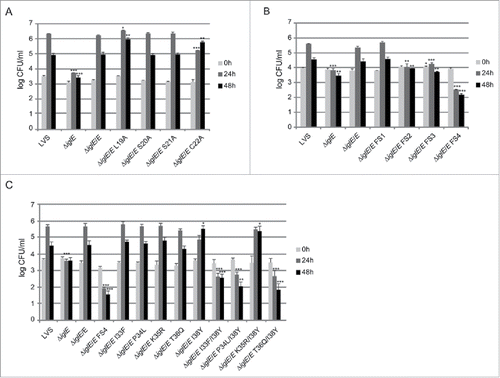
Figure 8. Cytopathogenicity of F. tularensis IglE mutant strains, including (A) lipobox mutants, (B) frameshift mutants, and (C) substitution mutants within the region overlapping with FS4. Culture supernatants of infected J774 cells were assayed for LDH activity at 0, 24 and 48 h and the activity was expressed as a percentage of the level of non-infected lysed cells (positive lysis control). Means and SD of triplicate wells from one representative experiment of 2 are shown. The asterisks indicate that the cytopathogenicity levels were significantly different from LVS-infected cells at a given time point as determined by a 2-sided t-test with equal variance, including the Bonferroni correction for multiple pair-wise comparisons (*, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001).
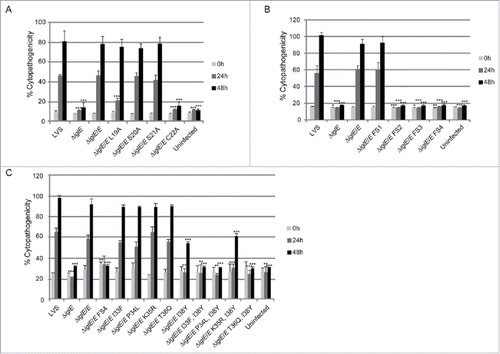
Figure 9. TNF-α secretion of F. tularensis infected macrophages. Uninfected J774 cells or cells infected with F. tularensis at an MOI of 500 for 2 h were washed and subsequently incubated in the presence of E. coli-derived LPS (50 ng/ml) for an additional 2 h. The average TNF-α secretion in % compared with LVS, which was set as 100 %, and the SD of quadruple samples (n = 4) from 2 or more representative experiments, are shown. The asterisks indicate that the cytokine levels were significantly different than those of LVS-infected cells as determined by a 2-sided t-test with equal variance, including the Bonferroni correction for multiple pair-wise comparisons (*, P ≤ 0.05; ***, P ≤ 0.001).
To determine whether functionality correlated with protein expression, we again used immunoblot analysis based on anti-GSK antiserum to detect the different IglE mutant forms in bacterial lysates. Out of the lipobox mutants, S20A and S21A were as abundant as the wild-type protein, but to our surprise we could not detect L19A or C22A (Fig. S6A, ). This was not expected since they showed partial complementation in the functionality assays (above), and hence must be expressed to some extent. We also tried to detect the mutant proteins using anti-IglE antiserum to exclude any problem with the GSK-tag, but with the same outcome (data not shown). Using qRT-PCR we detected a 7–8 times reduction in the mRNA levels of the L19A and C22A-encoding IglE-constructs, suggesting that the instability may be related to the mRNA levels. Notably, when isolated from Francisella, but not E. coli, these plasmids both showed partial degradation, which may explain the reduction of the mRNA levels. Importantly, previous studies have also revealed impaired expression of a C22G mutant, suggesting that substitutions of this amino acid affect protein levels, although not necessarily protein stability.Citation33,34 Out of the frameshift mutants, FS3 was not detected either, which may account for its aforementioned inability to complement the ΔiglE mutant (Fig. S6A, ). In contrast, FS1, FS2 and FS4 were all expressed at varying levels (Fig. S6A, ). Compared to wild-type IglE, FS1 was slightly less abundant, while FS4 was more abundant (Fig. S6A, ). The FS2 variant was poorly expressed and showed slower mobility in SDS-polyacrylamide gels compared with wild-type IglE, likely an effect from the altered sequence of codons 7–17 (Fig. S6A, ).
Out of the 3 frameshift mutants that failed to complement the ΔiglE mutant with respect to function (FS2 to FS4), FS4 was the only one that was expressed at high levels. Moreover, this mutant was avirulent in mice infected by the intradermal route (data not shown). We therefore decided to continue analyzing the contribution of individual residues to the defective phenotype of the FS4 mutant. Thus, each residue within the region incorporating amino acids 33 to 38 was individually exchanged to the counterpart of that of FS4, resulting in substitution mutants I33F, P34L, K35R, T36Q and I38Y. Since residue K37 was unaltered in the FS4 mutant, no substitution of this residue was made (). GSK-tagged variants were introduced into ΔiglE and their levels determined using anti-GSK antibodies. Notably, only K35R were produced at wild-levels. In contrast, the remaining mutant variants were more abundant, similar to the FS4 mutant protein (Fig. S6B, ). An ΔiglE mutant expressing either of I33F, P34L, K35R or T36Q exhibited efficient intramacrophage growth and induced an efficient cytopathogenic response similar to LVS and the wild-type-complemented mutant (, ). In contrast, ΔiglE expressing I38Y exhibited a minor but consistent growth defect at the 24 h time point and only demonstrated minor LDH release at 48 h (, ). This mutant also exhibited intermediate suppression of TNF-α secretion upon LPS stimulation, similar to the lipobox mutants L19A and C22A, while the other substitution mutants exhibited LVS-like suppression (, ). Hence, this pinpoints residue I38 as important for the IglE function. We then combined the I38Y mutation with the other single amino acid substitutions to look for additive effects on IglE function. The resulting double mutants, i.e., I33F/I38Y, P34L/I38Y, K35R/I38Y and T36Q/I38Y, were introduced in trans into the ΔiglE mutant. Similarly to FS4, all double mutants were expressed at elevated levels (Fig. S6B, ), but when tested for intracellular growth, LDH release and suppression of TNF-α secretion, a mixture of phenotypes were observed. Thus, similar to LVS and ΔiglE/E, the ΔiglE mutant expressing K35R/I38Y in trans demonstrated efficient growth (, ) but only minor LDH release, the latter similar to what observed for single mutant I38Y (, ). In the TNF-α secretion assay, this double mutant exhibited intermediate suppression (, ). Double mutants I33F/I38Y, P34L/I38Y and T36Q/I38Y failed to promote growth (, ). In fact, the numbers of CFU for ΔiglE expressing I33F/I38Y, and even more so P34L/I38Y or T36Q/I38Y, decreased over time, similar to what was observed for the FS4-complemented mutant (, ). None of these 3 double mutants were able to cause LDH release from infected J774 cells (, ), or suppress LPS-induced TNF-α secretion (, ). This suggests that all of the residues I33, P34, K35, T36 and I38 contribute to IglE function, as revealed by substituting one to 2 amino acids at a time.
IglC secretion promoted by mutated IglE variants in vitro
The inability of some of the generated IglE mutants to complement LVS ΔiglE may be a direct consequence of the loss of functional T6S. Thus, to determine their impact on IglC secretion, we again took advantage of the in vitro KCl secretion assay, introducing the GSK-tagged mutant variants into F. novicida ΔiglE and analyzing the amounts of secreted IglC in culture supernatants from strains grown in the presence of KCl. Overall, there was a strong correlation between the ability of any mutant IglE variant to promote IglC secretion and its ability to complement the LVS ΔiglE mutant in the various cell-assays (above). Thus, of the lipobox mutants, L19A and C22A were both defective for promoting IglC secretion, while the S20A and S21A mutants behaved similarly to the WT protein (, ). Similarly, out of the frameshift mutants FS1 to FS4, only FS1 could restore IglC secretion by the F. novicida ΔiglE mutant (, ). In addition, the in-frame deletion mutants which all were defective in the cell-based assays, were also defective for IglC secretion, whereas only minimal secretion was still maintained for ΔiglE expressing either of the 2 most C-terminal deletion mutants, i.e., Δ85–105 and Δ106–125 (). We also analyzed the ability of the various single- and double substitution mutants within the FS4 region (aa 33–38) to promote IglC secretion. Out of the 5 single amino acid mutants, only I38Y was defective in promoting IglC secretion (, ), correlating with a partially defective phenotype in the cell-based assays (above). All double mutants showed significantly reduced IglC secretion, K35R/I38Y being the least defective (, ). This mutant still maintained partial function in the cell assays, while the other ones behaved similarly to the severely defective FS4 mutant (above). Taken together, the results from this in vitro secretion assay suggest that IglE mutants that fail to promote efficient T6S are also defective for supporting essential processes, such as intracellular growth upon phagocytosis, phagosomal escape, cytopathogenicity and cytokine secretion.
