
ABSTRACT
Wound-colonizing microorganisms can form complex and dynamic polymicrobial communities where pathogens and commensals may co-exist, cooperate or compete with each other. The present study was aimed at identifying possible interactions between different bacteria isolated from the same chronic wound of a patient with the genetic blistering disease epidermolysis bullosa (EB). Specifically, this involved two different isolates of the human pathogen Staphylococcus aureus, and isolates of Bacillus thuringiensis and Klebsiella oxytoca. Particular focus was attributed to interactions of S. aureus with the two other species, because of the high staphylococcal prevalence among chronic wounds. Intriguingly, upon co-cultivation, none of the wound isolates inhibited each other's growth. Since the extracellular proteome of bacterial pathogens is a reservoir of virulence factors, the exoproteomes of the staphylococcal isolates in monoculture and co-culture with B. thuringiensis and K. oxytoca were characterized by Mass Spectrometry to explore the inherent relationships between these co-exisiting bacteria. This revealed a massive reduction in the number of staphylococcal exoproteins upon co-culturing with K. oxytoca or B. thuringiensis. Interestingly, this decrease was particularly evident for extracellular proteins with a predicted cytoplasmic localization, which were recently implicated in staphylococcal virulence and epidemiology. Furthermore, our exoproteome analysis uncovered potential cooperativity between the two different S. aureus isolates. Altogether, the observed exoproteome variations upon co-culturing are indicative of unprecedented adaptive mechanisms that set limits to the production of secreted staphylococcal virulence factors.
Introduction
Studies on pathogenic bacteria, either in vitro or in animal models, commonly address the respective bacteria as secluded species. Such studies do not consider the natural ecosystem of bacteria that colonize and invade the human body, which may happen in competition or cooperation with the existing microbiota and other pathogens. Furthermore, although bacteria-host interactions are the key to understanding the pathophysiology of infectious diseases, it is also important to scrutinize the parallel relationships between different bacterial species. This segment of microbial ecology may also elucidate how pathogens adjust their metabolism and other adaptive responses to survive and persist in the human body.
An example of a well-adapted bacterium associated with humans and animals, both as a commensal and as a pathogen, is Staphylococcus aureus [Citation1, Citation2]. This bacterium has an exceptional ability to adapt and evolve swiftly into multidrug resistant lineages, as shown since the clinical introduction of penicillin and subsequently developed antibiotics [Citation3, Citation4]. S. aureus is able to cause a wide range of afflictions ranging from impetigo and upper respiratory tract infections to osteomyelitis, endocarditis and sepsis. The fruitless attempts to develop a vaccine against S. aureus [Citation5, Citation6]. suggest the need for a better understanding of community interactions and the respective metabolic networks. This need is further underpinned by the high prevalence of infections caused by methicillin-resistant S. aureus (MRSA) in hospitals and communities worldwide [Citation7, Citation8]. Nevertheless, the vast majority of molecular studies on the virulence of S. aureus have investigated this pathogen in isolation.
In the present study, we explored the possible interactions between two phenotypically different S. aureus isolates from a chronic wound of a patient with epidermolysis bullosa (EB) and two other bacterial species, Bacillus thuringiensis and Klebsiella oxytoca, isolated from the same wound. EB is a genetic blistering disease characterized by the development of chronic wounds upon simple mechanical trauma. It was previously shown that S. aureus is an important wound colonizer in patients with EB, where a patient can carry several S. aureus types in a single wound, and autoinoculation from the upper respiratory tract can occur [Citation9–15]. Notably, next to S. aureus, also other bacteria are known to colonize the wounds of EB patients [Citation16]. Considering these colonization characteristics and the high prevalence of S. aureus in the chronic wounds of patients with EB [Citation15], we decided to study the interactions of S. aureus wound isolates with coexisting B. thuringiensis or K. oxytoca isolates. In particular, we focused attention on the extracellular proteomes (in short, exoproteomes) of these bacteria, because the exoproteome is a major reservoir of virulence factors [Citation17].
B. thuringiensis is a Gram-positive, spore-forming aerobic bacterium that is commonly used as a safe bioinsecticide since it produces pore-forming proteins that are toxic for insect larvae [Citation18]. It belongs to the same group as Bacillus anthracis and Bacillus cereus, the etiologic agents of anthrax and the “fried rice syndrome” (i.e. food poisoning), respectively [Citation19, Citation20]. B. thuringiensis, unlike the other two, is not known as a major human pathogen [Citation21]. However, it has been reported that this bacterium may be responsible for opportunistic pulmonary infections in susceptible subjects [Citation22, Citation23]. On the other hand, the Gram-negative bacterium K. oxytoca is clearly regarded as an opportunistic pathogen. It is commonly found on mucosal surfaces of mammals, including the nasopharynx and colon of humans [Citation24]. Klebsiella species are the second most common cause of nosocomial Gram-negative bacteremia after Escherichia coli, and they are often involved in urinary tract infections in risk group patients and neonatal sepsis [Citation25–28].
