
ABSTRACT
Candida albicans is an important etiological agent of superficial and life-threatening infections in individuals with compromised immune systems. To date, we know of several overlapping genetic networks that govern virulence attributes in this fungal pathogen. Classical use of deletion mutants has led to the discovery of numerous virulence factors over the years, and genome-wide functional analysis has propelled gene discovery at an even faster pace. Indeed, a number of recent studies using large-scale genetic screens followed by genome-wide functional analysis has allowed for the unbiased discovery of many new genes involved in C. albicans biology. Here we share our perspectives on the role of these studies in analyzing fundamental aspects of C. albicans virulence properties.
Introduction
Of the 1.5 million estimated fungal species on Earth, only about 300 are known to be harmful to their hosts and roughly 50 of these species are commonly isolated from clinical infections.Citation1,2 Candida albicans is a prominent fungal species that exists as both a pathogen and a commensal of humans.Citation3 This diploid fungus is able to undergo several important morphological transitions that contribute to its virulence, such as transitioning between the round yeast form to the elongated hyphal form, switching between two distinct cell types termed “white” and “opaque,” and developing community structures called biofilms, all of which are important for C. albicans to cause disease. These morphological transitions are also closely linked to host factors, including the status of the immune system due to HIV infection, corticosteroid, antibiotic, and chemotherapy use, and the use of anti-rejection therapeutics administered during organ transplantation.
Understanding how C. albicans is able to switch from a commensal to a pathogen at the molecular level may be the key to developing novel therapeutics to target this opportunistic pathogen. Screening existing C. albicans mutant libraries will thus enable and guide researchers in the discovery of novel antifungal drug targets. Since C. albicans is a eukaryote that shares several essential pathways with humans,Citation4-6 mutant libraries are needed to access non-conserved antifungal targets and to minimize side effects of antifungals to humans.
Genome-wide analyses have played – and will continue to play – important roles in discovering gene function, and to better understand the biology and pathogenesis of C. albicans.Citation8-9 In the current review, we discuss the roles of available mutant libraries in the genetic analysis of phenotypic switching, biofilm development, antifungal drug resistance and target discovery and host-pathogen interaction. This review will allow for a comprehensive and updated assessment of the functional data gleaned from the different C. albicans mutant libraries.
The review process
To begin the review, we conducted a keyword search on mutant screens which analyzed C. albicans phenotypic switching, biofilm development, antifungal drug and target discovery and host-pathogen interaction. Google Scholar was searched for English language peer-reviewed articles, followed by a manual search in various scientific journals to enrich the article search. The following key terms were used: “large-scale molecular genetic analysis,” “large-scale genetic screening” or “genetic screening,” “large-scale functional analysis” or “large-scale functional screening” and “mutant screening” or “mutant screens.” These key terms were major identifiers for libraries fitting the chosen criteria in this review. They were combined with other key terms such as “C. albicans phenotypic switching,” “C. albicans morphogenesis,” “C. albicans yeast-hypha switching,” “C. albicans white-opaque switching,” “C. albicans biofilm development,” “C. albicans biofilms,” “C. albicans drug discovery,” “C. albicans drug targets” and “host-pathogen interaction.” Articles retrieved were individually screened for relevance to the chosen criteria. From these articles designated as relevant, we excluded those published before the year 2000 and those that we could not translate to the English language. In total, this literature search resulted into 11 relevant mutant libraries, which were grouped into insertion, conditional expression or GRACE and deletion libraries (). The article search was finalized in August 2016.
Table 1. Candida albicans mutant libraries constructed from different background strains
Types of approaches used to generate C. albicans genetic libraries
The use of molecular tools in C. albicans gene function discovery is often directed toward understanding the impact of genetics over virulence of this species. Molecular tools are combined with whole genome sequencing and the use of the Candida genome database (http://www.candidagenome.org), and together they advance the implementation of large-scale molecular genetic analyses in C. albicans. Equally, genetic manipulation approaches described in literature have paved an important path for large-scale production of C. albicans mutant strains.Citation10-15 Below we discuss some of these genetic manipulation approaches adopted to generate genetic libraries.
Transposon insertion
Transposons are mobile genetic elements that integrate the bacterial and eukaryotic genomes, and induce mutations through a process termed transposition. In C. albicans, the use of bacterial Tn7 transposon is common. This transposon has been fused to elements required for selection and gene expression monitoring in C. albicans, as well as replication and antibiotic resistance in Escherichia coli.Citation16 Insertion mutants generated by the Tn7 system, containing elements as aforementioned, have been generated by combining Tn7 transposon with enzyme-cut C. albicans genomic DNA (gDNA), as well as transposases required to catalyze transposition reaction (). After this reaction, gDNA fragments integrated by the Tn7 cassette are generated such they contain the URA3 marker at different loci. Strains transformed with these Tn7-URA3-ORF fragments can then be selected as prototrophic URA+ isolates and form a C. albicans mutant library ().
Figure 1. Generation of C. albicans mutant libraries. (a) Wild type strain; no deletion or insertion, (b) Tn7-URA3 heterozygous insertion, (c) Tn7-UAU1 homozygous insertion, (d) Tagged Tn5-UAU1 homozygous insertion, (e) gene deletion and conditional gene expression, (f) heterozygous gene deletion, and (g) homozygous gene deletion. N/A – not applicable, DTag – down tag, UTag – up tag, C. d. – C. dubliniensis, C. m. – C. maltosa
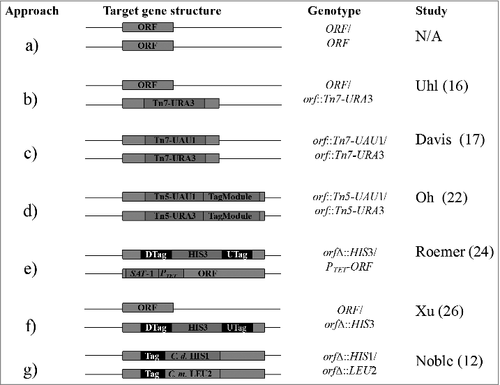
The Tn7 system adopted by Nobile and Mitchell contains the UAU1 (uracil-arginine-uracil) cassette in the place of URA3.Citation11,17 As originally described by Enloe et al., the UAU1 cassette generates homozygous mutants following a single integrative transformation and subsequent selections on Arginine and Uracil containing medium.Citation18 The insertion libraries of Blankenship et al.Citation19 and Finkel et al.Citation20 were derived using the Tn7 system based on UAU1-derived homozygosis. Mechanistically, two Transprimer transposons which confer resistance to antibiotics are incorporated into a Tn7-UAU1 transposition reaction. These Transprimers contain unique priming and restriction sites for sequence confirmation and restriction mapping of Tn7-UAU1 cassettes inserted into C. albicans gDNA, respectively. A slight modification to the Tn7 reaction is described by Nobile and Mitchell, and involves cloning of C. albicans targets into pGEM-T Easy plasmid vector.Citation11,17 Thus, a library of pGEM-T Easy-ORFs, which can be isolated and used for large-scale mutant construction, is generated using this modified version. Authors have recommended that pGEM-T Easy-ORF fragments with Tn7 insertions occurring near the middle of target ORFs be selected for large-scale mutagenesis. To confirm insertions, plasmids are subsequently digested with an enzyme that releases ORFs, as intact or as several pieces, from Tn7-UAU1 pGEM-T Easy backbone. The confirmed plasmids can then be used to generate a library of C. albicans insertion strains. Following this, insertion strains are subjected to selection, which induces recombination, resulting in an intact URA3 copy placed on one of the target alleles ().
A mutagenesis approach developed by Oh et al. is based on a Tagged Tn5 transposon,Citation21 tagged by 4280 synthetic Gateway-compatible TagModules that are flanked by universal priming sites. These TagModules are cloned into a Gateway entry vector flanked by recombination sequences required for cloning into a Gateway conversion cassette. The resultant constructs containing the different TagModules are eventually fused to the Tn5-UAU1 cassette, forming a tagged Tn5 transposon cassette. Universal primer binding sites which flank the up- and down-TagModules, correspond to C. albicans ORFs of interest. These primers allow for PCR amplification of all tags in a mutant library (). Following this, tagged PCR products can be hybridized to an Affymetrix TAG4 microarray containing the tag complements.Citation21,22 Signal intensity for each tag generated from this hybridization can be translated to fitness and sensitivity scores for each constructed mutant.
Overexpression/suppression
Among promoters designed for regulatable gene expression in C. albicans,Citation23 the Tet promoter is currently the only one used in the construction of the gene replacement and conditional expression (GRACE) libraries.Citation24,25 Construction of these libraries is demonstrated in . Here, conditionally constructed strains are generated by replacing one copy of the target gene using a tagged HIS3 auxotrophic marker. This marker is flanked by sequences homologous to the 5′- and 3′-untranslated regions of the target gene. Following this, the Tet promoter cassette modified with streptothricin acetyl transferase (SAT1) gene, is integrated upstream of the second allele, where it replaces the native promoter of the target gene. Since this promoter is ligated to the SAT1 gene, that is required for growth on nourseothricin drug, positive C. albicans isolates resistant to this drug can easily be selected.Citation24 Gene expression in the GRACE libraries occurs in a Tet-Off-dependent manner – in absence of the drug, tetracycline or doxycycline.Citation24,25 The GRACE method is valuable for the discovery and characterization of essential genes, as well as the discovery of novel antifungal targets producing terminal phenotypes.