Figure 10. IglC secretion promoted by F. novicida ΔiglE expressing various IglE mutants in trans, including (A) lipobox- and frameshift mutants, (B) deletion mutants, and (C) substitution mutants within the region overlapping with FS4. Indicated F. novicida strains were grown in TSB with 5 % KCl and FPI protein synthesis (pellet fractions) and secretion (cleared culture supernatants) were analyzed using SDS-PAGE and immunoblotting with anti-IglC antiserum. Immunoblotting for the inner membrane protein PdpB was included as a lysis control. The experiment was repeated 3 times and a representative example is shown.
Secretion of mutated IglE variants in vivo
Previously, we have shown that an IglE-TEM fusion is secreted by LVS during intracellular infection in a T6SS-dependent fashion.Citation29 To determine the efficiency of secretion of some of our IglE mutant variants, they were cloned into the TEM vector pJEB709 and the resulting plasmids transformed into LVS or ΔiglG. The latter control strain exhibits wild-type levels of replication in J774 macrophages, but only minor translocation of substrates during infection.Citation29 The resulting strains were used to infect J774 cells, and at 18 h, TEM substrate was added and the amounts of blue and green cells determined. Delivery of β-lactamase-tagged fusion proteins into the host cell cytosol will lead to cleavage of the TEM substrate, resulting in a change in fluorescence from green to blue emission. At this time point, LVS expressing wild-type IglE resulted in roughly 5% of blue cells, which was reduced below the cut-off of the assay (< 0.5%) when an ΔiglG mutant background was used (). When analyzing some of the lipobox mutants and frameshift mutants FS1 to FS4, 2 groups were distinguished; L19A, C22A, FS2, FS3 and FS4 were all secreted less efficiently than the wild-type protein (, ). In the case of L19A, C22A and FS3, the TEM fusion proteins appeared unstable, which likely accounts for these negative results (Fig. S7, ). In contrast, S21A, FS2 and FS4 were expressed to levels similar to the wild-type protein (FS4) or just below (S21A and FS2), but all exhibited impaired secretion (∼ 44%, ∼ 25% and ∼ 34% of WT-levels respectively) ( and S7, ). A TEM analysis using the single or double substitution mutants generated within the FS4 region suggested that the majority were expressed and secreted at levels very similar to WT IglE, with the exception of mutant P34L ( and S7, ). Thus, poor secretion of these mutant forms per se cannot explain the drastic defects that some of them exhibit in the various cell-based assays (above). To our surprise, frameshift mutant FS1 was secreted much more efficiently (∼ 15 times) than the wild-type protein. This occurred in an IglG-dependent fashion as the number of blue cells dropped by ∼ 98.5% when an ΔiglG mutant background was used instead of LVS ( and , ). This hyper-secretion phenotype does not, however, appear to be due to elevated levels of FS1 (Fig. S7). To verify the phenotype using a different subspecies of Francisella, we therefore expressed TEM fusions of WT IglE or FS1 in the F. novicida FTN1072 β-lactamase mutant. When the resulting strains were tested in the TEM assay, the FS1 variant was consistently better secreted than the WT TEM-fusion, resulting in ∼ 3 times more blue cells after infection (). Thus, the FS1 mutant form is clearly more efficiently secreted than the WT form of IglE in Francisella. We therefore decided to continue analyzing the region mutated in FS1 (residues 2–6), to try and further pinpoint the contribution of individual amino acids to this intriguing phenotype. Thus, we made additional sequential frameshift mutations called FS5 to FS13 within this region, mutating residue(s): 2 (FS5), 2–3 (FS6), 2–4 (FS7), 2–5 (FS8), 3–5 (FS9), 3–6 (FS10), 4–6 (FS11), 5–6 (FS12) and 6 (FS13) (for details, see ). For comparison, the resulting mutations were identical to the changes made in the FS1 clone. Importantly, GSK-tagged versions of all mutant variants behaved indistinguishably from the wild-type protein in their ability to restore intra-macrophage growth upon phagocytosis, LDH release and inhibition of TNF-α secretion of the LVS ΔiglE mutant (data not shown, ). When expressed as TEM fusions and introduced into LVS, 7 out of 9 frameshift mutants were hyper-secreted when compare with the WT protein. Of these, mutants FS6, FS7, FS8 and FS10 were secreted to the same degree as the FS1 fusion, while FS5, FS11 and FS12 were somewhat less hyper-secreted (, ). In contrast, FS9 was secreted to wild-type levels and secretion of FS13 was reduced (, ). When expressed in the ΔiglG background, secretion of all mutants was severely diminished (data not shown), showing a dependency on IglG for functional T6S. When bacterial lysates were analyzed, all mutants appeared to be expressed at more or less wild-type levels, with the exception of FS9 (Fig. S7) and this was also supported upon quantification using an ImageQuant LAS4000 imager. Thus, levels of FS9 was ∼ 64% of the wild-type IglE-TEM protein, while the levels of the remaining mutants were in the range of 96 to 115%. Thus, elevated protein levels cannot explain the hyper-secretion phenotype seen for several of the frameshift mutants. A bioinformatic-based analysis of the subtle differences in amino acid properties possessed by the frameshift mutants did not present any obvious pattern that could help distinguish the hyper-secreted variants from the non-hyper-secreted ones (data not shown). Additionally, since several of the frameshift mutants were generated to overlap, it is somewhat difficult to pinpoint the contribution of individual residues to this intriguing phenotype, although it should be noted that all mutants carrying the substitution Y2T (Tyrosine to Threonine) were hyper-secreted.
Figure 11. Secretion of IglE and the mutant variant FS1 into J774 macrophages. Macrophages were infected with LVS, ΔiglG or the F. novicida bla mutant FTN1072 expressing TEM fusions of wild-type IglE (WT) or the frameshift mutant FS1, carrying altered sequence of codons 2 to 6. After infection, cells were washed and loaded with CCF2/AM and analyzed using live cell microscopy. TEM β-lactamase activity is revealed by the blue fluorescence emitted by the cleaved CCF2 product, whereas uncleaved CCF2 emits a green fluorescence.
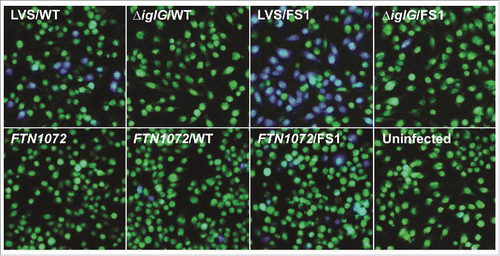
Figure 12. Secretion of IglE mutants into J774 macrophages. Macrophages were infected with LVS expressing TEM fusions of wild-type IglE (WT), lipobox mutants, frameshift mutants FS1 to FS13 or substitution mutants within the FS4 region (for details of the mutants, see ). After infection, cells were washed and loaded with CCF2/AM and analyzed using live cell microscopy. TEM β-lactamase activity is revealed by the blue fluorescence emitted by the cleaved CCF2 product, whereas uncleaved CCF2 emits a green fluorescence. The average secretion in % compared with the WT IglE protein, which was set as 100 %, and the SD from 2 samples (n = 2) from multiple experiments, in which 10,000 - 15,000 cells were counted in each experiment, are shown. The asterisks indicate that the secretion levels were significantly different than those of WT IglE-infected cells as determined by a 2-sided t-test with equal variance, including the Bonferroni correction for multiple pair-wise comparisons (***, P ≤ 0.001).