Altogether, the present study was aimed at exploring the potentially synergistic or competitive relationships between S. aureus and two different classes of bacteria that are, in principle, capable of colonizing and invading the human body, thereby causing disease. Our specific objective was to unravel the bacterial response patterns using the exoproteome as a read-out. To this purpose, two different S. aureus isolates were, respectively, co-cultured with B. thuringiensis (Bt) or K. oxytoca (Ko) isolates from the same wound environment, and the exoproteomes of individually or co-cultured bacteria were analyzed by Mass Spectrometry (MS).
Results
S. aureus grows unimpaired when co-cultured with B. thuringiensis or K. oxytoca on solid agar
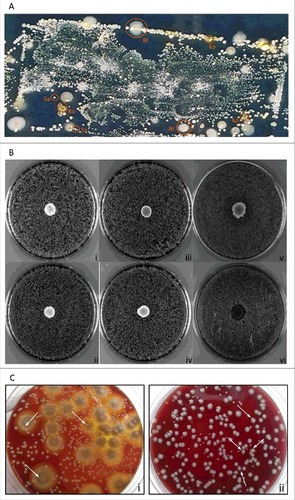
In a previous study, we applied replica plating of used wound dressings to assess the topography of distinct S. aureus types in chronic wounds of patients with EB [Citation13]. Notably, this previous study was focused on S. aureus only, and it did not take into consideration other co-existing microbial species that are clearly evident upon replica plating of wound dressings (). Such co-existing species were collected in the present study, using the dressing of a chronic wound from a patient with Junctional Epidermolysis Bullosa. This led to the identification of two S. aureus isolates with spa types t111 and t13595 that grew in close proximity to B. thuringiensis (Bt) and K. oxytoca (Ko) isolated from the same wound. A zone inhibition experiment was employed to assess possible competitive or bactericidal interactions between these four wound isolates. As shown for the S. aureus t111 and t13595 isolates in , there was no growth inhibition detectable when, respectively, Bt or Ko were spotted onto blood agar plates that were confluently inoculated with S. aureus. The same was essentially observed when Bt was spotted onto a confluent inoculated plate with Ko. Additionally, B. subtilis strain 168 was chosen as control based on the known growth inhibition that B. subtilis 168 exerts on S. aureus due to production of the bacteriocin sublancin [Citation29]. Indeed, the spotting of B. subtilis 168 onto the S. aureus cells caused a clear growth inhibition zone (). Furthermore, it was noted that the growth of Bt and Ko occurred on top of the S. aureus spread. To confirm this observation, serial dilution experiments were performed with mixed cultures of S. aureus and Bt, or S. aureus and Ko. As marked by the arrows in , colonies of S. aureus were not only able to grow in the proximity of Bt and Ko colonies, but some were observed to touch or even grow on top of each other (). This is fully consistent with the situation encountered in the wound environment from which the bacteria were isolated and where they thrive in close proximity ().
Figure 1. Growth characteristics of the isolated bacteria on agar plates. (A) Microbiome topography of culturable bacteria from the wound dressing of a patient with Junctional EB. The used dressing from a chronic wound was replica plated onto CLED agar for isolation of the bacteria present. (B) Zone inhibition experiments. Unimpaired staphylococcal growth upon spotting of B. thuringiensis or K. oxytoca: (i) t111+Bt, (ii) t13595+Bt, (iii) t111+Ko, and (iv) t13595+Ko. Unimpaired growth of K. oxytoca upon spotting of B. thruingiensis (v). S. aureus growth inhibition halo caused by spotting of B. subtilis 168 onto a lawn of staphylococcal cells (vi; both S. aureus strains exhibited the same effect). (C) Dilution and subsequent plating: (i) S. aureus colonies growing on top of larger colonies of B. thuringiensis; (ii) K. oxytoca and S. aureus colonies growing in close proximity and occasionally touching each other. Typical S. aureus colonies are marked with arrows.
S. aureus isolates t111 and t13595 are highly related but not identical
As a first step in the characterization of the selected S. aureus t111 and t13595 isolates, their antibiotic resistance profiles were determined. Both isolates were shown to be resistant to tetracycline, ciprofloxacin and fusidic acid. In addition, isolate t111 was found to be resistant to trimethoprim and sulfamethoxazole (Supplemental Table S1). These findings suggested that both isolates are very similar, but not identical. This was confirmed by whole-genome sequencing, revealing that the respective core genomes differed only by 192 nucleotides. Genome sequencing further revealed that both S. aureus isolates belong to the sequence type 5 (ST5). Thus, the closest related reference genome is the one of S. aureus strain N315 which was, therefore, used for comparative genomic analyses and annotation.