Gene deletion
Two strategies, shown in , used to generate C. albicans heterozygous and homozygous gene deletion libraries have been described by Noble and Johnson Citation12 and Xu et al.Citation26 These deletion strategies share certain crucial requirements. Firstly, before mutant generation can commence, target genes are selected using a set of criteria such as being essential and conserved in close and distant fungi, sharing of homology with higher eukaryotes and uniqueness to C. albicans.Citation26-28 In addition, functional motifs of the selected genes must relate to virulence of C. albicans or other fungal pathogens.Citation28 Secondly, deletion cassette(s) used to disrupt these genes must carry neutral auxotrophic markers. However, disruption cassettes may differ slightly. For instance, the 5′ and 3′-untranslated ends of the marker gene may be interrupted by unique up- and down-tags and primer binding regions common for amplification of these tags.Citation26 By default, each mutant generated using these tagged cassettes will be represented by two isolates marked by a unique up- and down-barcode. At the same time, common priming sites in generated tagged strains will facilitate the use of independent strains in pooled assays and, later, determination of relative abundances. Therefore, the use of this tagged strategy allows for the identification of fitness genes which can be targeted for antifungal drug development. Although this deletion strategy shares some similarities with the strategy described by Noble and Johnson,Citation12 relatively short sequences of about 43-bp homologous to sequences flanking target C. albicans ORFs are used.Citation26 These short sequences can impact negatively on the binding of the deletion cassettes to the target locus. In an effort to limit this non-specific binding, Noble and Johnson proposed and used 350-bp sequences that are homologous to sequences flanking the gene of interest.Citation12 These sequences are long enough to increase specificity of the deletion cassettes used during mutant construction; other authors have also proposed the use of even longer sequences of up to 1-kb.Citation14 The 350-bp long sequences used by Noble and Johnson, however, also flank C. dubliniensis HIS1 and C. maltosa LEU2 auxotrophic markers.Citation12 Thus, gene disruption efficiency is further improved.
Large-scale genetic analysis of phenotypic switching
Filamentous growth
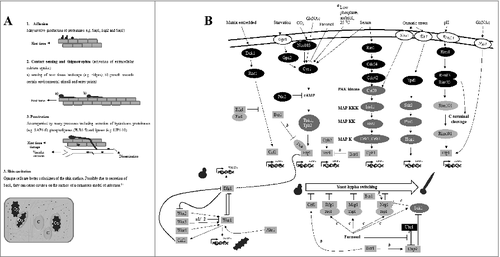
Transcriptional regulation of yeast to filament transition is important for C. albicans to thrive in host-specific niches, each with its own set of changing microenvironments. Filamentous growth in C. albicans is triggered by a plethora of conditions (e.g. CO2 availability, pH fluctuations, serum and starvation) and is co-regulated with other biological processes such as secretion of hydrolases (). Thus, in responding to its environment, C. albicans can activate several virulence factors which will enable it to adhere, colonize and penetrate important host tissues (). Large-scale genetic screens coupled with phenotypic analyses and the use of infection models, are important investigative tools used to explore gene functions underlying filamentous growth. Numerous libraries listed in and discussed below have contributed to the current knowledge of how filamentation is controlled at the molecular level.
Figure 2. C. albicans' response to the surrounding environment. A. Virulence factors attributable to host tissue damage. Op, opaque; C, cavities. B. Regulation of morphogenesis by varied environmental stimuli. Proteins are colored as follows: white, environmental sensors; gray, transcription regulators; dark gray, protein kinases; black, other genes. a, conditions required for the activation of opaque cell filamentation; b, interactions during the regulation of opaque cell filamentation; c, interactions during the regulation of the response to high cell density. HSG's, hyphal-specific genes; OpSG's, opaque-specific genes. Kinase modules are indicated as follows: MAPKK, MAPK kinase; MAPKKK, MAPKK kinase; PAK, p21-activated kinase. Black arrows indicate signal flow. T-shaped bars indicate inhibition
Haploinsufficiency gene insertion library
Uhl et al. constructed a random Tn7-URA3 insertion library consisting of 18,000 mutant C. albicans strains, and performed haploinsufficiency screens to identify phenotypically altered disruption mutants.Citation16 Tn7 insertions in these strains, herein referred to as haploinsufficient mutants, were verified by sequencing. Generated haploinsufficient mutants were screened for defects in filamentous growth on yeast-extract-peptone-dextrose (YPD) agar supplemented with 1% serum, and Spider medium agar. These media are enriched with nutrient supplements suitable to mimic the nutrient-rich and nutrient-poor host environment that C. albicans is often exposed to; the former media type is a potent inducer of filamentation in C. albicans, while the latter induces a more moderate filamentous growth response. A total of 325 of the 18,000 Tn7 insertion strains (1.8%) displayed altered filamentous growth on at least one of the two media types, when compared to a wild-type control (). These 325 Tn7 insertion strains correspond to a total of 146 unique genes involved in yeast-hypha switching. Only 6 out of the 146 genes (4%) had been previously identified as being involved in filamentous growth of C. albicans. Notably, 32% of identified genes in this screen lacked significant sequence similarity (based on BLAST scores less than -15) to genes in currently available databases. Therefore, this study identified putative functions for many genes that are unique to C. albicans morphogenesis which may serve as potential antifungal targets.
Homozygous gene insertion library
The insertion library developed by Davis et al. was derived using the Tn7-UAU1 cassette.Citation17 Using in vitro Tn7-mediated transposition, Davis generated and cloned 756 insertions, which yielded disruptions in 353 unique ORFs.Citation17 Two hundred and fifty-three of these sequence-verified disruption cassettes were transformed into C. albicans and subsequently screened for homozygous gene disruptions, yielding 217 unique homozygous gene disruptants (). However, this screen failed to identify homozygous disruptions for 36 of the 253 constructs. The 36 target loci were found to be triplicated, with one wild-type allele maintained. These loci were also regarded as representing essential genes. Homozygous disruptions of the SLA2, RIM13, and MDS3 gene were identified in a screen for strains that are defective in pH-induced filamentation. Unlike Sla2 and Rim13, Mds3 was previously not implicated in filamentation or pH response. It was subsequently determined to be required for normal alkaline-induced filamentation, but not for serum or N-acetylglucosamine (GlcNAc)-induced filamentation. Mds3 appears to independently activate a pathway parallel to the Rim8p-Rim101p complex.Citation17 Therefore, this study has provided fresh insights into how C. albicans RIM proteins and Mds3 may potentially coordinate pH-responses inside the human host.
Transcription regulator gene deletion library
Regulatory genes are of central importance due to their influence on microbial response to environmental stimuli. Transcriptional regulators or TRs can alter cellular development and routine biological functions of cells through promoter binding of many genes, including other TRs. Homann et al. developed a target list of 184 known or predicted TRs, with an emphasis on sequence-specific DNA binding proteins, and excluding general transcription factors that influence the transcription of most genes in the cell.Citation27 Using the flanking-homology disruption method (),Citation12 Homann et al. successfully created 143 high-confidence homozygous mutant strains, with two independent isolates of each homozygous deletion strain.Citation27 An additional 23 low-confidence mutants represented by a single homozygous knockout isolate was also constructed. The 143 high-confidence strains formed the official TRKO library, with each strain phenotypically profiled based on 55 independent growth conditions.
The C. albicans TRKO library was not developed for exclusive analysis of filamentous growth. Nevertheless, some 20 TRs that were previously uncharacterized were associated with this phenotype. These TRs were grouped with other previously characterized TRs as general growth regulators, 30˚C negative regulators and complex colony morphology regulators. Homann et al. have thus presented a comprehensive set of functional TRs with a range of roles in C. albicans biological processes.Citation27 As shown later in this review, several studies have established the TRKO library as a valued resource for the C. albicans research community.
Protein kinase gene insertion library
C. albicans can respond to quorum sensing sesquiterpene compounds. Some of these compounds, mainly farnesol, can inhibit yeast-to-hypha transition in dense populations of C. albicans.Citation28,29 The homozygous Tn7-UAU1 library of PK (protein kinase) strainsCitation19 has been useful in screening for response to cell density changes in the presence of farnesol.Citation30 Screening of this insertion library revealed that farnesol-mediated inhibition of yeast-hyphal transition is distinct from the cAMP-mediated pathway. A kinase gene named SOK1 was identified as a vital component required for this unique mechanism. This kinase gene was found to be transiently expressed in cells inoculated into fresh YPD medium, which represent low density culture settings. However, expression of Sok1 was not detected in YPD medium supplemented with farnesol, suggesting that farnesol may inhibit expression of Sok1 and associated repressors of yeast-hyphal transition. Among repressors that could potentially be inhibited include Nrg1 and Tup1.Citation30,31 These TRs may be targeted by Sok1, and they are likely repressed in low density cultures in C. albicans since switching to the hyphal state is observed under these culture conditions. Likewise, Sok1 may positively regulate Nrg1 and Tup1 when C. albicans cells become densely populated, thereby preventing switching to the hyphal state (). An earlier study found that Tup1 is steadily expressed in farnesol treated cells.Citation31 Whether this expression is linked to Sok1 remains a matter of speculation. However, Lu et al. could correlate elevated mRNA transcript and protein levels of NRG1 with the lack of expression of Sok1 in the presence of farnesol.Citation30 Therefore, Sok1 could be phosphor-regulating Nrg1 more than it does Tup1 and other negative regulator genes of yeast-hypha switching (). Other proteins included in this regulatory system were discovered following screening of the TRKO library. These include Cup9, targeted by E3 Ubiquitin ligase, and a negative regulator of Sok1 and Nrg1 ().