Discussion
The ability of F. tularensis to replicate intracellularly within host cells is intimately linked to the function of the Francisella Pathogenicity Island demonstrated to encode a T6SS. There are numerous publications demonstrating that a majority of the FPI proteins are essential for the function of this T6SS.Citation18,20,33-35,38,41,48-54 The FPI secretion system is phylogenetically distinct from all other described T6SSs, although some FPI gene products demonstrate homologies to conserved T6SS components, e.g., VipA (IglA), VipB (IglB), Hcp (IglC), IcmF (PdpB), IglG, VgrG, and DotU, of which IglA and IglB are the most conserved.Citation38,50 Their homologues of V. cholerae, VipA and VipB, are considered to constitute the sheath core structure of the T6S apparatus, since they spontaneously form tubular structures with cogwheel-like cross sections, all of which share remarkable structural similarities with the bacteriophage T4 sheath.Citation55 Recently, cryoelectron microscopy revealed a mesh-like architecture of the F. novicida T6SS sheath, consisting of IglA/IglBCitation44. Still, the structure of this T6SS is largely unknown. Presumably, many of the proteins constitute essential integral components of the machinery and, therefore, are required for cytosolic escape and secretion of effectors and, indirectly, intracellular replication. Previously, we have assessed the role of the FPI for secretion of FPI proteins and demonstrated by use of the TEM β-lactamase reporter assay that each of the components DotU, VgrG, and IglC are necessary for detectable secretion of the 8 FPI proteins secreted, one of which is IglE, during infection with F. tularensis LVS.Citation29 This suggests that the former encode structural components of the translocation machinery and clearly demonstrate that secretion of FPI proteins is indeed T6SS-dependent.Citation29 In the present study, we observed that the ΔiglE mutant was confined to the phagosome by confocal and electron microscopy and, as demonstrated for other F. tularensis mutants with the same confined phenotype, also incapable to replicate intracellularly upon phagocytosis, but not upon direct microinjection into the macrophage cytosol.Citation31 Analogously to the delayed phagosomal escape, we also observed that the cytopathogenic response was very distinct between LVS- and ΔiglE-infected cells, since the former cells demonstrated marked morphological effects and high levels of LDH release, while the latter cells were essentially indistinguishable from uninfected cells. These findings demonstrate key roles for IglE in the interaction of F. tularensis with phagocytic cells. Moreover, the loss of IglE also resulted in loss of virulence of the mutant in mice, a phenotype consistent with previously described FPI mutants that lack phagosomal escape and, hence, intracellular replication, thereby corroborating the intracellular nature of F. tularensis.Citation33-35,38,41,52,53,56
Despite that several of the FPI proteins are being secreted, bacterial fractionation experiments suggest that several of them display a membrane localization. This indicates that some FPI members may have multiple functions and serve as integral cell wall components, as well as integral components of the T6S machinery, and also secreted effectors. One such example is IglE, which not only is an integral membrane protein, but also a lipoprotein, and, in addition, was found to be the most effectively secreted protein out of the 8 FPI proteins secreted by F. tularensis LVS.Citation29 IglE demonstrates no obvious similarity to proteins other than F. tularensis homologs in Genbank, thus providing no clues as to its function, although in silico analysis has revealed that it possesses features typical of bacterial lipoproteins. This is evidenced by a 21-amino acid signal peptide with a positively charged N-terminus, followed by a hydrophobic region and a conserved lipobox motif. These predicted features have been validated experimentally by phase-partitioning and fatty acid-labeling and these studies also demonstrated that the lipidated protein upon fractionation is exclusively localized to the outer membrane.Citation33,34 Besides the validation of IglE as a lipoprotein, the essential role of the cysteine at position 22 was demonstrated as a substitution to glycine resulted in a protein incapable of complementation in both SCHU S4 and F. novicida. In addition to an altered localization from the outer membrane to both soluble and inner membrane fractions, the mutant protein was less stable and the total levels reduced.Citation33,34 In contrast, we observed that several mutations in the lipobox, i.e., L19A, S20A and S21A, resulted in preserved function, at least with regard to intracellular replication and cytopathogenicity, although the L19A mutant demonstrated an intermediate phenotype with regard to suppression of TNF-α secretion. Interestingly, efficient replication was also observed for the C22A, C22G, and C22S mutants, although they were distinct from the wild-type protein in that they, similar to the L19A mutant, were not observed in Western blot analysis and, in addition, they induced no cytotoxicity. This indicates that low levels of IglE are enough to promote intracellular replication and that lipidation is not necessary for the replication. Notably, the C22A mutant demonstrated no secretion of IglC, thus showing dissociation between intracellular growth and secretion. Intriguingly, such dissociation was also observed for IglG, since the N-terminus of the protein is critical for intracellular growth and virulence in mice, but not for T6SS-mediated secretion,Citation30 demonstrating that discrete parts of the FPI proteins have distinct functions.
Bacterial lipoproteins possess signal peptides and after processing, become N-terminally fatty acylated. It is generally believed that lipoproteins of Gram-negative bacteria are localized in the inner membrane, or in the inner surface of the outer membrane. For example, TssJ, the core constituents of the prototypic T6SS Sci-1 locus of E. coli is anchored to the periplasmic part of the outer membrane where it forms a complex with TssL and TssM that contacts the peptidoglycan layer.Citation57 TssJ was suggested by Nguyen et al to be a functional homolog of IglE.Citation33,34 However, unlike this prototypic, non-surface-localized T6SS lipoprotein, recent studies suggest that also surface-exposed lipoproteins may be rather prevalent.Citation58 In fact, there is such an example in F. tularensis, FipB, which is a lipoprotein and a virulence factor with both oxidase and isomerase activity.Citation59 Although IglE has a predominant outer membrane localization, there is no evidence in previous studies, or in the present study, that it is surface-localized.Citation33,34 and, moreover, it is secreted by both LVS and F. novicida during infection Citation29,32 Secretion of IglE is T6S-dependent, since no secretion was observed when T6S was abrogated.Citation29 These findings appear paradoxical in view of its outer membrane localization, but a likely explanation is that IglE may have dual roles; both as an outer membrane-localized lipoprotein and as a non-processed T6S effector. It is possible that processing may not occur for secreted T6SS lipoproteins, since it is executed by the membrane-localized signal peptidase II, whereas, presumably, proteins are selected for secretion and sorted already in the cytosol. In support of this assumption, we observed that the amino acid composition of the proximal part of the IglE N-terminus greatly affected its secretion in macrophages, which indicates that the T6S machinery recognizes this region before the proteins are secreted. However, our bioinformatic analysis did not provide any evidence as to what guides the machinery and did not identify any similarity between the FS1 region and regions of other secreted FPI proteins. Therefore, it is likely not a conserved amino acid sequence that serves to modulate secretion, but rather subtle changes in protein conformations. This is an area that requires much more experimental work on multiple effectors before testable hypotheses can be generated regarding the mechanisms modulating T6S in Francisella.
There are several studies demonstrating an AIM2-, ASC-, and pyrin-dependent activation of the inflammasome in F. tularensis-infected cells, resulting in cleavage of caspase-1 and efficient IL-1β release.Citation39,40,60,61 Inefficient IL-1β secretion has been observed by numerous FPI mutants, all with the common phenotype of being confined to the phagosome.Citation25,34,35,38,41,52 Accordingly, we observed much reduced IL-1β secretion by the phagosomally confined LVS ΔiglE mutant upon macrophage infection. Previously, we and others have demonstrated that the LVS infection renders the infected cells incapable to respond to TLR2 or TLR4 stimuli, such as bacterial lipoprotein or E. coli LPS.Citation24,35,41,62 It has been hypothesized that retention of F. tularensis in the phagosome prolongs the interaction with TLR2, thereby enhancing TLR2-dependent gene expression.Citation25,63 We investigated if this phenotype also applied to the ΔiglE mutant after stimulation with E. coli LPS. The resulting TNF-α secretion, an inflammasome-independent event, was followed and from cells infected with the mutant, high levels were secreted, whereas the LVS infection completely muted the response. Thus, in agreement with previous findings, the phagosomal location of the ΔiglE mutant renders it incapable of interfering with the LPS-induced signaling and indicates that the phagosomal escape is a necessary prerequisite for the perturbation.