Although there are not many differences in the core genomes of the t111 and t13595 isolates, a few of these differences are noteworthy. For example, the fnbA and fnbB genes of the t111 isolate were recombined into a single gene. Isolate t13595 contained a point mutation in agrA specifying the replacement of Gly148 with Asp, and this mutation was correlated with an agr negative phenotype (i.e. unlike the t111 isolate, the t13595 isolate displayed no α, β or δ-hemolysis). Frame shift mutations were also observed in various other genes of the t13595 isolate, including the mutL gene for a DNA mismatch repair protein, and genes such as sbi, araC, geh and fnbA. For a more detailed comparison of the S. aureus isolates with respect to different mobile genetic elements, please refer to Supplemental Table S2.
Total numbers of S. aureus exoproteins are reduced when S. aureus is co-cultured with Bt or Ko
Since the S. aureus, Bt and Ko isolates addressed in this study were co-existent in the same wound environment and are capable of growing in close contact in an in vitro setting, we wanted to know whether and, if so, how they influence each other. To address this question, we selected a proteomics approach in which we focused our attention on the extracellular proteome. Further, to simulate the conditions in the human body, we selected the tissue culture medium RPMI as the growth medium for our studies, because S. aureus cells grown on RPMI or human plasma display very similar genome-wide transcript profiles Citation[30]. Thus, the S. aureus isolates t111 and t13595 were cultured in three different conditions, namely in pure culture, or in combination with either Bt or Ko. As expected from the plating experiments, we observed only slight differences in the logarithmic growth rates of the mixed cultures as assessed by OD600 readings. For example, when S. aureus was co-cultured with Bt, the overall growth seemed slightly slower, while the opposite happened when S. aureus was co-cultured with Ko (Figure S1). This can be explained by the particular growth rates of the Bt and Ko isolates on RPMI, where the growth rate of Bt in monoculture is relatively slow, while that of Ko is relatively fast. Nevertheless, the overall growth patterns of the co-cultures were similar to those of the S. aureus pure cultures and, importantly, they were highly reproducible. Hence, in all the experiments, our reference for sampling was the S. aureus growth curve in monoculture.
To obtain an overview of the behavior of the cultures, we compared the total numbers of proteins detected by LC-MS/MS in all conditions (,) and verified the reproducibility of the biological replicates (Supplemental Figure S2). This analysis revealed clear differences between the pure and mixed cultures. Particularly, a total of 64 extracellular proteins were detected in the S. aureus t111 monoculture, while in combination with Bt only 26 extracellular staphylococcal proteins were detected, and merely 16 when the t111 isolate was cultured with Ko. Essentially the same was observed for the exoproteome of the strain t13595, where 49 extracellular proteins were found in the monoculture, and only 29 and 12 in the co-cultures with Bt or Ko, respectively (). We also assessed the numbers of extracellular proteins detected in the monocultures of Bt and Ko in comparison to the co-cultures. The results indicate a decrease of ∼25% in the number of Ko proteins found in the co-cultures with S. aureus (), but this effect was clearly not as prominent as the effect of Ko on the staphylococcal exoproteome. On the other hand, the number of exoproteins of Bt decreased only in the presence of the S. aureus t111 isolate but not in the presence of the t1359 isolate (). Of note, the exoprotein differences among the cultures were also reflected in the absolute amounts of protein detectable in the growth medium (Figure S3).
Figure 2. Total numbers of extracellular proteins identified per culture. (A) Numbers of S. aureus t111 and t13595 exoproteins identified in monocultures or upon co-culturing with B. thuringiensis (+Bt) or K. oxytoca (+Ko). (B) Numbers of B. thuringiensis and K. oxytoca exoproteins identified in monocultures or upon co-culturing with S. aureus t111 (+t111) or t13595 (+t13595) isolates. (C-F) Predicted subcellular localization of proteins per strain in mono- or co-cultures. (C) 11, S. aureus t111 exoproteins; 11B, S. aureus t111 exoproteins upon co-culture with B. thuringiensis; 11K, S. aureus t111 exoproteins upon co-culture with K. oxytoca. (D) 13, S. aureus t13595 exoproteins; 13B, S. aureus t13595 exoproteins upon co-cultures with B. thuringiensis; 13K, S. aureus t13595 exoproteins upon co-culture with K. oxytoca. (E) K, K. oxytoca exoproteins; K11, K. oxytoca exoproteins upon co-culture with S. aureus t111; K13, K. oxytoca exoproteins upon co-culture with S. aureus t13595. (F) B, B. thuringiensis exoproteins; B11, B. thuringiensis exoproteins upon co-culture with S. aureus t111; B13, B. thuringiensis exoproteins upon co-culture with S. aureus t13595.