Therefore, this study has provided important leads regarding C. albicans' response toward changing cell-densities. Understanding these mechanisms in detail is of particular relevance given that C. albicans co-exists with other species secreting quorum sensing molecules inside the human host.
Homozygous gene deletion library
Studying C. albicans genetics was previously complicated by the diploid nature and the use of auxotrophic markers involved in virulence of this organism.Citation32 Over the years, the Candida research community has improved gene disruption protocols () to study the diploid genome of C. albicans. Among milestones that emerged from these developments was the construction of the homozygous gene deletion library containing up to 3000 auxotrophic C. albicans strains.Citation28 These strains are disrupted for a total of 674 non-essential genes, and they cover about 11% of the C. albicans genome ().
Phenotypic profiling of deletion strains in the homozygous library was conducted by incubating C. albicans cells on Spider medium agar for 14 days at 30 ˚C. Following this long incubation period, colonies from 133 deletion strains were found to be either more or less wrinkled or filamentous than the wild type strain. Oddly, most strains (115) exhibiting enhanced colony filamentation also exhibited defects in infectivity in a mouse model. The majority of these strains (89) exhibited normal proliferative ability in vitro. An additional 46 deletion strains with normal colony morphology and proliferative ability also displayed infectivity defects. While some of the affected genes in the 46 strain set have undescribed functions, some genes are involved in cell wall biogenesis (e.g., PGA32, CHS4, CHS7 and CHT2), lipid biosynthesis (e.g., CYB1 and HET1), nutrient acquisition and metabolism (e.g., CDC19, HGT8 and HGT19) as well as signaling and transcription (e.g., CZR1, RGA2, KAR2, and MTLA1). This implies that C. albicans infection machinery is driven by many presiding genetic factors that are in part or entirely unlinked to morphogenesis.Citation28 Thus, Noble et al's assessment of colony morphogenesis, infectivity and proliferation can provide an informed analysis of C. albicans infection machinery useful for routine morphological tests conducted in the clinical setting.Citation28
White-opaque switching
The transformation of C. albicans from the default form to the mating competent form is called white-opaque switching. This switching system came to light during analysis of the wild type clinical isolate, WO-1, which lacks lacks the a1 gene that corresponds to the mating-type-like or MTL locus. White-opaque switching was later observed in other Candida species.Citation33-35 It is apparent from a number of seminal papers that C. albicans white-opaque switching is highly modulated by the MTL locus, following which activation of genes in this locus induces interconnecting feedback loops that are regulated by EFG1, CZF1 and Wor genes (WOR1-4) ().Citation36-43 However, studying white opaque switching in C. albicans mutants requires that diploid MTL heterozygous strains (MTLa/α) be converted from being mating and switching incompetent to being competent for these processes. To convert these strains, targeted gene disruption of either MTLa or MTLα is conducted, which will result in diploid strains homozygous for the a or α allele.Citation41 In this homozygous state, C. albicans a-specific and α-specific genes encoded in the MTL locus cannot form a heterodimer complex (a1-α2) that inhibits the Wor1 gene required for the expression of phase-specific genes ().Citation41-43 Understanding how this mechanism operates makes it possible to analyze white and opaque cells in existing C. albicans mutant libraries using various conditions. To date, there's a body of knowledge regarding distinct genetic mechanisms used by white and opaque cell types to thrive under distinct conditions. Citation40,41,44-48 Below, we discuss the contribution of the TRKO library to understanding C. albicans white-opaque switching.
Regulation of CO2-induced white-opaque switching
In addition to other inducers such as GlcNAc, carbon dioxide (CO2) is one of the potent inducers of white-to-opaque switching in C. albicans ().Citation36,50 Screening the TRKO library revealed Flo8 as a regulator of CO2-induced switching under incubation in 5%CO2 ().Citation57 In these conditions, switching competent C. albicans cells lacking Flo8 formed mostly white cells when cultivated on Lee's glucose (Glc) medium. At the same time, the wild-type strain formed <1% white colonies under the same conditions. Likewise, opaque colonies of the Flo8 double knockout isolate were significantly reduced (<0.4%) compared with opaque colonies of a wild-type control (>86%). No difference in switching was observed between flo8Δ/Δ and the wild type strain on Lee's GlcNAc medium. As indicated in , GlcNAc activates Ras1-cAMP-dependent white-opaque switching in C. albicans. Therefore, CO2 could induce Flo8-dependent switching via a distinct pathway. Previous studies have indicated the induction of yeast-hypha switching by CO2 via Flo8-dependent signaling.Citation55,56 Combined with these studies, the study of Du et al.Citation57 demonstrate how different switching systems and the underpinning signaling paths may develop in response to similar environmental cues in human fungal pathogens.
Regulation of opaque cell filamentation
Filamentous growth pathways in white and opaque cells overlap in C. albicans.Citation59 Given their operation in distinct cell types, these pathways are activated by distinct environmental cues. For instance, opaque cells filament efficiently in low phosphate and sorbitol, and at 25 °C via the cAMP-dependent signaling pathway ().Citation59 Filamentation in these cells is also possible with conditions that induce white cell filamentation such as 37 °C in GlcNAc. However, unlike in white cells where filamentation is efficiently induced by these conditions, opaque cells require an incubation period of up to five days to filament.Citation59
Lessons learned from studies of yeast-hyphal switching in white cells suggest that studying yeast-hypha-specific TRs better advances understanding of regulatory mechanisms of this switching process. This is consistent with the fact that a number of studies opt to analyze the TRKO library to deeply understand regulation of filamentation.Citation20,60 Guan et al. have screened this library and identified Bcr1 as a major regulator of opaque cell filamentation ().Citation60 Bcr1 was found to exert its control upstream of TR genes such as CUP9 and CZF1 (), and to strongly bind promoters of these TRs in opaque cells than in white cells.Citation60 Also apparent from this screen was the fact that Bcr1 is a global regulator of opaque cell filamentation, as demonstrated by failure of its deletion strain to filament under a wide variety of culture conditions. The general importance of this study is the finding that networks governing switching overlap, giving C. albicans enough plasticity and capacity to rapidly adapt to changing host microenvironments.
Large-scale genetic analysis of C. albicans biofilm development
Biofilms are a densely compact microbial community of cells encased in a matrix of extracellular polymers (). They usually form in vitro consisting mostly of yeast and hyphal cells, or in vivo as polymicrobial cultures, co-existing with bacterial species of the gut micribiota.Citation52,61,62 Structural appearance of C. albicans biofilms is biologically unique from the appearance of suspended yeast cells, and this allows C. albicans to thrive under harsh environmental conditions (). For instance, biofilms overexpress efflux pumps which oftentimes increase tolerance of biofilms to antifungal drug treatment.Citation63 On the other hand, cell-surface adhering capacity of biofilms presents a major clinical challenge by enabling biofilms to colonize implanted, and life-saving medical devices such as pacemakers. Thus, adherence can cause serious complications and even lead to death of patients with implanted devices. The extracellular matrix of matured biofilms forms a rigid and impermeable layer against antifungal drugs and, as a result, stabilizes the structure of mature biofilms ().Citation63,64 Studies have also shown that C. albicans ‘persisters’, which are derived from a small subpopulation of dormant cells, are highly tolerant to antifungal treatment and they may develop into a more drug tolerant biofilm.Citation63,64 Due to these traits, C. albicans biofilms are responsible for increased hospital-associated infections and resulting mortality rates. Here we discuss the complex networks governing biofilm developmental stages with lessons learned from the screens of C. albicans libraries.