The essential role of IglE and most other FPI proteins for the phagosomal escape, intracellular replication, and virulence have been documented numerous times, in almost all instances based on the use of mutants generated in LVS or F. novicida backgrounds.Citation20,33-35,38,41,48,50,52-54,64 Additionally, the importance of IglE has been well-documented in several genetic screens for F. tularensis virulence factors, e.g., it was identified, together with all other FPI proteins, in an F. novicida screen using a murine infection model Citation65 and also found to be essential for intracellular replication of F. novicida in murine RAW264.7 Citation66 and J774 macrophage-like cells.Citation50 Moreover, IglE was essential in a virulence screen in Drosophila melanogaster.Citation67 A caveat with many of the aforementioned studies has been the possibility that transposon insertions yield polar effects and; therefore, there is high likelihood that identification of individual FPI components as virulence factors instead depends on accidental inactivation of downstream factors. Thus, to pinpoint the roles of individuals FPI proteins, more thorough studies are required based on targeted in-frame deletion mutagenesis and complementation of the phenotypes. The corresponding mutants have rarely been generated in virulent F. tularensis strains, although there are exceptions and the relevance of the findings obtained by use of the mutants generated in low virulent strains have been corroborated predominantly in the SCHU S4 background.Citation30,34,52,53,56,68 There are 2 notable discrepancies, though; the deletion of PdpC did not affect the virulence of F. novicida, whereas the ΔpdpC mutant of LVS and SCHU S4 showed very marked attenuation.Citation50,52,53 Moreover, IglG is essential for intracellular replication of F. novicida, whereas the ΔiglG mutants of LVS and SCHU S4 replicate well, although with delayed kinetics.Citation30 In other cases, the phenotypes of mutants appear to be similar whether or not the mutations have been generated in attenuated or virulent F. tularensis strains.Citation33,Citation35,38,48,52,56,64,68 In agreement with the findings herein on the ΔiglE mutant, a similar phenotype was also noted for the SCHU S4 and F. novicida ΔiglE mutants.Citation33,34 Notably, the SCHU S4 ΔiglE mutant showed a clearly attenuated phenotype, but unlike the LVS ΔiglE mutant and some of the previously described FPI SCHU S4 mutants confined to the phagosome, e.g., ΔiglC, as much as ∼ 40% of the SCHU S4 ΔiglE bacteria escaped into the cytoplasm.Citation34,56 Thus, it is possible that the LVS mutant has more defective phagosomal escape than the corresponding SCHU S4 mutant.
Our use of multiple IglE mutants demonstrated a high correlation between IglC protein being secreted in the KCl assay and the ability of the corresponding mutants to complement the biological functions tested, the most important being intracellular replication. There was one notable exception to this correlation, though; the C22A mutant did not support secretion, but replicated efficiently intracellularly. Although it is possible that the high concentration of KCl is not a true physiological stimulus, our findings still validate the use of the KCl assay as a convenient means to analyze the effect of specific mutations on T6SS-mediated secretion in Francisella. A region incorporating residues 33 to 38 was found to be essential for IglE function as revealed by the phenotype of the FS4 frameshift mutant. When expressed in ΔiglE, the resulting strain was unable to support IglC secretion, elicit a cytopathogenic response, inhibit LPS-induced TNF-α secretion and cause disease in mice. Interestingly, the number of mutant bacteria dropped over time, suggesting that the mutant was sensitive to macrophage killing. Single amino acid mutants highlighted a role for residue I38 in all of the above mentioned processes, as the mutant (I38Y) showed a small, but consistent growth defect, defective IglC secretion, intermediate TNF-α secretion, and only a minor cytopathogenic response. Strikingly, by combining the I38Y mutation with mutations in either of I33, P34 or T36, mutants with a phenotype similar to the FS4 frameshift mutant was generated. Together this suggests that this region of IglE plays a critical role for its function. Intriguingly, the FS4 mutation, as well as all but one of the single and double substitution mutations generated within this region, resulted in increased protein levels compared with the WT-protein. Still, elevated levels, suggestive of increased protein stability, was not enough to rescue the iglE mutant bacteria. Future work will demonstrate how this complex region relates to the IglE structure, which may provide an explanation to all of these interesting observations.
The findings regarding IglE as well as several other FPI proteins provide indirect clues as to their functions. Most FPI mutants demonstrate a uniform null-mutant phenotype as evidenced by lack of phagosomal egress, intracellular replication and a cytopathogenic response. This has been interpreted to mean that the gene products all play a part as structural components in the common T6SS apparatus. More detailed, recent studies have, however, revealed a more nuanced picture and identified FPI mutants, e.g., ΔpdpE, ΔiglG, ΔiglI and ΔpdpC, with retained or partially retained functions with respect to the above mentioned processes.Citation30,35,53,69 These proteins all perform specific functions that are not essential for T6S as such, but still contribute to the virulence of the bacterium, PdpE being an exception. The role of IglE in this context appears not to be clear cut, on one hand its lipidation and outer membrane localization argue for a structural role, however, in view of its efficient secretion, it is not farfetched to consider that it also could execute effector functions. For this reason, the identification of putative eukaryotic targets during host cell infection will give an important clue as to the function of this unique protein.
Materials and methods
Bacterial strains, plasmids and growth conditions
Bacterial strains and plasmids used in this study are listed in . Escherichia coli strains were cultured in Luria Bertani broth (LB) or on Luria agar plates at 37°C. F. tularensis was grown on modified GC-agar base or in liquid Chamberlain's medium Citation74 at 37°C. When necessary, carbenicillin (Cb; 100 μg/ml), tetracycline (Tet; 10 μg/ml), kanamycin (Km; 50 μg/ml for E. coli, 10 μg/ml for F. tularensis), or chloramphenicol (Cm; 25 μg/ml for E. coli, 2.5 μg/ml for F. tularensis) were used.
Table 3. Strains and plasmids used in this study.
Construction of an ΔiglE null mutant in F. tularensis LVS and F. novicida U112
Primer combinations used to construct the iglE null mutants are listed in . All amplified fragments were first cloned into pCR4-TOPO TA cloning vector (450030, Invitrogen AB) to facilitate sequencing (Eurofins MWG Operon) before proceeding with the cloning. The iglE deletion construct was made as follows: upstream and downstream flanking regions of ∼1,200 bp were sequentially cloned into pBluescript SK+ (212205, Stratagene) using the XhoI/BamHI and BamHI/SacI sites, respectively, thereby generating a fragment encoding IglE Δ6–119 with flanking regions joined by a BamHI site. The deletion fragment was cloned into XhoI/SacI-digested pDM4, generating pJEB755 (pΔiglE). Conjugal mating experiments using S17–1λ pir as the donor strain and sucrose-selection allowed for the allelic exchange of the suicide plasmids within regions of complementary sequence on the chromosome of LVS or U112.Citation64 To remove both copies of the iglE gene in LVS, the procedure was repeated. PCR screening and western blot analysis was used to verify that the anticipated genetic event had occurred.
Table 4. Oligonucleotides used in this study.
Construction of expression vectors
Plasmids used in this study are listed in . Primer combinations and restriction enzymes used to generate the plasmids are listed in . All amplified fragments were first cloned into pCR4-TOPO TA cloning vector (450030, Invitrogen AB) to facilitate sequencing (Eurofins MWG Operon). PCR or overlap PCR was used to introduce deletion-, frameshift- or substitution mutations within iglE. To optimize expression, substitutions were adapted according to the codon usage preferences of Francisella tularensis according to http://www.kazusa.or.jp/codon. LVS was commonly used as template in the PCR reaction with the exception of when FS4 double substitution mutants were generated. In this case, the mutant iglE allele encoding I38Y was used as template. C-terminally GSK-tagged or TEM-tagged versions of iglE and mutated variants thereof were constructed by introducing non-tagged PCR amplified alleles into the NdeI/KpnI sites of pMOL52, a derivative of pKK289Km expressing GSK under the groE promoter Citation48 or into pJEB709, a derivative of pKK289Km expressing TEM under the groE promoter.Citation29 For expression of the FS1-TEM fusion in F. novicida, the whole FS1-TEM fusion was cut out as a single NdeI/EcoRI fragment from plasmid pJEB953 and subsequently cloned under the groE promoter of pKK214,Citation72 resulting in plasmid pJEB1039. Plasmids used for the Bacterial 2-hybrid analysis were constructed by introducing NdeI/NotI fragments of mutated iglE or pdpB/icmF into either pACTR-AP-Zif or pBRGPω.Citation75 Plasmids were transferred into F. tularensis or E. coli by electroporation.
Quantitative real-time PCR
Protocols for isolation of bacterial RNA, cDNA synthesis and qPCR have been described in detail elsewhere.Citation48 Primer sequences are given upon request. For all samples, controls were made with either template or reverse transcriptase omitted during cDNA synthesis. All reactions were performed in triplicate on 3 independent RNA preparations, with a 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA) using the Sequence Detection System software. Samples were normalized against the F. tularensis 17-kDa house-keeping gene tul4 (FTL0421) and compared with respective genes in LVS. Results were analyzed using the delta delta Ct method of analysis and converted to relative expression ratio (2−ΔΔCt) for statistical analysis.Citation76 Paired two-tailed t-tests were used to compare means.