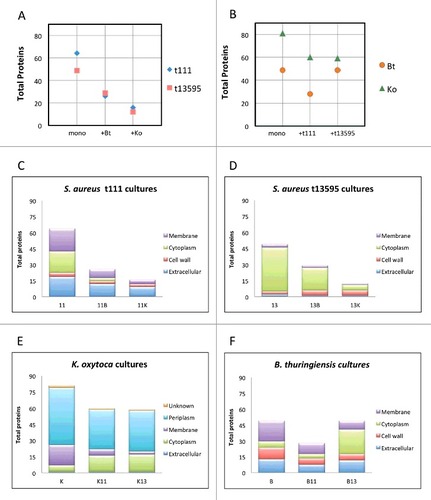
As a first approach to characterize the identified extracellular proteins, we assessed their predicted subcellular localization. For the two S. aureus isolates and the Bt isolate, which are all three Gram-positive bacteria, altogether 150 (50%) predicted cytoplasmic proteins, 26 (9%) cell wall proteins, 74 (25%) membrane proteins and 47 (16%) extracellular proteins were identified in the exoproteome (Supplemental Figure S4). For the Gram-negative Ko isolate, we detected 50 (34%) predicted cytoplasmic, 25 (17%) membrane, 1 (1%) extracellular, and 67 (46%) periplasmic proteins (Supplemental Figure S4). Because of the acute changes in the S. aureus exoproteome upon co-culturing, we were interested in knowing which predicted subcellular protein fractions were most strongly affected in the various conditions. For both S. aureus isolates, the fraction of predicted cytoplasmic proteins sharply decreased upon co-culturing, and such proteins were even completely missing from the co-culture of the t111 isolate with Ko (,). On the contrary, the fraction of predicted cytosolic Bt proteins in the extracellular proteome sharply increased when co-cultured with S. aureus t13595, while the numbers of predicted cytoplasmic proteins of Ko in co-cultures with either of the two S. aureus isolates were similar, showing a relatively slender increase when S. aureus was present (,).
Co-cultured S. aureus, B. thuringiensis and K. oxytoca isolates display species-specific exoproteome changes in terms of biological processes
Although both S. aureus isolates are closely related, particular variations in their exoproteomes were observed. For example, the core exoproteome of the t111 isolate included only 14 proteins (i.e. proteins constant in all cultures), while the core exoproteome of the t13595 isolate included only nine proteins ( and Table S3). Likewise, the biological processes in which these proteins are involved were also different. The core exoproteome of the t111 isolate was characterized by functions in peptidoglycan catabolic processes, pathogenesis and cobalamin transport, whereas the core exoproteome of the t13595 isolate is involved in cell redox homeostasis, cell wall organization, glucose metabolic processes, pathogenesis, cell adhesion and glycine betaine biosynthetic processes from choline.
Figure 3. Relationships among cultures. The diagrams show the number of proteins identified in all cultures and those proteins shared in the different conditions. (A) Total number of S. aureus t111 proteins in monoculture (11) and in co-culture with B. thuringiensis (11B) or K. oxytoca (11K); (B) Total number of S. aureus t13595 proteins in monoculture (13) and in co-culture with B. thuringiensis (13B) or K. oxytoca (13K); (C) Total number of B. thuringiensis proteins in monoculture (B) and in co-culture with S. aureus t111 (B11) or S. aureus t13595 (B13); (D) Total number of K. oxytoca proteins in monoculture (K) and in co-culture with S. aureus t111 (K11) or S. aureus t13595 (K13); (E) Total number of S. aureus proteins in monoculture (11) or (13) and in co-culture (11+13).