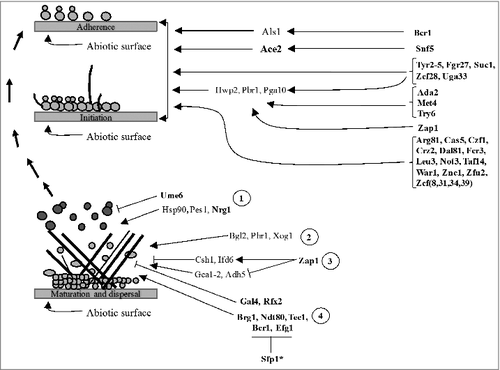
Figure 3. Development of C. albicans community structures (biofilms) on the surface of a medical device such as a pace maker. Left pane indicates steps leading to biofilm formation. Right pane indicates the underlying regulatory mechanism involved in each step. Bolded proteins are transcription regulators while non-bolded proteins are some of the downstream components. 1. Regulation of dispersion. Dispersed cells from matured biofilms may restart the process of forming biofilms or cause disseminated infections; 2. Delivery and arrangement of β-1,3 glucans in the matrix 3. Regulation of extracellular matrix accumulation; 4. Regulation of biofilm development. #Only Bcr1 and Efg1 are targeted for inhibition by Sfp1. Black arrows indicate signal flow or activation. T-shaped bars indicate inhibition
TR gene insertion and TRKO library in studying biofilm regulation
Nobile and Mitchell identified 83 putative TR genes associated with biofilm formation.Citation65 Using the split-marker transposon insertion method (), authors designed a library of C. albicans TR insertion strains from the 83 putative genes. All of the 83 generated insertion strains were assayed on a silicone substrate suspended in Spider medium. This screen identified Bcr1 and Tec1 as key biofilm regulators, for which insertions in ORFs of these TRs gave rise to mutants with underdeveloped biofilms, consisting mostly of yeast cells. By contrast, biofilms of the wild type strain formed a biofilm with a typical density consisting of yeast and hyphal cells. Tec1 is a known hypha-specific regulator (see ) while Bcr1 was identified for the first time in this mutant screen as the biofilm master regulator. Genome-wide expression profile of the BCR1 strain indicated that the Bcr1 TR controls expression of cell-surface proteins and adhesins, including Als1 (). Therefore, appearance of underdeveloped biofilms following the insertion mutation is caused by altered production of cell-surface proteins and adhesins. Disruption in the TEC1 gene, like disruption in other hypha-specific TR genes such as EFG1, is expected to yield biofilm defects in C. albicans.Citation66 In the mutant strain of the TEC1 gene, the BCR1 gene is downregulated, suggesting that its expression is Tec1-dependent.Citation65 This study has thus provided one of the first direct links between the biofilm and yeast-hypha switching network.
The biofilm network has since been expanded through screening of the TRKO library, by introducing four additional TRs genes (EFG1, NDT80, ROB1 and BRG1) also essential to biofilm regulation ().Citation67 Deletion mutants of these TR genes displayed abnormal biofilm growth under standard in vitro growth conditions and in the in vivo murine model, suggesting that they positively regulate biofilm development.Citation67 The four TRs, together with Bcr1 and Tec1, regulate more than 1000 target genes, some of which are also TRs.Citation68,69 Several downstream targets in this circuit have contributory roles relevant to typical biofilm characteristics.Citation62,67 Although not initially identified during characterization of the interconnected biofilm network,Citation67 additional screening of the TRKO library introduced more TRs (Flo8, Gal4 and Rfx2) which also form part of the biofilm regulatory network ().Citation69 By comparing in vitro and in vivo test conditions, Fox et al. could show how mutant strains of these TRs exhibit defects similar for both set of conditions.Citation69 Consistent with binding patterns displayed between typical biofilm TRs, Flo8, Gal4 and Rfx2 bound the promoter regions of Bcr1, EFG1, Tec1, Ndt80, Rob1 and Brg1. Together with the in vitro and in vivo analyses, these findings suggest that these TRs are also master regulators of the biofilm network, with Gal4 and Rfx2 involved as negative regulators ().Citation69
Transposon gene insertion library
Finkel et al. assayed 197 Tn7-UAU1 insertion strains for their involvement in cell surface adherence.Citation20 Thirty TR strains represented positive regulators of adherence, as they displayed significantly reduced adherence relative to the wild-type strain. Affected TRs in these strains formed a large cell surface regulatory circuit ever to be described in C. albicans (). One of analyzed strains, representing SWI/SNF chromatin remodeling complex or Snf5, showed cell separation defects and hypersensitivity to cell wall perturbing agents. These traits are reminiscent to those of the Ace2 mutant. Ace2 plays a major role in biofilm formation and is also an effector of the RAM (Regulation of Ace2 Morphogenesis) signaling network.Citation70 In the screen of Finkel et al.,Citation20 this TR shared targets with Snf2, suggesting that Snf2 has a functional relationship with the RAM network. Other strains analyzed were those disrupted for TRs regulating hyphal- and virulence-associated genes, targets of Zap1 and cell surface targets of adherence regulators. These groups of genes formed part of the large cell surface regulatory network.Citation20 Together with the RAM targets, these groups of genes represent a quarter of all cell surface protein genes regulated by TRs core to the cell surface circuitry. Therefore, this work expands the knowledge for early biofilm development and its underlying comprehensive circuitry. Dissecting the C. albicans cell surface adherence network aids the development of strategies that could prevent biofilm formation or destabilize the structure of already formed biofilms.Citation62
Large-scale genetic screening of antifungal drug resistance and possible targets
C. albicans' resistance to a wide range of antifungal drugs rests upon changes in its phenotypic and genetic states. Common causes of resistance are mutations causing repression or overexpression of drug targets. However, antifungal drug resistance is further favored by the expression of multidrug resistance efflux pumps by C. albicans (e.g., Cdr1 and Mdr1; ).Citation63,71 Studies have also found these efflux pumps to have a profound antifungal resistance effect in other human fungal pathogens.Citation71 Therefore, finding newer and effective antifungal drug targets will help ameliorate treatment action plans against fungal infections. This section discusses the role of the GRACE and Haploinsufficiency libraries in identifying antifungal targets and compounds with antifungal activities (). The mechanisms through which different antifungal compounds produce their antimycotic effects are beyond the scope of this review; the reader is thus guided to several reviews on this topic (refs Citation4-6).
Table 2. Antifungal targets and S. cerevisiae homologues, and their cognate compounds studied through library screening in C. albicans
GRACE libraries
Roemer et al. implemented a comparative and phenotypic approach to analyze 823 essential genes as priority broad- and narrow-spectrum targets.Citation24 In the comparative approach, genes essential in C. albicans were compared to orthologues in other fungi and humans. This analysis revealed 33 targets that are conserved between C. albicans and Saccharomyces cerevisiae. All the 33 targets were absent in humans and 27 of them non-conserved in Schizosaccharomyces pombe and Neurospora crassa. Therefore, the 27 identified targets can advance development of narrow-spectrum antifungal drugs. Phenotypic analyses of the GRACE strains identified 212 essential genes, for which strains were inoculated in Tetracycline pretreated and untreated mice. The pretreated mice, therefore, imposed Tet-off conditions, for which suppression of expression of genes in inoculated GRACE strains is supported. These pretreated mice survived seven days following inoculation. By contrast, the untreated mice representing Tet-on conditions injected with the same set of strains were infected within 7-10 days following inoculation. Among strains that were infective were those disrupted for a copy of ALG7, RHO1 and YBR070c (). However, only the Rho1 affected GRACE strain was infective five days following inoculation. This GRACE strain also caused systemic C. albicans infection, while the remaining strains caused mild infections.Citation24 This suggests that Rho1 is a candidate protein target for developing treatment against systemic Candidiasis.Citation24 Therefore, Roemer has demonstrated the use of conditional expression systems in prioritizing and validating essential genes as potential antifungal targets.Citation24
Using multiple hyphal inducing cues, O'Meara et al. screened 18 GRACE strains disrupted for homologues of S. cerevisiae ergosterol biosynthetic genes.Citation25 These strains were exposed to sub-inhibitory concentrations of certain antifungal compounds, which targeted C. albicans yeast-to-hyphal switching.Citation25 This analysis revealed importance of transcriptionally repressing ergosterol biosynthetic genes () to suppress filamentation, instead of killing C. albicans cells. Using antifungals to target virulence factors has some benefits, such as imposing a reduced selection pressure for drug resistance mutations, a recurring problem in C. albicans.Citation73 O'Meara also identified four essential genes with no human or murine homologs.Citation25 These fungal-specific essential genes represent potential antifungal targets.
Tagged haploinsufficiency deletion and insertion library
Xu et al. used a reverse genetics approach and target-specific inhibitory compounds to analyze drugs, their targets and mechanism of action (MOA).Citation26 Induced hypersensitivity in 2,868 heterozygous tagged strains of C. albicans identified several antifungal targets. Among these were target proteins of fluconazole, Erg11, and its accessory protein, Ncp1, both of which are not necessarily essential in S. cerevisiae (). Authors have also analyzed enzymes and protein complexes, such as the fatty acid synthase subunit, Fas1, and α-tubulin, Tub1, as potential antifungal targets ().Citation26 Although genes for these protein complexes are nonessential in C. albicans, Fas1 and Tub1 haploinsufficient strains displayed reproducible hypersensitivity toward their cognate compounds (). Sensitivity induced by target-specific inhibitory compounds can identify corresponding drug targets and MOA–related genes in C. albicans. Compounds such as 5-fluorocytosine and 5-fluorouracil, as well as tubercidin were useful in studying MOAs using haploinsufficient strains. Furthermore, screening of the heterozygous tagged strains has uncovered previously undescribed antifungal compounds which caused dose-dependent microtubular defects in the Tub1 haploinsufficient strain.