Western blot analysis
Unless stated otherwise, lysates from bacteria grown on modified GC agar were prepared in Laemmli sample buffer and boiled prior to separation on 12–15% sodium dodecyl sulfate (SDS)-polyacrylamide gels (161–0175, Bio-Rad). Proteins were transferred onto nitrocellulose membranes (170–4158, Bio-Rad) using a semidry blotter (Bio-Rad laboratories, Hercules, CA). Membranes were probed with mouse monoclonal antibodies recognizing IglC or PdpB at 1/2,000 and 1/1,000 dilutions respectively (NR-3196 and NR-3198, BEI Resources), rabbit polyclonal antibodies recognizing VgrG at 1/1,000 dilution (Inbiolabs, Tallinn, Estonia), mouse monoclonal antibodies recognizing Tul4 (1/3,000 dilution) (kind gift from Dr J. Stulik, Czech Republic), rat polyclonal antibodies recognizing IglE Citation34 (1/2,000 dilution). GSK-tagged variants of IglE were detected using rabbit anti-GSK antibodies (1/1,000 dilution) (9336S, Cell Signaling Technology). The secondary horseradish peroxidase (HRP)-conjugated antibodies used were: goat anti-mouse and goat anti-rat (S-2005 and S-2032, Santa Cruz Biotechnology) at 1/2,000 and 1/5,000 dilutions respectively, and donkey anti-rabbit (NA934V, GE Healthcare) at 1/13,300 dilution. For detection, the Enhanced Chemiluminescence system (ECL; RPN 2232, GE Healthcare) was used.
Fractionation of F. tularensis
A detailed protocol for fractionation of F. tularensis bacteria into soluble, inner membrane and outer membrane fractions has been described in details elsewhere.Citation35 The purity of soluble, inner- and outer membrane fractions was determined using antiserum against IglC, PdpB/IcmF, and Tul4, respectively. Protein fractions, 5 µg, were separated by SDS-PAGE and analyzed using standard Western blot techniques together with ECL detection.
In vitro secretion assay
F. novicida strains grown O/N on MC agar were subcultured to OD600 = 0.15 into TSB medium supplemented with filter sterilized 0.1% Glucose, 0.1% Cysteine and, when appropriate, 5% KCl. Antibiotics were added when appropriate. Strains were grown until OD600 ∼ 1.5, upon which pellet samples as well as filter-sterilized supernatant samples containing secreted proteins were harvested. The supernatants, ∼ 7 - 8 ml, were TCA precipitated for 1 h on ice and centrifuged at 2,800 × g at 4°C for 30 minutes. The obtained precipitate was dissolved in 0.5 ml 2% SDS and 3 volumes of freeze cold acetone were added. After 30 min of incubation at -20°C, samples were centrifuged at 20,200 × g for 10 min, and the obtained pellet dried and dissolved in Sample buffer.
Bacterial 2-hybrid analysis
As the reporter strain for the bacterial-2-hybrid experiments, the E. coli strain KDZif1ΔZ was used.Citation73 It harbors an F9 episome containing the lac promoter-derivative placZif1–61 driving expression of a linked lacZ reporter gene. Cells were grown with aeration at 37°C in LB supplemented with 0.4 - 1 mM IPTG (I-6758, Sigma-Aldrich). Cells were permeabilized with SDS-CHCl3 and assayed for β-galactosidase activity as described previously.Citation75 Assays were performed at least 3 times in triplicate on separate occasions.
Cultivation and infection of macrophages
J774 macrophages (ATCCTIB-67) were used in all cell infection assays, except for the IL-1β secretion assay where PECs were used. J774 macrophages were cultured and maintained in DMEM (31966–047, GIBCO BRL) with 10% heat-inactivated FBS (10270098, GIBCO). PECs were isolated from 8- to 10-week-old C57BL/6 mice 3 d after intraperitoneal injection of 2 ml of 10% proteose peptone (82450, Sigma-Aldrich) as previously described.Citation77 The day before infection, macrophages were seeded in tissue culture plates in DMEM with 10% FBS. Following incubation overnight, cells were washed, reconstituted with fresh culture medium and allowed to recover for at least 30 min prior to infection. A multiplicity of infection (MOI) of 200 was used in all infection experiments, with the exception of the TNF-α secretion assay where we used a MOI of 500 Citation26 and for the TEM microscopy study, where a MOI of 1000 was used.Citation35
Intracellular replication in macrophages
To determine the ability of F. tularensis or F. novicida to grow within macrophages, cells were infected for 2 h, washed 3 times, and incubated in the presence of 5 μg/ml gentamicin for 30 min (corresponds to time zero). At 0, 24 and 48 h, the macrophage monolayers were lysed in PBS with 0.1% deoxycholate (D6750, Sigma-Aldrich), serially diluted in PBS and plated on modified GC-agar base plates for determination of viable counts. A two-sided t-test with equal variance was used to determine whether the growth of a strain differed significantly from that of LVS or U112.
Microinjection assay
The microinjection procedure was executed as described previously Citation31 and was repeated at least 3 times. In brief, LVS and isogenic mutants were suspended in PBS and mixed with rhodamine dextran (R9379, Sigma-Aldrich) and injected with 40 hPa into the cytosol of J774 cells. The injection medium contained gentamicin and cytochalasin D (C8273, Sigma-Aldrich) to eliminate extracellular bacteria and their phagocytosis. Approximately, 20 - 30 cells per strain and experiment were injected. Data are presented as means of the approximate number of bacteria per cell representing no/little, moderate or extensive bacterial replication at 24 h, respectively. Significant difference between the mean numbers of bacteria for each strain at each time point was determined by the chi-square test.
LDH release assay
The LDH release assay has been described in detail elsewhere.Citation35 In short, cells were infected as described in “Intracellular replication in macrophages” and supernatants were sampled at 0, 24 or 48 h and assayed for the presence of released Lactate dehydrogenase (LDH). Data are means ± SD of 3 wells from one representative experiment of 3. Uninfected cells lysed in PBS with 0.1% deoxycholate served as a positive control, and the value for this control was arbitrarily considered 100% cell lysis. Sample absorbance was expressed as the percentage of the positive control value.
Intracellular immunofluorescence assay
To assess phagosomal escape, GFP-expressing F. tularensis were used in the cell infections as described previously.Citation48 Cells were then stained for the LAMP-1 glycoprotein as described previously.Citation49 Colocalization of GFP-labeled F. tularensis and LAMP-1 was analyzed with an epifluorescence microscope (ZeissAxioskop2; Carl Zeiss MicroImaging, Jena, Germany) and a confocal microscope (Nikon Eclipse 90i, Nikon Instruments, Amsterdam, Netherlands). From two separate experiments, each with a total number of 5 glass slides per strain, 50 bacteria/slide were scored.
Transmission electron microscopy
The protocol describing the infection and sample preparation for TEM has been described in details elsewhere.Citation35 Sections were viewed with a JEOL JEM 1230 Transmission Electron Microscope (JEOL Ltd., Tokyo, Japan). To examine membrane integrity, at least 100 bacteria from 2 different sections were analyzed and categorized as having: (i) an intact phagosomal membrane, (ii) a slightly damaged phagosomal membrane (< 50% of membrane integrity affected), (iii) a highly damaged phagosomal membrane (> 50% of membrane integrity affected) or (iv) no residual membrane.
TEM secretion assay
The TEM secretion assay has been described in detail elsewhere.Citation29 In short, at 18 h after infection cells were washed twice with PBS before loading them with CCF2-AM in the presence of Probenecid (P36400, Invitrogen) (f. c 2.5 mM) at RT according to the manufacturer's instructions (Invitrogen, CA, USA). Translocation of β-lactamase fusions into CCF2-loaded cells was determined after 60 min of incubation, by counting the number of blue fluorescent cells in images taken with a live-cell imaging microscope (Nikon Eclipse Ti-E) equipped with a Nikon DS-U2/L2 camera, using a Chroma β-lactamase double filter. Loaded cells that did not exhibit secretion of TEM fusions appeared green. Images were assembled using Adobe Photoshop CS2 (Adobe Systems, San Jose, CA). Each strain, freshly transformed with the TEM expression vector, was tested on at least 4 separate occasions using duplicate wells. For statistical analysis of blue vs green fluorescent cells, an average of 10,000– 15,000 cells that included pictures from all separate experiments for a particular strain was counted. Each picture was considered an independent observation and used to calculate the average % of blue fluorescent cells and the SD for a particular strain. A TEM-fusion was considered to be non-secreted if it resulted in less than 1 blue fluorescent cell per 200 cells, i.e., the cut-off of the assay was < 0.5%. Student´s t-test was used to determine whether the % of blue fluorescent cells differed significantly between strains harboring TEM fusions with wild-type or mutant forms of IglE.
TNF-α secretion assay
To measure TNF-α secretion upon 2 h of LPS stimulation of J774 cells, we followed our previously established protocols,Citation35 using the BD OptEIA Mouse TNF-α Elisa Set (555268, BD Biosciences) according to the manufacturer's instructions.