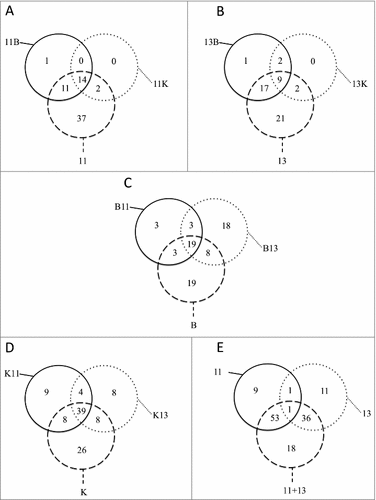
Remarkably, there were not many staphylococcal proteins expressed uniquely in the co-cultures. In fact, there were no distinctive proteins detected in the co-cultures with Ko for both S. aureus isolates (,). For the co-cultures with Bt, we identified only the translocase subunit SecG as a distinctive feature in the growth medium of the t111 isolate, and an uncharacterized distinctive protein was identified in the growth medium of the t13595 isolate. In contrast, in the presence of the t111 or t13595 isolates, Ko displayed 9 and 8 unique extracellular proteins, respectively (). The latter 9 proteins produced in the presence of the t111 isolate were related, by their gene ontologies, to chromosome condensation, regulation of transcription, the phosphoenolpyruvate-dependent sugar phosphotransferase system, and glycerol ether metabolic processes. Similarly, Bt displayed 18 distinctive extracellular proteins in the presence of the t13595 isolate (). These proteins relate to iron-sulphur cluster assembly, protein transport, cell cycle, glucose and glycerol ether metabolic processes.
To further categorize the identified extracellular proteins, we classified them according to their function and categorized them in one of the following six groups: (1) cell envelope and cellular processes (including cell wall/membrane, transport/binding proteins and lipoproteins, signal transduction, membrane bioenergetics, motility and chemotaxis, protein secretion, cell division, cell adhesion, response to stress); (2) information pathways (including DNA restriction/modification and repair, transcription and regulation, ribosomal proteins, protein synthesis/modification/folding); (3) intermediary metabolism; (4) cell redox homeostasis, (5) virulence factors; and (6) unknown functions (see Supplemental Table S3).
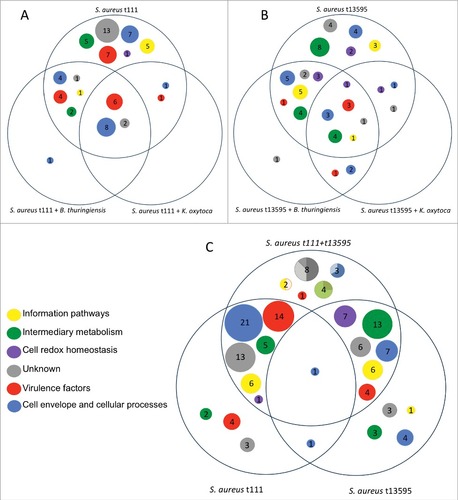
In the diagrams of , the differences between the S. aureus t111 and t13595 isolates in terms of the functions of extracellular proteins are clearly evident; while the t111 isolate seems to be more virulent, the extracellular proteins of the t13595 are more involved in processes related to cell redox homeostasis and intermediary metabolism. Further, the core functions of the t13595 isolate appear more diverse than those of the t111 isolate. In the case of Ko, the overall picture indicates that this bacterium's exoproteome is strongly involved in cell envelope and cellular processes, especially processes relating to the transport and binding of proteins and lipoproteins (Supplemental Figure S5A). For Bt, we observed that the response towards the co-cultured S. aureus isolates was considerably different. In particular, the extracellular Bt proteins related to many more different functions when Bt was co-cultured with the t13595 isolate than when Bt was co-cultured with the t111 isolate. Interestingly, like the S. aureus t13595 isolate co-cultured with Bt, Bt also exhibited many extracellular proteins related to redox homeostasis when co-cultured with the t13595 isolate (Supplementary Figure S5B).
Figure 4. Overview of the predicted functions of the exoproteins found in all staphylococcal cultures. Diagram (A) depicts only S. aureus t111 proteins. The area on the top shows the proteins detected only in monoculture, while the lower left area shows a single protein detected when t111 was co-cultured with B. thuringiensis. Likewise, diagram (B) shows exclusively the proteins that belong to S. aureus t13595 in monoculture and co-culture with B. thuringiensis or K. oxytoca. Diagram (C) depicts the proteins of S. aureus t111 and t13595 grown in monoculture in the bottom areas, whilst the upper area shows the proteins identified upon co-culturing both S. aureus isolates. In the latter area, the circles are represented as pie charts. The charts show the percentage of proteins that belong to each isolate. For the unknown proteins (in grey), 50% belong to t111, 37% to t13595, and 12.5% to both of them; for the cell envelope proteins (blue) 67% belong to t111 and 33% to both isolates; for proteins involved in intermediary metabolism (green), 25% belong to t111 and 75% to t13595; and for information pathways proteins (yellow), 50% belong to t111 and 50% to both isolates.