Oh et al. assessed a panel of 1,521 chemical compounds categorized in terms of targets shared between C. albicans and S. cerevisiae as well as in terms of varying MOAs or unique side effects against these species.Citation22 These chemical compounds were assessed against a library of 3,633 tagged C. albicans heterozygous transposon insertion strains (). Nutrient- and drug-dependent haploinsufficiency competitive assays revealed 40 such compounds with 20-90% inhibition in C. albicans than in S. cerevisiae. This suggests that these compounds display MOAs that are unique for C. albicans and S. cerevisiae. A genetic screen was conducted and identified a strain haploinsufficient for the SEC7 gene as most sensitive to brefeldin A (). This compound inhibits intracellular protein transport, while Sec7 is a guanine nucleotide exchange factor which mediates formation of transport vesicles. SEC7 has also been targeted using this compound in a previous haploinsufficiency screen ().Citation26 However, Sec7 was not identified as a target corresponding to brefeldin A in S. cerevisiae, suggesting that it is unique to C. albicans. Oh et al. also characterized two previously undescribed antifungal agents which target two nonessential genes, ORF19.2411 and TFP1.Citation22 ORF19.2411 is a homolog of S. cerevisiae SYN8, while TFP1 lacks an ortholog in this species.
Therefore, the above studies have interrogated an inventory of chemically diverse inhibitory compounds with MOAs () using tagged mutant strains of C. albicans. They have consequently increased the number of priority targets to be considered in future to combat C. albicans infections.
The interplay between C. albicans and host immune response
C. albicans frequently colonizes mucosal surfaces in healthy and immune compromised individuals. Since this pathogen can readily switch from yeast to hyphal cells, the mammalian innate immune system has adopted the ability to discriminate between the two cell types.Citation74 The mammalian innate immune system represents the first line of defense, alongside physical barriers such as the skin and mucosal epithelium. An arsenal of defense mechanisms including the inflammasome (e.g., NLRP3) activation, nutritional immunity, phagolysogenesis, antigen presentation and T helper cell differentiation, supplement this immune response.Citation74-79 However, this surveillance system may severely be weakened or prone to manipulations by pathogens in immune compromised hosts. Consequently, this will afford C. albicans the opportunity to penetrate important organ tissues and cause systemic Candidiasis. Therefore, understanding molecular mechanisms utilized by C. albicans to breach innate host immunity is important to undertake precautionary measures against emerging infections. So far, a few C. albicans libraries have advanced knowledge of pathogen-host immune interaction.
Wellington et al. showed that ahr1Δ/Δ and upc2Δ/Δ, screened from the TRKO library, were not only defective in filamentation but also defective in sufficiently inducing the production of interleukin ß (IL-1ß).Citation78 In essence, these mutants cannot inhibit caspase-1, an enzyme required to convert pro-IL-1ß to IL-1ß (the biologically active form) during inflammatory programmed cell death or pyroptosis.Citation78 Therefore, the AHR1 and UPC2 genes are implicated in controlling this process. Another study which analyzed the same strains (ahr1Δ/Δ and upc2Δ/Δ) confirmed the previous findings,Citation78 but strongly implicated Upc2 in controlling processes leading to pyroptosis.Citation81
A survey of the GRACE strains conducted by O'Meara et al. suggests that filamentous growth is insufficient for C. albicans to escape from mammalian macrophages.Citation25 Indicative of this was the observation that previously internalized heat-killed C. albicans mutant strains with defects in filamentation were able to lyse macrophages. When these GRACE strains were treated with endoglycosidase that removes glycoproteins from the cell wall, they failed to induce macrophage lysis. This implicates glycosylation of surface proteins in macrophage lysis. Interestingly, O'Meara have confirmed that this glycosylation-dependent macrophage lysis also occurs in Crypt. neoformans but not in S. cerevisiae.Citation25 Perhaps this is a process shared among human fungal pathogens.
Taken together, these studies have expanded the knowledge surrounding conserved fungal processes that govern host cell-pathogen interactions and immunity.Citation25,80,81 In future, this interaction may represent a potential target for the development of drugs and chemicals that can enhance endocytosis, prophylaxis of invasive Candidiasis and other invasive mycoses, as well as the development of novel therapeutic agents.
The impact of C. albicans libraries
In order to understand the extent to which C. albicans libraries shaped the understanding of fungal pathobiology, a number of scientific reports citing these libraries were searched from the literature. The key search terms described previously (see The review process section) facilitated the search process. At the end of this search, the number of articles that were retrieved when the key search terms were combined, were analyzed for relevance. For instance, such key terms as “Large-scale molecular genetic analysis” were combined with “C. albicans phenotypic switching,” to unify them into one search term which translated into a single meaningful sentence (e.g., “Large-scale molecular genetic analysis in C. albicans phenotypic switching”). Within papers retrieved using the combination of terms as explained above, we counted the number of papers containing specific terms such as “Large-scale genetic analysis” and “Mutant screening.” These papers were designated as relevant and were further analyzed (). Follow up analysis was conducted by browsing within the relevant articles, searching for reports that have specifically analyzed C. albicans virulent properties and other aspects of pathogenicity. We found that libraries reviewed in this paper have been cited by 1772 reports relevant to C. albicans. These were the reports used to measure the scientific impact of libraries. Two scenarios were considered; 1) the extent to which each library was cited by papers dealing with the relevant aspect(s) that were previously discussed in this review (e.g., phenotypic switching and biofilm development) (and the number of times each library was cited each year from the year in which it was released ().
Figure 4. A. Citations retrieved using a combination of key search terms. >, just above; <, just below; k, counting was done in thousands. B. Citations with respect to specific aspects of virulence. Relevant studies are indicated as RS and other studies are indicated as OS. C. Citations observed per year since release of mutant libraries. Bolded numbers represent sum of citations for that year
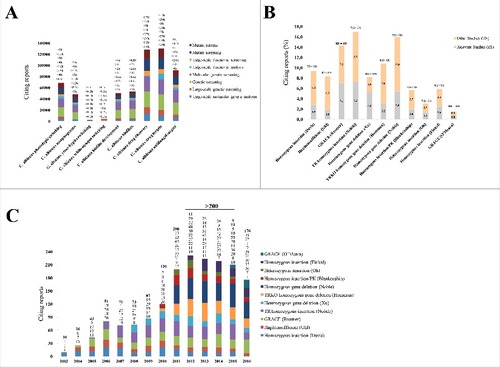
Mutant libraries were in fact cited by publications analyzing aspects of interest, such as phenotypic switching, biofilm development, drug and target discovery and host-pathogen interactions. However, libraries such as the GRACE,Citation24 TR homozygous insertion,Citation17 heterozygous and homozygous gene deletion librariesCitation26,28 showed more literature citations of between 5% and 7% than other libraries within their respective aspects. We also discovered that for a number of libraries, citing papers that are relevant (designated as RS for relevant studies in ) appear to be equal or greater than citing papers that are not relevant (designated as OS for other studies in ) to the discussed aspect. This was true for the GRACE libraries where the RS citations matched the OS citations, and the heterozygous gene deletion library, where the RS citations surpassed the OS citations (). A conclusion can be drawn from this analysis of relevant and non-relevant citing papers; that is, the use and recognition of C. albicans libraries does not only revolve around aspects of virulence, but may also appeal to other purposes, and this may likely broaden the scope of applicability of C. albicans mutant libraries.
As illustrated in , we performed citations-per-year assessment beginning with reports citing libraries, for which their release dates ranged from 2003 to 2016. This analysis has thus been based on a 14 years' timeline, and could illuminate a long standing contribution of C. albicans libraries on understanding virulence aspects. An emerging trend from this 14 years' timeline is that of an increasing number of citations that is consistent with an increase in released libraries (). The highest number of citations (>100) was recorded between 2010 and 2016. A number of factors may have driven this increase in citations. Specifically, we noted several important libraries such as the TRKO homozygous deletion library published by Homann et al. and the homozygous gene deletion library published by Noble et al.Citation27,28 Not to dismiss other crucially relevant factors, we believe that there are certain attributes which may play a role more important than others in promoting the public use of these mutant libraries in mechanistically studying C. albicans virulence factors. These include the following: Deletion strains in the TRKO library display phenotypes pertaining to underlying regulatory mechanisms. This allows researchers to readily address regulatory mechanisms of C. albicans virulence, and hence its use is appealing to many researchers in the field. A large number of deletion strains in the homozygous gene library have infectivity defects. This helps identification of novel virulence factors that may serve as potential targets for disease management strategies. Both homozygous gene deletion libraries are deposited with the Fungal Genetics Stock Center (http://www.fgsc.net/). This further potentiates public use and concomitantly increase recognition of these libraries, which seen expressed here in terms of high citation numbers. Another emerging trend from the yearly citation data was that of older libraries (e.g., the haploinsufficient,Citation16 GRACE and TR homozygous insertion library)Citation17,24 being cited each year from the time they were released until 2016. Although the number of reports citing these libraries is not as high as those citing the TRKO homozygous deletion library and the homozygous gene deletion library, their citation trend has been stable over time. This suggests that these libraries have addressed very critical aspects of C. albicans virulence, a contribution which is still being esteemed by the research community.