IL-1β secretion assay
The IL-1β secretion assay was performed as described previously.Citation35 Samples were taken at 24 h and analyzed using the BD OptEIA Mouse IL-1β Elisa Set (559603, BD Biosciences) according to the manufacturer's instructions.
Mouse infections
For determination of the virulence of each strain, C57BL/6 female mice (n = 5) were infected intradermally. Aliquots of the diluted cultures were also plated on GC-agar to determine the number of CFU injected. The virulence of the ΔiglE mutant and the complemented strain was tested in 3 separate experiments with consistent results. For one experiment in which approximately 4 × 107 or 6 × 108 CFU of F. tularensis were injected, the actual doses were: 5.0 × 107 (LVS), 6.9 × 108 (ΔiglE), 3.0 × 107 (ΔiglE/E) and 7.0 × 108 (ΔiglE/E), 3.0 × 108 (ΔiglE/FS4). Mice were examined twice daily for signs of severe infection and euthanized by CO2 asphyxiation as soon as they displayed signs of irreversible morbidity. In our experience, such mice were at most 24 h from death, and time to death of these animals was estimated on this premise. All animal experiments were approved by the Local Ethical Committee on Laboratory Animals, Umeå, Sweden (no. A113–08, A99–11, and A67–14).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KVIR_S_1258507.pdf
Download PDF (775.5 KB)Acknowledgements
We are indebted to Dr Marc Röhm for assistance with the confocal microscopy, Dr G. Robertson for kindly providing the anti-IglE antiserum, Dr J. Stulik for providing the Tul4 antiserum, Drs Moa Lavander and Athar Alam for constructing the pMOL53 and pALA009 vectors, respectively, and Lena Lindgren for valuable technical assistance.
Funding
Financial support was obtained from the Swedish Research Council, K2013–4581 and K2013–8621, and a grant from the Medical Faculty, Umeå University, Umeå, Sweden. The work was performed in part at the Umeå Center for Microbial Research (UCMR).
References
- Bingle LE, Bailey CM, Pallen MJ. Type VI secretion: a beginner's guide. Curr Opin Microbiol 2008; 11:3-8; PMID:18289922; http://dx.doi.org/10.1016/j.mib.2008.01.006
- Boyer F, Fichant G, Berthod J, Vandenbrouck Y, Attree I. Dissecting the bacterial type VI secretion system by a genome wide in silico analysis: what can be learned from availabel microbial genomic resources? BMC Genomics 2009; 10:104; PMID:19284603; http://dx.doi.org/10.1186/1471-2164-10-104
- Lery LM, Frangeul L, Tomas A, Passet V, Almeida AS, Bialek-Davenet S, Barbe V, Bengoechea JA, Sansonetti P, Brisse S, et al. Comparative analysis of Klebsiella pneumoniae genomes identifies a phospholipase D family protein as a novel virulence factor. BMC Biol 2014; 12:41; PMID:24885329; http://dx.doi.org/10.1186/1741-7007-12-41
- Suarez G, Sierra JC, Erova TE, Sha J, Horneman AJ, Chopra AK. A type VI secretion system effector protein, VgrG1, from Aeromonas hydrophila that induces host cell toxicity by ADP ribosylation of actin. J Bacteriol 2010; 192:155-68; PMID:19880608; http://dx.doi.org/10.1128/JB.01260-09
- Pukatzki S, Ma AT, Revel AT, Sturtevant D, Mekalanos JJ. Type VI secretion system translocates a phage tail spike-like protein into target cells where it cross-links actin. Proc Natl Acad Sci U S A 2007; 104:15508-13; PMID:17873062; http://dx.doi.org/10.1073/pnas.0706532104
- Toesca IJ, French CT, Miller JF. The Type VI secretion system spike protein VgrG5 mediates membrane fusion during intercellular spread by pseudomallei group Burkholderia species. Infect Immun 2014; 82:1436-44; PMID:24421040; http://dx.doi.org/10.1128/IAI.01367-13
- Hood RD, Singh P, Hsu F, Guvener T, Carl MA, Trinidad RR, Silverman JM, Ohlson BB, Hicks KG, Plemel RL, et al. A type VI secretion system of Pseudomonas aeruginosa targets a toxin to bacteria. Cell Host Microbe 2010; 7:25-37; PMID:20114026; http://dx.doi.org/10.1016/j.chom.2009.12.007
- Russell AB, Singh P, Brittnacher M, Bui NK, Hood RD, Carl MA, Agnello DM, Schwarz S, Goodlett DR, Vollmer W, et al. A widespread bacterial type VI secretion effector superfamily identified using a heuristic approach. Cell Host Microbe 2012; 11:538-49; PMID:22607806; http://dx.doi.org/10.1016/j.chom.2012.04.007
- Ma LS, Hachani A, Lin JS, Filloux A, Lai EM. Agrobacterium tumefaciens deploys a superfamily of type VI secretion DNase effectors as weapons for interbacterial competition in planta. Cell Host Microbe 2014; 16:94-104; PMID:24981331; http://dx.doi.org/10.1016/j.chom.2014.06.002
- Russell AB, Hood RD, Bui NK, LeRoux M, Vollmer W, Mougous JD. Type VI secretion delivers bacteriolytic effectors to target cells. Nature 2011; 475:343-7; PMID:21776080; http://dx.doi.org/10.1038/nature10244
- English G, Trunk K, Rao VA, Srikannathasan V, Hunter WN, Coulthurst SJ. New secreted toxins and immunity proteins encoded within the Type VI secretion system gene cluster of Serratia marcescens. Mol Microbiol 2012; 86:921-36; PMID:22957938
- Whitney JC, Chou S, Russell AB, Biboy J, Gardiner TE, Ferrin MA, Brittnacher M, Vollmer W, Mougous JD. Identification, structure, and function of a novel type VI secretion peptidoglycan glycoside hydrolase effector-immunity pair. J Biol Chem 2013; 288:26616-24; PMID:23878199; http://dx.doi.org/10.1074/jbc.M113.488320
- Pukatzki S, Ma AT, Sturtevant D, Krastins B, Sarracino D, Nelson WC, Heidelberg JF, Mekalanos JJ. Identification of a conserved bacterial protein secretion system in Vibrio cholerae using the Dictyostelium host model system. Proc Natl Acad Sci U S A 2006; 103:1528-33; PMID:16432199; http://dx.doi.org/10.1073/pnas.0510322103
- Salomon D, Kinch LN, Trudgian DC, Guo X, Klimko JA, Grishin NV, Mirzaei H, Orth K. Marker for type VI secretion system effectors. Proc Natl Acad Sci U S A 2014; 111:9271-6; PMID:24927539; http://dx.doi.org/10.1073/pnas.1406110111
- Liang X, Moore R, Wilton M, Wong MJ, Lam L, Dong TG. Identification of divergent type VI secretion effectors using a conserved chaperone domain. Proc Natl Acad Sci U S A 2015; 112:9106-11; PMID:26150500
- Sjöstedt A. Intracellular survival mechanisms of Francisella tularensis, a stealth pathogen. Microbes Infect 2006; 8:561-7; PMID:16239121; http://dx.doi.org/10.1016/j.micinf.2005.08.001
- Clemens DL, Lee BY, Horwitz MA. Virulent and avirulent strains of Francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect Immun 2004; 72:3204-17; PMID:15155622; http://dx.doi.org/10.1128/IAI.72.6.3204-3217.2004
- Golovliov I, Baranov V, Krocova Z, Kovarova H, Sjöstedt A. An attenuated strain of the facultative intracellular bacterium Francisella tularensis can escape the phagosome of monocytic cells. Infect Immun 2003; 71:5940-50; PMID:14500514; http://dx.doi.org/10.1128/IAI.71.10.5940-5950.2003
- Lai XH, Golovliov I, Sjöstedt A. Francisella tularensis induces cytopathogenicity and apoptosis in murine macrophages via a mechanism that requires intracellular bacterial multiplication. Infect Immun 2001; 69:4691-4; PMID:11402018; http://dx.doi.org/10.1128/IAI.69.7.4691-4694.2001
- Mariathasan S, Weiss DS, Dixit VM, Monack DM. Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J Exp Med 2005; 202:1043-9; PMID:16230474; http://dx.doi.org/10.1084/jem.20050977