Particular exoproteins of S. aureus, K. oxytoca and B. thuringiensis show quantitative changes in mono- or co-cultures
Conceivably not all the differences in the exoproteome composition of mono- and co-cultured S. aureus, Ko and Bt are absolute, but also quantitative differences may exist for particular exoproteins. Therefore, we comparatively assessed significant fold-changes in the exoproteins detected in mono- and co-cultures by mapping their spectral counts from all replicates using volcano plots (Supplemental Figure S6). The results of this analysis, presented in , indicate that there were 11 exoproteins significantly ‘upregulated’ in the S. aureus monocultures compared to the co-cultures with Bt or Ko. These included S. aureus proteins, such as elongation factor Ts, triosephosphate isomerase, thioredoxin reductase, 2,3-bisphosphoglycerate-dependent phosphoglycerate mutase and a secreted β-lactamase. In the case of Ko, 22 exoproteins were present in significantly different amounts when mono and co-cultures were compared. Here, 17 exoproteins of Ko were significantly upregulated in the monocultures, whereas 5 other exoproteins of Ko were significantly upregulated in the co-cultures with S. aureus. The latter proteins are mostly involved in carbohydrate metabolism. Also in the case of Bt, 22 exoproteins were present in significantly different amounts when mono and co-cultures were compared (). Interestingly, 21 of these Bt exoproteins were significantly ‘upregulated’ in the monocultures compared to the co-cultures with S. aureus. The functions of these proteins were rather diverse, including the cell wall hydrolase LytE, three different proteases, and the protein folding catalyst PrsA.
Table 1. Summary of differentially detected extracellular proteins among the different cultures. Volcano plots (Supplemental Figure S6) displayed P values (-log10) versus fold changes (log2) of the normalized spectral counts. Folds ≤1 represent proteins that were significantly up-regulated in the co-cultures and folds >1 represent proteins that were significantly up-regulated in monoculture.
Discussion
Our present co-culturing approach represents an opportunity to study changes in protein diversity as a result of inter-bacterial relationships that can, to a certain extent, reflect the natural situation in a wound. In principle, the in vitro growth characteristics of S. aureus colonies over B. thuringiensis or K. oxytoca colonies suggest adaptive mechanisms for all species. From the proteome perspective, one mechanism that stands out is the depletion of staphylococcal cytoplasmic proteins from the exoproteomes of co-cultures upon co-culture with Bt or Ko. Proteomic analysis also revealed two uniquely detectable exoproteins in the co-cultures with Bt. The exoprotein found in the co-culture of S. aureus t111+Bt was SecG, a subunit of the heterotrimeric complex SecYEG. This complex serves as a membrane channel for translocation of secretory proteins in an unfolded state [Citation31]. In S. aureus, SecG is of importance since secG deletion mutants displayed impaired secretion of several wall-bound proteins and abundant exoproteins [Citation32]. Despite its known function in protein secretion, the reason for the unique detection of SecG in the t111-Bt exoproteomes is presently not clear. Additionally, in S. aureus t13595+Bt co-cultures an uncharacterized protein that resembles a control factor of the competence regulator ComK, named YlbF/YmcA, was found. This protein is known to be involved in biofilm formation by other Gram-positive and archaeal organisms. Further characterization of these two proteins might help us to understand why they are present in the exoproteome when S. aureus is co-cultured with Bt.
While the exoproteome of S. aureus in monoculture was previously found to contain numerous typically cytoplasmic proteins [Citation17, Citation33–38], this is not an exclusive trait of S. aureus. The exoproteomes of other Gram-positive bacteria, such as B. subtilis, Clostridium difficile,Corynebacterium diphtheria, Group A Streptococcus, Mycobacterium tuberculosis and Streptomyces lividans, but also Gram-negative bacteria like Porphyromonas gingivalis, have been associated with high numbers of such typically cytoplasmic proteins.[Citation36, Citation39–43] In B. subtilis, this observation has been explained by a combination of high cell lysis propensity, increased cell wall turnover and/or autolysin activity [Citation44]. It has also been proposed that the ‘liberation’ of typical cytoplasmic proteins that lack the prototype signal peptides of proteins exported via the general Sec pathway for protein secretion could be facilitated by as yet unidentified alternative pathways, also referred to as ‘non-classical secretion systems’ [Citation45–50]. For example, it is conceivable that, besides the well-characterized endolysins, the expression of prophage-encoded holins or holin-like proteins might have a role in the liberation of otherwise cytosolic proteins [Citation42,Citation50]. The release of cytosolic proteins due to the large pores formed by holins is still somewhat debated, since it has not been extensively studied in Gram-positive organisms. Nevertheless, the presence of additional holins could, thus, at least partially explain the elevated number of cytoplasmic proteins found in the growth medium of the S. aureus t13595 isolate compared to the t111 isolate. Yet, it has been shown for B. subtilis that engineered strains lacking all prophages still release typical cytoplasmic proteins into their growth medium [Citation52]. Clearly, for the bacterial isolates investigated in the present study, it remains to be defined which particular mechanisms contribute, individually or synergistically, to the observed non-classical secretion phenomenon and to what extent.