Limitations
A number of limitations halts the frequent usage of large-scale genetic screens for analyses of C. albicans genetics. These limitations range from those that perpetuate reconstruction of mutants in wild type clinical isolates, to those enforcing verification of roles of genes presented by library screens. These include laboratory wild type isolates which display a high degree of chromosomal abnormalities,Citation82 and the use of auxotrophic wild type strains within most constructed libraries. Some limiting factors rest on the fact that certain phenotypic processes, such as the yeast-hypha transitioning, observed at cellular and colonial level, do not always associate with in vivo virulence.Citation25,28,80,81 This enforces the use of in vivo models of infection to validate the roles of genes alleged to have contributed to C. albicans virulence by culture-based methods. The mouse model itself, although shown many times to be useful in establishing in vivo roles of virulence genes in order to address infectivity aspects and to put them into a medical context, is surrounded by some limiting factors. For one, this model requires strict adherence to set ethical standards and accordingly, skilled personnel. Thus, it potentially imposes monetary restrictions, especially for laboratories in developing countries which will use it for the first time in screening mutant libraries. On the other hand, there are currently strict regulations put in place for interstate or intercontinental movement of mutant libraries. These are imposed by federal agencies and are often accompanied by permits which aim to enforce biosafety, deter potential risks to the environment as well as to minimize misuse of biological specimen. However, these processes could potentially delay projects underway since they may drag on for months or even years, and would likely discourage researchers interested in using mutant libraries. Another major setback, which has been shown to be prevalent among libraries in fungal phytopathogens (plant fungal pathogens),Citation83 is that of screening a large number of isolates but only to end up with only a small number of virulence genes identified (). Improving mutant construction techniques and implementing sensitive phenotypic screens will enhance gene and function discovery in such libraries.
Conclusions and future outlook
Mutant libraries have made substantial contributions to C. albicans biology. Many genes involved in phenotypic switching, biofilm development, host-pathogen interaction, and those that are presented as antifungal targets, have been identified. In addition, a lot of other genes with in vivo roles have been validated and linked to C. albicans virulence. These milestones have, however, resulted from the analysis of a large number of mutant libraries, each with a large number of strains constructed in different wild type backgrounds. Having to deal with this large set of mutant isolates requires a couple of important questions be asked: Have all the strains in these libraries been represented in terms of gene function? Since there are hundreds of libraries that have emerged from different laboratories world-wide, how do we effectively preserve and utilize the huge amount of function-based data coming from these libraries? Importantly, how do we translate this data into new and more effective treatment plans for C. albicans emerging infections and concomitant diseases?
To provide answers to some of these questions we can direct from work conducted by multiple dedicated research groups across the world. Some of these research groups have shown that tagging of mutant strains can have great potential in testing large groups of strains. Thus, this can enable rapid analysis of previously unexamined strains present in C. albicans libraries. The capacity to analyze large quantities of data has been put to the test by emergence of high throughput sequencing, which resulted in the development of plenty of online platforms to process such data. The same approach could be implemented to process the amount of function-based data generated from screening mutant libraries. The use of animal infection models, for which the animal used shares a large part of its biology with humans, ensures that the function-based discoveries coming from library screening have medical and health implications. In so far, many C. albicans mutant strains have been tested for the ability to colonize the gastrointestinal tract and to cause systemic or disseminated infections using the murine model of infection. However, the greatest challenge is to ensure there is an increase in the rate at which transfer of knowledge occurs between healthcare practitioners and molecular geneticists. Undoubtedly, as a research community, we stand more to gain if all libraries are made publically available and easily accessible. This will enforce collaboration between library designers and researchers across different disciplines, and further promote knowledge dissemination.
Abbreviations
| Ca | = | C. albicans |
| H | = | heterozygous or haploinsufficient strain |
| n.r. | = | Not reported |
| N | = | null mutant strain |
| SANARE | = | Soluble NSF (N-ethylmaleimide-sensitive factor) attachment protein Receptor |
| Sc | = | S. cerevisiae |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We gratefully acknowledge Nobile CJ and Hernday AD (University of California, Merced, USA) for discussions and useful suggestions during preparation of this manuscript.
Funding
TEM is supported by the Scarce Skills Fellowship Program of the South African National Research Foundation (NRF, Grant no. SFP14070774252). The financial assistance of the NRF toward this research is hereby acknowledged. Opinions expressed and conclusions arrived at, are those of the author and not necessarily to be attributed to the NRF.
Related Research Data
References
- Agrios GN. Plant Pathology, 5th edn. San Francisco, CA: Elsevier Academic Press 2005.
- Tylor LH, Latham SM, Woolhouse MEJ. Risk factors for human disease emergence. Phil Trans R Soc L and B 2001; 356:983-989; PMID:11516376; https://doi.org/https://doi.org/10.1098/rstb.2001.0888
- Hallen-Adams HE, Suhr MJ. Fungi in the healthy human gastrointestinal tract. Virulence 2016; 0:1-7; https://doi.org/https://doi.org/10.1080/21505594.2016.1247140
- Chang Y-L, Yu S-J, Heitman J, Wellington M, Chen Y-L. New facets of antifungal therapy. Virulence 2016; 0:1-15; https://doi.org/https://doi.org/10.1080/21505594.2016.1257457
- DiDomenico D. Novel antifungal drugs. Curr Opin Microbiol 1999; 2:509-515; PMID:10508731
- Odds FC, Brown AJP, Gow NAR. Antifungal agents: mechanisms of action. Trends Microbiol 2003; 11:272-279; PMID:12823944
- Galagan JE, Henn MR, Ma L-J, Cuomo CA, Birren B. Genomics of the fungal kingdom: Insights into eukaryotic biology. Genome Res 2005; 15:1620-1631; PMID:16339359; https://doi.org/https://doi.org/10.1101/gr.3767105
- Bruno VM, Mitchell AP. Large-scale gene function analysis in Candida albicans. Trends Microbiol 2004; 12:157-161; PMID:15051065; https://doi.org/https://doi.org/10.1016/j.tim.2004.02.002
- Noble SM, Johnson AD. Genetics of Candida albicans, a diploid human fungal pathogen. Annu Rev Genet 2007; 41:193-211; PMID:17614788; https://doi.org/https://doi.org/10.1146/annurev.genet.41.042007.170146
- Berman J1, Sudbery PE. Candida Albicans: a molecular revolution built on lessons from budding yeast. Nat Rev Genet 2002; 3:918-30; PMID:12459722; https://doi.org/https://doi.org/10.1038/nrg948
- Nobile CJ, Mitchell AP. Large-scale gene disruption using the UAU1 cassette. Methods Mol Biol 2009; 499:175-94; PMID:19152049; https://doi.org/https://doi.org/10.1007/978-1-60327-151-6_17
- Noble SM, Johnson AD. Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans. Eukaryot Cell 2005; 4:298-309; PMID:15701792; https://doi.org/https://doi.org/10.1128/EC.4.2.298-309.2005
- Samaranayake DP, Hanes SD. Milestones in Candida albicans gene manipulation. Fungal Genet Biol 2011; 48:858-65; PMID:21511047 https://doi.org/https://doi.org/10.1016/j.fgb.2011.04.003
- Motaung TE, Albertyn J, Pohl CH, Köhler G. Candida albicans mutant construction and characterization of selected virulence determinants. J Microbiol Methods 2015; 115:153-65; PMID:26073905; https://doi.org/https://doi.org/10.1016/j.mimet.2015.06.004