- Wickstrum JR, Bokhari SM, Fischer JL, Pinson DM, Yeh HW, Horvat RT, Parmely MJ. Francisella tularensis induces extensive caspase-3 activation and apoptotic cell death in the tissues of infected mice. Infect Immun 2009; 77:4827-36; PMID:19703976; http://dx.doi.org/10.1128/IAI.00246-09
- Hazlett KR, Cirillo KA. Environmental adaptation of Francisella tularensis. Microbes Infect 2009; 11:828-34; PMID:19524059; http://dx.doi.org/10.1016/j.micinf.2009.06.001
- Rajaram MV, Ganesan LP, Parsa KV, Butchar JP, Gunn JS, Tridandapani S. Akt/Protein kinase B modulates macrophage inflammatory response to Francisella infection and confers a survival advantage in mice. J Immunol 2006; 177:6317-24; PMID:17056562; http://dx.doi.org/10.4049/jimmunol.177.9.6317
- Telepnev M, Golovliov I, Sjöstedt A. Francisella tularensis LVS initially activates but subsequently down-regulates intracellular signaling and cytokine secretion in mouse monocytic and human peripheral blood mononuclear cells. Microb Pathog 2005; 38:239-47; PMID:15925273; http://dx.doi.org/10.1016/j.micpath.2005.02.003
- Cole LE, Santiago A, Barry E, Kang TJ, Shirey KA, Roberts ZJ, Elkins KL, Cross AS, Vogel SN. Macrophage proinflammatory response to Francisella tularensis live vaccine strain requires coordination of multiple signaling pathways. J Immunol 2008; 180:6885-91; PMID:18453609; http://dx.doi.org/10.4049/jimmunol.180.10.6885
- Telepnev M, Golovliov I, Grundström T, Tärnvik A, Sjöstedt A. Francisella tularensis inhibits Toll-like receptor-mediated activation of intracellular signalling and secretion of TNF-alpha and IL-1 from murine macrophages. Cell Microbiol 2003; 5:41-51; PMID:12542469; http://dx.doi.org/10.1046/j.1462-5822.2003.00251.x
- Bosio CM, Dow SW. Francisella tularensis induces aberrant activation of pulmonary dendritic cells. J Immunol 2005; 175:6792-801; PMID:16272336; http://dx.doi.org/10.4049/jimmunol.175.10.6792
- Bröms JE, Sjöstedt A, Lavander M. The role of the Francisella tularensis pathogenicity island in Type VI secretion, intracellular survival, and modulation of host cell signaling. Front Microbiol 2010; 1:136; PMID:21687753; http://dx.doi.org/10.3389/fmicb.2010.00136
- Bröms JE, Meyer L, Sun K, Lavander M, Sjöstedt A. Unique substrates secreted by the type VI secretion system of Francisella tularensis during intramacrophage infection. PLoS One 2012; 7:e50473; PMID:23185631; http://dx.doi.org/10.1371/journal.pone.0050473
- Rigard M, Bröms JE, Mosnier A, Hologne M, Martin A, Lays C, Lindgren L, Punginelli C, Walker O, Charbit A, et al. Francisella tularensis IglG belongs to a novel family of PAAR-like T6SS proteins and harbors a unique N-terminal extension required for virulence. PLoS Pathogens 2016; 12:e1005821; PMID:27602570; http://dx.doi.org/10.1371/journal.ppat.1005821
- Meyer L, Bröms JE, Liu X, Rottenberg ME, Sjöstedt A. Microinjection of Francisella tularensis and Listeria monocytogenes reveals the importance of bacterial and host factors for successful replication. Infect Immun 2015; 83:3233-42; PMID:26034213; http://dx.doi.org/10.1128/IAI.00416-15
- Hare RF, Hueffer K. Francisella novicida pathogenicity island encoded proteins were secreted during infection of macrophage-like cells. PLoS One 2014; 9:e105773; PMID:25158041; http://dx.doi.org/10.1371/journal.pone.0105773
- Nguyen JQ, Gilley RP, Zogaj X, Rodriguez SA, Klose KE. Lipidation of the FPI protein IglE contributes to Francisella tularensis ssp. novicida intramacrophage replication and virulence. Pathog Dis 2014; 72:10-8; PMID:24616435
- Robertson GT, Child R, Ingle C, Celli J, Norgard MV. IglE is an outer membrane-associated lipoprotein essential for intracellular survival and murine virulence of type A Francisella tularensis. Infect Immun 2013; 81:4026-40; PMID:23959721; http://dx.doi.org/10.1128/IAI.00595-13
- Bröms JE, Lavander M, Meyer L, Sjöstedt A. IglG and IglI of the Francisella pathogenicity island are important virulence determinants of Francisella tularensis LVS. Infect Immun 2011; 79:3683-96; PMID:21690239; http://dx.doi.org/10.1128/IAI.01344-10
- Chong A, Celli J. The Francisella intracellular life cycle: toward molecular mechanisms of intracellular survival and proliferation. Front Microbiol 2010; 1:138; PMID:21687806; http://dx.doi.org/10.3389/fmicb.2010.00138
- Goetz M, Bubert A, Wang G, Chico-Calero I, Vazquez-Boland JA, Beck M, Slaghuis J, Szalay AA, Goebel W. Microinjection and growth of bacteria in the cytosol of mammalian host cells. Proc Natl Acad Sci U S A 2001; 98:12221-6; PMID:11572936; http://dx.doi.org/10.1073/pnas.211106398
- Barker JR, Chong A, Wehrly TD, Yu JJ, Rodriguez SA, Liu J, Celli J, Arulanandam BP, Klose KE. The Francisella tularensis pathogenicity island encodes a secretion system that is required for phagosome escape and virulence. Mol Microbiol 2009; 74:1459-70; PMID:20054881; http://dx.doi.org/10.1111/j.1365-2958.2009.06947.x
- Cremer TJ, Ravneberg DH, Clay CD, Piper-Hunter MG, Marsh CB, Elton TS, Gunn JS, Amer A, Kanneganti TD, Schlesinger LS, et al. MiR-155 induction by F. novicida but not the virulent F. tularensis results in SHIP down-regulation and enhanced pro-inflammatory cytokine response. PLoS One 2009; 4:e8508; PMID:20041145; http://dx.doi.org/10.1371/journal.pone.0008508
- Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O'Rourke K, Chan S, Dong J, Qu Y, Roose-Girma M, et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci U S A 2010; 107:9771-6; PMID:20457908; http://dx.doi.org/10.1073/pnas.1003738107
- Bröms JE, Meyer L, Lavander M, Larsson P, Sjöstedt A. DotU and VgrG, core components of type VI secretion systems, are essential for Francisella LVS pathogenicity. PLoS One 2012; 7:e34639; PMID:22514651; http://dx.doi.org/10.1371/journal.pone.0034639
- Lindgren M, Eneslätt K, Bröms JE, Sjöstedt A. Importance of PdpC, IglC, IglI, and IglG for modulation of a host cell death pathway induced by Francisella tularensis. Infect Immun 2013; 81:2076-84; PMID:23529623; http://dx.doi.org/10.1128/IAI.00275-13
- Forslund AL, Kuoppa K, Svensson K, Salomonsson E, Johansson A, Byström M, Oyston PC, Michell SL, Titball RW, Noppa L, et al. Direct repeat-mediated deletion of a type IV pilin gene results in major virulence attenuation of Francisella tularensis. Mol Microbiol 2006; 59:1818-30; PMID:16553886; http://dx.doi.org/10.1111/j.1365-2958.2006.05061.x
- Clemens DL, Ge P, Lee BY, Horwitz MA, Zhou ZH. Atomic structure of T6SS reveals interlaced array essential to function. Cell 2015; 160:940-51; PMID:25723168; http://dx.doi.org/10.1016/j.cell.2015.02.005
- Qin A, Zhang Y, Clark ME, Moore EA, Rabideau MM, Moreau GB, Mann BJ. Components of the type six secretion system are substrates of Francisella tularensis Schu S4 DsbA-like FipB protein. Virulence 2016:1-13; PMID:27028889; http://dx.doi.org/10.1080/21505594.2016.1168550
- Robb CS, Nano FE, Boraston AB. Cloning, expression, purification, crystallization and preliminary X-ray diffraction analysis of intracellular growth locus E (IglE) protein from Francisella tularensis subsp. novicida. Acta Crystallogr Sect F Struct Biol Cryst Commun 2010; 66:1596-8; PMID:21139203; http://dx.doi.org/10.1107/S1744309110034378
- Charity JC, Costante-Hamm MM, Balon EL, Boyd DH, Rubin EJ, Dove SL. Twin RNA polymerase-associated proteins control virulence gene expression in Francisella tularensis. PLoS Pathog 2007; 3:e84; PMID:17571921; http://dx.doi.org/10.1371/journal.ppat.0030084