Interestingly, the common denominator between the presently identified S. aureus exoproteome and that of its ‘wound mates’ is the presence of housekeeping enzymes, particularly glycolytic enzymes. For instance, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) found in the S. aureus t13595, Bt and Ko cultures, has also been located on the surface of other pathogenic bacteria like group A streptococci, and enteropathogenic E. coli where it can serve either as a binding protein or as signal transduction initiator in epithelial cells [Citation53–55]. Cell wall-associated proteins like enolase (displayed in the same cultures) have been reported to bind to plasminogen both in Gram-positive (Streptococcus pneumoniae) and Gram-negative (Actinobacillus actinomycetemcomitans) bacteria [Citation56, Citation57]. Other multifunctional ‘moonlighting’ proteins like fructose-bisphosphate aldolase, identified in S. aureus t111 cultures, are recognized to play a role in adhesion to mammalian cells and binding of human plasminogen by several bacterial species, like S. pneumoniae, M. turberculosis, and Neisseria meningitidis [Citation58]. Similarly, the elongation factor (EF) Tu, a guanosine nucleotide binding protein important for protein synthesis, was detected on the surface of Mycoplasma pneumoniae and Lactobacillus johnsonii, where it was involved in fibronectin- and mucin-binding, respectively [Citation59, Citation60]. Furthermore, an immunoproteomic study of bacteremia showed that six housekeeping enzymes and EF-Tu were linked to leukopenia caused by Klebsiella pneumonia [Citation61]. Altogether, these findings imply that proteins, which were originally considered to be restricted to the cytoplasm, may also be important for virulence enhancement and invasive disease [Citation50, Citation62]. Indeed, for S. aureus, such proteins have previously been implicated in virulence, epidemiology and intracellular survival [Citation46–48, Citation50, Citation63].
By tracking the S. aureus-specific exoproteome, we observed that especially the cytoplasmic protein fractions were depleted from the exoproteome in co-cultures. This suggests that there might be either an altered localization, decreased cell lysis, enhanced proteolysis or consumption of these proteins by the other organism in the respective co-culture. In the literature, a loss of the cytoplasmic exoproteome fraction was recently associated to the latter. When studying marine bacteria in mixed cultures, Christie-Oleza et al. showed that extracellular cytoplasmic proteins from Synechococcus are notably reduced when Roseobacter strains are present [Citation64]. Their study also reported an abundance of active transporters and flagellar proteins of Roseobacter during co-culturing. Such a situation could potentially also apply to K. oxytoca and B. thuringiensis as the present data suggests that, when grown in a nutrient-limited environment, these bacteria might act as scavengers of carbon and nitrogen sources, particularly by degrading and assimilating extracellular staphylococcal proteins. If so, this would suggest that, at least in a closed system like the one we established in vitro, nothing goes to waste and that the carbon and nitrogen fixing characteristics of Bt and Ko are perhaps a distinguishable reflection of short-term interspecies interactions. Nevertheless, we cannot exclude the possibility that there could also be less S. aureus cell lysis during co-culture. One way to verify the idea that Bt and Ko consume extracellular proteins of S. aureus, may involve the metabolic labeling of S. aureus proteins with particular isotopes, providing these proteins to Bt or Ko cultures, and investigating the incorporation of the isotopes into Bt or Ko proteins or metabolites. Likewise, interactions between S. aureus and Bt or Ko could be investigated by transcriptomics and metabolomics. Irrespective of the molecular background of our observations, the depletion of S. aureus proteins from the exoproteome upon co-culturing has an important implication for the virulence of this bacterium as exoproteins should be considered as the prime reservoir of virulence factors [Citation17, Citation50].
Beyond the known proteome heterogeneity [Citation33, Citation34, Citation37]. and the genetic diversity of the colonizing staphylococcal strains [Citation15, Citation65], we found a mutL mutation indicative of impaired mismatch repair and other hypermutable traits in our S. aureus t13595 isolate [Citation66]. This finding allows us to place the relationships between the investigated S. aureus isolates in a broader perspective, where it is of note that the presently investigated bacterial isolates from an EB patient were obtained upon one-time sampling of a >8 months existing chronic wound. Importantly, it has been hypothesized that hypermutable strains play roles in species persistence and chronic infection [Citation67, Citation68] and they could perhaps also favor the development of antibiotic resistance [Citation69–71]. In the context of the long-term colonization of chronic wounds of EB patients by S. aureus, the influence of mutations on the intra-species heterogeneity should be considered in view of microbial social behavior and the consequences for fitness that particular mutations may provide [Citation72]. Interestingly, with respect to the observed proteomic profiles, the S. aureus t111 isolate seems to foster a committed production of metabolically costly siderophores, virulence factors and cell adhesion molecules, while the t13595 isolate shows a more ‘recalcitrant’ performance focusing only on its metabolism and homeostasis. Moreover, during co-culturing of the t111 and t13595 isolates, we did not observe any protein overexpression, contrary to what was observed for the monocultures. This outcome is suggestive of a cooperative behavior. Therefore, it is tempting to hypothesize that the genetic variability as displayed by the ‘selfish’ t13595 isolate may be beneficial for the spread or persistence of S. aureus as a wound-colonizing species [Citation73].