- Vyas VK, Barrasa MI, Fink GR. A Candida albicans CRISPR system permits genetic engineering of essential genes and gene families. Sci Adv 2015; 1:e1500248; PMID:25977940; https://doi.org/https://doi.org/10.1126/sciadv.1500248
- Uhl MA, Biery M, Craig N, Johnson AD. Haploinsuficiency-based large-scale forward genetic analysis of filamentous growth in the diploid human fungal pathogen C. albicans. EMBO J 2003; 22:2668-2678; PMID:12773383; https://doi.org/https://doi.org/10.1093/emboj/cdg256
- Davis DA, Bruno VM, Loza L, Filler SG, Mitchell AP. Candida albicans Mds3p, a conserved regulator of pH responses and virulence identified through insertional mutagenesis. Genetics 2002; 162:1573-1581; PMID:12524333
- Enloe B, Diamond A, Mitchell AP. A single-transformation gene function test in diploid Candida albicans. J Bacteriol 2000; 182:5730-36; PMID:11004171
- Blankenship JR, Fanning S, Hamaker JJ, Mitchell AP. An extensive circuitry for cell wall regulation in Candida albicans. PLoS Pathog 2010; 6:e1000752; PMID:20140194; https://doi.org/https://doi.org/10.1371/journal.ppat.1000752
- Finkel JS, Xu W, Huang D. Portrait of Candida albicans adherence regulators. PLoS Pathog 2012; 8:e1002525; PMID:22359502; https://doi.org/https://doi.org/10.1371/journal.ppat.1002525
- Oh J, Fung E, Price MN, Dehal PS, Davis RW, Giaever G, Nislow C, Arkin AP, Deutschbauer A. A universal TagModule collection for parallel genetic analysis of microorganisms. Nucleic Acids Res 2010; 38:e146; PMID:20494978; https://doi.org/https://doi.org/10.1093/nar/gkq419
- Oh J, Fung E, Schlecht U, Davis RW, Giaever G, St. Onge RP, Deutschbauer A, Nislow C. Gene annotation and drug target discovery in Candida albicans with a tagged transposon mutant collection. PLoS Pathog 2010; 6:e1001140; PMID:20949076; https://doi.org/https://doi.org/10.1371/journal.ppat.1001140
- Eckert SE, Mühlschlegel FA. Promoter regulation in Candida albicans and related species. FEMS Yeast Res 2009; 9:2-15; PMID:19054124; https://doi.org/https://doi.org/10.1111/j.1567-1364.2008.00455.x
- Roemer T, Jiang B, Davison J, Ketela T, Veillette K, Breton A, Tandia F, Linteau A, Susan Sillaots S, Catarina Marta C, et al. Large-scale essential gene identification in Candida albicans and applications to antifungal drug discovery. Mol Microbiol 2003; 50:167-181; PMID:14507372
- O'Meara TR, Veri AO, Ketela T, Jiang B, Roemer T, Cowen LE. Global analysis of fungal morphology exposes mechanisms of host cell escape. Nat Com 2015; 6:6741; PMID:25824284; https://doi.org/https://doi.org/10.1038/ncomms7741
- Xu D, Jiang B, Ketela T, Lemieux S, Veillette K, Martel N, Davison J, Sillaots S, Trosok S, Bachewich C, et al. Genome-wide fitness test and mechanism-of-action studies of inhibitory compounds in Candida albicans. PLoS Pathog 2007; 3:e92; PMID:17604452; https://doi.org/https://doi.org/10.1371/journal.ppat.0030092
- Homann OR, Dea J, Noble SM, Johnson AD. A phenotypic profile of the Candida albicans regulatory network. PLoS Genet 2009; 5:e1000783; PMID:20041210; https://doi.org/https://doi.org/10.1371/journal.pgen.1000783
- Noble SM, French S, Kohn LA, Chen V, Johnson AD. Systematic screens of a Candida albicans homozygous deletion library decouple morphogenetic switching and pathogenicity. Nat Genet 2010; 42:590-599; PMID:20543849; https://doi.org/https://doi.org/10.1038/ng.605
- Han T-L, Cannon RD, Villas-Bôas SG. The metabolic basis of Candida albicans morphogenesis and quorum sensing. Fungal Genet Biol 2011; 48:747-763; PMID:21513811; https://doi.org/https://doi.org/10.1016/j.fgb.2011.04.002
- Lu Y, Su C, Unoje O, Liu H. Quorum sensing controls hyphal initiation in Candida albicans through Ubr1-mediated protein degradation. Proc Natl Acad Sci 2014; 11:1975-1980; PMID:24449897; https://doi.org/https://doi.org/10.1073/pnas.1318690111
- Kebaara BW, Langford ML, Navarathna DHMLP, Dumitru R, Nickerson KW, Atkin AL. Candida albicans Tup1 is involved in farnesol-mediated inhibition of filamentous-growth induction. Eukaryot Cell 2008; 7:980-987; PMID:18424510; https://doi.org/https://doi.org/10.1128/EC.00357-07
- Brand A, MacCallum DM, Brown AJP, Gow NAR, Odds FC. Ectopic expression of URA3 can influence the virulence phenotypes and proteome of Candida albicans but can be overcome by targeted reintegration of URA3 at the RSP10 locus. Eukaryot Cell 2004; 3:900-909; PMID:15302823; https://doi.org/https://doi.org/10.1128/EC.3.4.900-909.2004
- Porman AM, Alby K, Hirakawa MP, Bennett RJ. Discovery of a phenotypic switch regulating sexual mating in the opportunistic fungal pathogen Candida tropicalis. Proc Natl Acad Sci USA 2011; 108:21158-63; PMID:22158989; https://doi.org/https://doi.org/10.1073/pnas.1112076109
- Porman AM, Hirakawa MP, Jones SK, Wang N, Bennett RJ. MTL-independent phenotypic switching in Candida tropicalis and a dual role for Wor1 in regulating switching and filamentation. PLoS Genet 2013; 9:e1003369; PMID:23555286; https://doi.org/https://doi.org/10.1371/journal.pgen.1003369
- Pujol C, Daniels KJ, Soll DR. Comparison of Switching and Biofilm Formation between MTL-Homozygous Strains of Candida albicans and Candida dubliniensis. Eukaryot Cell 2015; 14:1186-1202; https://doi.org/https://doi.org/10.1128/EC.00146-15
- Huang G. Regulation of phenotypic transitions in the fungal pathogen Candida albicans. Virulence 2012; 3:251-261; PMID:22546903; https://doi.org/https://doi.org/10.4161/viru.20010
- Slutsky BM, Staebell JA, Risen L, Pfaller M, Soll DR. “White-opaque transition”: a second high frequency switching system in Candida albicans. J Bacteriol 1987; 169:189-197; PMID:3539914
- Hull CM, Johnson AD. Identification of a mating type-like locus in the asexual pathogenic yeast Candida albicans. Science 1999; 285:1271-1275; PMID:10455055
- Lohse MB, Johson AD. Identification and characterization of wor4, a new transcriptional regulator of white-opaque switching. Genes Genom Genet 2016; 6:721-729; PMID:26772749; https://doi.org/https://doi.org/10.1534/g3.115.024885
- Lohse MB, Johson AD. Identification and characterization of wor4, a new transcriptional regulator of white-opaque switching. Genes Genom Genet 2016; 6:721-729; PMID:26772749; https://doi.org/https://doi.org/10.1534/g3.115.024885
- Miller MG, Johnson AD. White-Opaque switching in Candida albicans is controlled by mating-type locus homeodomain proteins and allows efficient mating. Cell 2002; 110:293-302; PMID:12176317
- Zordan RE, Galgoczy DJ, Johnson AD. Epigenetic properties of white-opaque switching in Candida albicans are based on a self-sustaining transcriptional feedback loop. Proc Natl Acad Sci USA 2006; 103:12807-12812; PMID:16899543; https://doi.org/https://doi.org/10.1073/pnas.0605138103
- Zordan RE, Miller MG, Galgoczy DJ, Tuch BB, Johnson AD. Interlocking transcriptional feedback loops control white-opaque switching in Candida albicans. PLoS Biol 2007; 5:e256; PMID:17880264; https://doi.org/https://doi.org/10.1371/journal.pbio.0050256
- C, Lachke SA, Srikantha T, Daniels K, Mccoy J, Soll DR. Misexpression of the opaque-phase-specific gene PEP1 (SAP1) in the white phase of Candida albicans confers increased virulence in a mouse model of cutaneous infection. Infect Immun 1998; 67:6652-6662; PMID:10569787
- Soll DR. Gene regulation during high-frequency switching in Candida albicans. Microbiology 1997; 143:279-88; PMID:9043104; https://doi.org/https://doi.org/10.1099/00221287-143-2-279
- Geiger J, Wessels D, Lockhart SR, Soll DR. Release of a potent polymorphonuclear leukocyte chemoattractant is regulated by white-opaque switching in Candida albicans. Infect Immun 2004; 72:667-677; PMID:14742507
- Sasse C, Hasenberg M, Weyler M, Gunzer M, Morschhäuser J. White-opaque switching of Candida albicans allows immune evasion in an environment-dependent fashion. Eukaryot Cell 2013; 12:50-58; PMID:23125350; https://doi.org/https://doi.org/10.1128/EC.00266-12
- Xie J, Tao L, Nobile CJ, Tong, Y, Guan, G, Sun Y, Cao C, Hernday AD, Johnson AD, Zhang L, Bai F-Y, et al. White-opaque switching in natural MTLa/α; isolates of Candida albicans: Evolutionary implications for roles in host adaptation, pathogenesis, and sex. PLoS Biol 2013; 11:e1001525; PMID:23555196; https://doi.org/https://doi.org/10.1371/journal.pbio.1001525