- Bröms JE, Lavander M, Sjöstedt A. A conserved alpha-helix essential for a type VI secretion-like system of Francisella tularensis. J Bacteriol 2009; 191:2431-46; PMID:19201795; http://dx.doi.org/10.1128/JB.01759-08
- Bönquist L, Lindgren H, Golovliov I, Guina T, Sjöstedt A. MglA and Igl proteins contribute to the modulation of Francisella tularensis live vaccine strain-containing phagosomes in murine macrophages. Infect Immun 2008; 76:3502-10; PMID:18474647; http://dx.doi.org/10.1128/IAI.00226-08
- de Bruin OM, Duplantis BN, Ludu JS, Hare RF, Nix EB, Schmerk CL, Robb CS, Boraston AB, Hueffer K, Nano FE. The biochemical properties of the Francisella pathogenicity island (FPI)-encoded proteins IglA, IglB, IglC, PdpB and DotU suggest roles in type VI secretion. Microbiology 2011; 157:3483-91; PMID:21980115; http://dx.doi.org/10.1099/mic.0.052308-0
- Lindgren H, Golovliov I, Baranov V, Ernst RK, Telepnev M, Sjöstedt A. Factors affecting the escape of Francisella tularensis from the phagolysosome. J Med Microbiol 2004; 53:953-8; PMID:15358816; http://dx.doi.org/10.1099/jmm.0.45685-0
- Lindgren M, Tancred L, Golovliov I, Conlan W, Twine SM, Sjöstedt A. Identification of mechanisms for attenuation of the FSC043 mutant of Francisella tularensis SCHU S4. Infect Immun 2014; 82:3622-35; PMID:24935978; http://dx.doi.org/10.1128/IAI.01406-13
- Long ME, Lindemann SR, Rasmussen JA, Jones BD, Allen LA. Disruption of Francisella tularensis Schu S4 iglI, iglJ, and pdpC genes results in attenuation for growth in human macrophages and in vivo virulence in mice and reveals a unique phenotype for pdpC. Infect Immun 2013; 81:850-61; PMID:23275090; http://dx.doi.org/10.1128/IAI.00822-12
- Santic M, Molmeret M, Klose KE, Jones S, Kwaik YA. The Francisella tularensis pathogenicity island protein IglC and its regulator MglA are essential for modulating phagosome biogenesis and subsequent bacterial escape into the cytoplasm. Cell Microbiol 2005; 7:969-79; PMID:15953029; http://dx.doi.org/10.1111/j.1462-5822.2005.00526.x
- Basler M, Pilhofer M, Henderson GP, Jensen GJ, Mekalanos JJ. Type VI secretion requires a dynamic contractile phage tail-like structure. Nature 2012; 483:182-6; PMID:22367545; http://dx.doi.org/10.1038/nature10846
- Twine S, Bystrom M, Chen W, Forsman M, Golovliov I, Johansson A, Kelly J, Lindgren H, Svensson K, Zingmark C, et al. A mutant of Francisella tularensis strain SCHU S4 lacking the ability to express a 58-kgdalton protein is attenuated for virulence and is an effective live vaccine. Infect Immun 2005; 73:8345-52; PMID:16299332; http://dx.doi.org/10.1128/IAI.73.12.8345-8352.2005
- Silverman JM, Brunet YR, Cascales E, Mougous JD. Structure and regulation of the type VI secretion system. Annu Rev Microbiol 2012; 66:453-72; PMID:22746332; http://dx.doi.org/10.1146/annurev-micro-121809-151619
- Wilson MM, Bernstein HD. Surface-exposed lipoproteins: an emerging secretion phenomenon in Gram-negative bacteria. Trends Microbiol 2016; 24:198-208; PMID:26711681; http://dx.doi.org/10.1016/j.tim.2015.11.006
- Qin A, Zhang Y, Clark ME, Rabideau MM, Millan Barea LR, Mann BJ. FipB, an essential virulence factor of Francisella tularensis subsp. tularensis, has dual roles in disulfide bond formation. J Bacteriol 2014; 196:3571-81; PMID:25092026; http://dx.doi.org/10.1128/JB.01359-13
- Gavrilin MA, Bouakl IJ, Knatz NL, Duncan MD, Hall MW, Gunn JS, Wewers MD. Internalization and phagosome escape required for Francisella to induce human monocyte IL-1beta processing and release. Proc Natl Acad Sci U S A 2006; 103:141-6; PMID:16373510; http://dx.doi.org/10.1073/pnas.0504271103
- Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006; 440:228-32; PMID:16407890; http://dx.doi.org/10.1038/nature04515
- Abplanalp AL, Morris IR, Parida BK, Teale JM, Berton MT. TLR-dependent control of Francisella tularensis infection and host inflammatory responses. PLoS One 2009; 4:e7920; PMID:19936231; http://dx.doi.org/10.1371/journal.pone.0007920
- Cole LE, Laird MH, Seekatz A, Santiago A, Jiang Z, Barry E, Shirey KA, Fitzgerald KA, Vogel SN. Phagosomal retention of Francisella tularensis results in TIRAP/Mal-independent TLR2 signaling. J Leukoc Biol 2010; 87:275-81; PMID:19889726; http://dx.doi.org/10.1189/jlb.0909619
- Golovliov I, Sjöstedt A, Mokrievich A, Pavlov V. A method for allelic replacement in Francisella tularensis. FEMS Microbiol Lett 2003; 222:273-80; PMID:12770718; http://dx.doi.org/10.1016/S0378-1097(03)00313-6
- Weiss DS, Brotcke A, Henry T, Margolis JJ, Chan K, Monack DM. In vivo negative selection screen identifies genes required for Francisella virulence. Proc Natl Acad Sci U S A 2007; 104:6037-42; PMID:17389372; http://dx.doi.org/10.1073/pnas.0609675104
- Llewellyn AC, Jones CL, Napier BA, Bina JE, Weiss DS. Macrophage replication screen identifies a novel Francisella hydroperoxide resistance protein involved in virulence. PLoS One 2011; 6:e24201; PMID:21915295; http://dx.doi.org/10.1371/journal.pone.0024201
- Åhlund MK, Ryden P, Sjöstedt A, Stöven S. A directed screen of Francisella novicida virulence determinants using Drosophila melanogaster. Infect Immun 2010; 78:3118-28; PMID:20479082
- Kadzhaev K, Zingmark C, Golovliov I, Bolanowski M, Shen H, Conlan W, Sjöstedt A. Identification of genes contributing to the virulence of Francisella tularensis SCHU S4 in a mouse intradermal infection model. PLoS One 2009; 4:e5463; PMID:19424499; http://dx.doi.org/10.1371/journal.pone.0005463
- Lindgren M, Bröms JE, Meyer L, Golovliov I, Sjöstedt A. The Francisella tularensis LVS DeltapdpC mutant exhibits a unique phenotype during intracellular infection. BMC Microbiol 2013; 13:20; PMID:23356941; http://dx.doi.org/10.1186/1471-2180-13-20
- Simon R, Priefer U, Pühler A. A broad host range mobilisation system for in vivo genetic engineering: transposon mutagenesis in Gram-negative bacteria. Biotechnology 1983; 1:787-96
- Milton DL, O'Toole R, Horstedt P, Wolf-Watz H. Flagellin A is essential for the virulence of Vibrio anguillarum. J Bacteriol 1996; 178:1310-9; PMID:8631707
- Kuoppa K, Forsberg A, Norqvist A. Construction of a reporter plasmid for screening in vivo promoter activity in Francisella tularensis. FEMS Microbiol Lett 2001; 205:77-81; PMID:11728719; http://dx.doi.org/10.1111/j.1574-6968.2001.tb10928.x
- Vallet-Gely I, Donovan KE, Fang R, Joung JK, Dove SL. Repression of phase-variable cup gene expression by H-NS-like proteins in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 2005; 102:11082-7; PMID:16043713; http://dx.doi.org/10.1073/pnas.0502663102
- Chamberlain RE. Evaluation of live tularemia vaccine prepared in a chemically defined medium. Appl Microbiol 1965; 13:232-5; PMID:14325885
- Dove SL, Hochschild A. A bacterial two-hybrid system based on transcription activation. Methods Mol Biol 2004; 261:231-46; PMID:15064462
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25:402-8; PMID:11846609; http://dx.doi.org/10.1006/meth.2001.1262
- Lindgren H, Stenman L, Tärnvik A, Sjöstedt A. The contribution of reactive nitrogen and oxygen species to the killing of Francisella tularensis LVS by murine macrophages. Microbes Infect 2005; 7:467-75; PMID:15788155; http://dx.doi.org/10.1016/j.micinf.2004.11.020