- Huang G, Srikantha T, Sahni N, Yi S, Soll DR. CO2 regulates white-to-opaque switching in Candida albicans. Curr Biol 2009; 19:330-334; PMID:19200725; https://doi.org/https://doi.org/10.1016/j.cub.2009.01.018
- Huang G, Yi S, Sahni N, Daniels KJ, Srikantha T, Soll DR. N-Acetylglucosamine induces white to opaque switching, a mating prerequisite in Candida albicans. PLoS Pathog 2010; 6:e1000806; PMID:20300604; https://doi.org/https://doi.org/10.1371/journal.ppat.1000806
- Lohse MB, Hernday AD, Fordyce PM. Identification and characterization of a previously undescribed family of sequence-specific DNA-binding domains. Proc Natl Acad Sci 2013; 110:7660-7665; PMID:23610392; https://doi.org/https://doi.org/10.1073/pnas.1221734110
- Fox EP, Cowley ES, Nobile CJ, Hartooni N, Newman DK, Johnson AD. Anaerobic bacteria grow within Candida albicans biofilms and induce biofilm formation in suspension cultures. Curr Biol 2014; 24:1-6; PMID:25308076; https://doi.org/https://doi.org/10.1016/j.cub.2014.08.057
- Daniels KJ, Srikantha T, Lockhart SR, Pujol CC, Soll DR. Opaque cells signal white cells to form biofilms in Candida albicans. EMBO J 2006; 25:2240-2252; PMID:16628217; https://doi.org/https://doi.org/10.1038/sj.emboj.7601099
- Tao L, Cao C, Liang W, Guan G, Zhang Q, Nobile CJ, Huang G. White cells facilitate opposite- and same-sex mating of opaque cells in Candida albicans. PLoS Genet 2014; 10:e1004737; PMID:25329547; https://doi.org/https://doi.org/10.1371/journal.pgen.1004737
- Ene IV, Bennett RJ. The cryptic sexual strategies of human fungal pathogens. Nat Rev Microbiol 2014; 12:239-251; PMID:24625892; https://doi.org/https://doi.org/10.1038/nrmicro3236
- Huang G, Srikantha T, Sahni N, Yi S, Soll DR. CO2 regulates white-to-opaque switching in Candida albicans. Curr Biol 2009; 19:330-334; PMID:19200725; https://doi.org/https://doi.org/10.1016/j.cub.2009.01.018
- Du H, Guana G, Xiea J, Cottier F, Sun Y, Jia W, Mühlschlegel FA, Huang G. The transcription factor Flo8 mediates CO2 sensing in the human fungal pathogen Candida albicans. Mol Biol Cell 2012; 23:2692-2701; PMID:22621896; https://doi.org/https://doi.org/10.1091/mbc.E12-02-0094
- Cao F, Lane S, Raniga PP, Lu Y, Zhou Z, Ramon K, Chen J, Liu H. The Flo8 transcription factor is essential for hyphal development and virulence in Candida albicans. Mol Biol Cell 2006; 17:295-307; PMID:16267276; https://doi.org/https://doi.org/10.1091/mbc.E05-06-0502
- Si H, Hernday AD, Hirakawa MP, Johnson AD, Bennett RJ. Candida albicans white and opaque cells undergo distinct programs of filamentous growth. PLoS Pathog 2013; 9:e1003210; PMID:23505370; https://doi.org/https://doi.org/10.1371/journal.ppat.1003210
- Guan G, Xie J, Tao L, Nobile CJ, Sun Y, Cao C, Tong Y, Huang G. Bcr1 plays a central role in the regulation of opaque cell filamentation in Candida albicans. Mol Microbiol 2013; 89:732-750; PMID:23808664; https://doi.org/https://doi.org/10.1111/mmi.12310
- Kumamoto CA. Candida biofilms. Curr Opin Microbiol 2002; 5:608-611; PMID:12457706
- Nobile CJ, Johnson AD. Candida albicans biofilms and human disease. Annu Rev Microbiol 2015; 69:71-92; PMID:26488273; https://doi.org/https://doi.org/10.1146/annurev-micro-091014-104330
- Mateus C, Crow SA Jr Ahearn DG. Adherence of Candida albicans to silicone induces immediate enhanced tolerance to fluconazole. Antimicrob Agents Chemother 2004; 48:3358-3366; PMID:15328097; https://doi.org/https://doi.org/10.1128/AAC.48.9.3358-3366.2004
- Dawson CC, Intapa C, Jabra-Rizk MA. “Persisters”: Survival at the cellular level. PLoS Pathog 2011; 7:e1002121; PMID:21829345; https://doi.org/https://doi.org/10.1371/journal.ppat.1002121
- Nobile CJ, Mitchell AP. Regulation of cell-surface genes and biofilm formation by the C. albicans transcription factor Bcr1p. Curr Biol 2005; 15: 1150–1155; https://doi.org/https://doi.org/10.1016/j.cub.2005.05.047.
- Ramage G, VandeWalle K, Lopez-Ribot JL, Wickes BL. The filamentation pathway controlled by the Efg1 regulator protein is required for normal biofilm formation and development in Candida albicans. FEMS Microbiol Lett 2002; 214:95-100; PMID:12204378.bb
- Nobile CJ, Fox EP, Nett JE, Sorrells TR, Mitrovich TM, Hernday AD, Tuch BB, Andes DR, Johnson AD. A recently evolved transcriptional network controls biofilm development in Candida albicans. Cell 2012; 148:126-138; PMID:22265407; https://doi.org/https://doi.org/10.1016/j.cell.2011.10.048
- Fox EP, Nobile CJ. A sticky situation: untangling the transcriptional network controlling biofilm development in Candida albicans. Transcription 2012; 3:315-22; https://doi.org/https://doi.org/10.4161/trns.22281
- Fox EP, Bui CK, Nett JE, Hartooni N, Mui MC, Andes DR, Nobile CJ, Johnson AD. An expanded regulatory network temporally controls Candida albicans biofilm formation. Mol Microbiol 2015; 96:1226-39; https://doi.org/https://doi.org/10.1111/mmi.13002
- Kelly MT, MacCallum DM, Clancy SD, Odds FC, Brown AJ, Butler G. The Candida albicans CaACE2 gene affects morphogenesis, adherence and virulence. Mol Microbiol 2004; 53:969-983; PMID:15255906; https://doi.org/https://doi.org/10.1111/j.1365-2958.2004.04185.x
- Morschhäuser J. Regulation of multidrug resistance in pathogenic fungi. Fungal Genet Biol 2010; 47:94-106; PMID:19665571; https://doi.org/https://doi.org/10.1016/j.fgb.2009.08.002
- Morschhäuser J, Barker KS, Liu TT, Blaß-Warmuth J, Homayouni R, Rogers PD. The transcription factor Mrr1p controls expression of the MDR1 efflux pump and mediates multidrug resistance in Candida albicans. PLoS Pathog 2007; 3:e164; PMID:17983269; https://doi.org/https://doi.org/10.1371/journal.ppat.0030164
- Vila T, Romo JA, Pierce CG, McHardy SF, Saville SP, Lopez-Ribot JL. Targeting Candida albicans filamentation for antifungal drug development. Virulence 2016; 7:1-9; PMID:27268130; https://doi.org/https://doi.org/10.1080/21505594.2016.1197444
- Gow NA, van de Veerdonk FL, Brown AJ, Netea MG. Candida albicans morphogenesis and host defence: discriminating invasion from colonization. Nat Rev Microbiol 2011; 10:112-122; PMID:22158429; https://doi.org/https://doi.org/10.1038/nrmicro2711
- da Silva Dantas A, Lee KK, Raziunaite I, Schaefer K, Wagener J, Yadav B, Gow NA. Cell biology of Candida albicans-host interactions. Curr Opin Microbiol 2016; 34:111-118; PMID:27689902; https://doi.org/https://doi.org/10.1016/j.mib.2016.08.006
- Dühring S, Germerodt S, Skerka C, Zipfel PF, Dandekar T, Schuster S. Host-pathogen interactions between the human innate immune system and Candida albicans-understanding and modeling defense and evasion strategies. Front Microbiol 2015; 6:625; PMID:26175718; https://doi.org/https://doi.org/10.3389/fmicb.2015.00625
- Netea MG, Brown GD, Kullberg BJ, Gow NA. An integrated model of the recognition of Candida albicans by the innate immune system. Nat Rev Microbiol 2008; 6:67-78; PMID:18079743; https://doi.org/https://doi.org/10.1038/nrmicro1815
- Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 2009; 7:99-109; PMID:19148178; https://doi.org/https://doi.org/10.1038/nrmicro2070
- Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 2015; PMID:26121197 21:677-87; https://doi.org/https://doi.org/10.1038/nm.3893
- Wellington M, Koselny K, and Krysan DJ. Candida albicans morphogenesis is not required for macrophage interleukin 1ß production. mBio 2013; 4:e00433-12; PMID:23269828; https://doi.org/https://doi.org/10.1128/mBio.00433-12
- Wellington M, Koselny K, Sutterwala FS, Krysan DJ. Candida albicans triggers NLRP3-mediated pyroptosis in macrophages. Eukaryot Cell 2014; 13:329-40; PMID:24376002; https://doi.org/https://doi.org/10.1128/EC.00336-13
- Selmecki A, Bergmann S, Berman J. Comparative genome hybridization reveals widespread aneuploidy in Candida albicans laboratory strains. Mol Microbiol 2005; 55:1553-1565; PMID:15720560; https://doi.org/https://doi.org/10.1111/j.1365-2958.2005.04492.x.b
- Motaung TE, Saitoh H, Tsilo TJ. Large-scale molecular genetic analysis in plant pathogenic fungi: A decade of genome-wide functional analysis. Mol Plant Pathol 2016; PMID:27733021; https://doi.org/https://doi.org/10.1111/mpp.12497
- Fonzi WA, Irwin MY. Isogenic strain construction and gene mapping in Candida albicans. Genetics 1993; 134:717-728; PMID:8349105
- Wilson RB, Davis D, Mitchell AP. Rapid hypothesis testing with Candida albicans through gene disruption with short homology regions. J Bacteriology 1999; 181:1868-1874; PMID:10074081