To the best of our knowledge, our research represents the first explanatory analysis of S. aureus exoproteomic shifts while in co-culture with wound-coexisting bacteria. Collectively, our findings point out the importance of studying bacteria in a broader biological context. It is evident that, for example, the growth rates of the four isolates explored here are not necessarily the same, and therefore it is not possible to compare the exoproteome of all investigated isolates at exactly the same growth stage. However, this situation is also true in a wound environment where not all co-existing microbes are completely synchronized or exist in equal numbers. Hence, the present data should be interpreted as one of many possible scenarios in which bacteria interact with each other with no apparent antagonistic relationships.
Because of the complexity of the wound environment and the peculiarities of each wound, it is difficult to establish a standard definition of the nutrients present in a wound. For example, temperature in a wound can influence local vasodilation and therefore the availability of nutrients. Furthermore, the biomolecules are primarily at the disposal of host cells rather than bacteria that have to compete for the nutrient restriction [Citation74]. In the present investigations, we have presented a scenario that simulates the conditions in human plasma, which relies on bacteria cultivated under shaking and not static conditions. Accordingly, the present culture conditions may not exactly reflect the situation in chronic wounds of an EB patient. However, it seems reasonable to assume that in the wound environment, cytosolic bacterial proteins could represent an important foundation for microbiome cross talk and homeostasis. Altogether, our experiments introduce an attractive avenue to explore possible pre- and probiotic targets, such as quorum sensing- or quorum quenching-related factors or bacterial growth factors that can either hinder the invasion by pathogenic bacteria, or enhance the presence of non-pathogenic microbes that may prevent infections and could even enhance wound healing.
Material and methods
Ethical approval
The local medical ethics committee (METc) of the University Medical Center Groningen (UMCG) approved of the collection of non-invasive samples from patients with EB on the basis of obtained written informed patient consent. The experiments were conducted in accordance with the guidelines of the Declaration of Helsinki. All samples were anonymized.
Bacterial isolation, identification and culture conditions
The used bandage from a chronic wound of a Dutch patient with Junctional Epidermolysis Bullosa was collected. The bandage was replica-plated with gentle pressure for 10 seconds onto CLED agar (Oxoid) using bioassay plates (245 × 245 × 245 ;mm; Nunc). After 48 h of incubation at 37°C, colonies were selected and replated onto blood agar plates for identification. Bacterial colonies were identified by matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) MS, using a MALDI Biotyper® (Bruker Corporation, Billerica, USA) as previously described [Citation75]. All isolates were stored with 10% glycerol at −80°C.
To investigate whether the studied bacteria secrete compounds that interfere with each other's growth, blood agar plates were spread with pure overnight cultures of S. aureus isolates with an optical density at 600 nm (OD600) of 0.5 to obtain a confluent lawn of cells. Then, after 5 min, 10 µL aliquots of pure overnight cultures of B. thuringiensis, K. oxytoca or Bacillus subtilis 168 (OD600 0.5) were applied to the center of the agar plate. For viable plate counting and the preparation of extracellular protein extracts, bacterial strains were grown overnight on Tryptic Soy Broth (TSB) under vigorous shaking at 37°C. The cultures were then diluted to an OD600 of 0.05 in pre-warmed RPMI medium without phenol red (GE Healthcare). Cultivation continued in a water bath under constant shaking (80–85 rpm) at 37°C until cultures reached an OD600 of ±0.5. Main cultures were again diluted in 120 mL pre-warmed RPMI medium under the same conditions. Monocultures were started with an initial OD600 of 0.05 while co-cultures were inoculated with an OD600 of 0.025 of each isolate to a total of 0.05. Given that an OD600 of 0.5 contained a total of ∼200 × 106 colony forming units per mL (CFU/mL) for t111, t13595 and Ko, and ∼15 × 106 CFU/mL for Bt, inoculated cultures corresponded to ∼20 × 106 CFU/mL for t111, t13595 and Ko and to ∼1.5 × 106 CFU/mL for Bt.
For proteome analyses and determination of CFU/mL, samples were collected at 90 min within the stationary growth phase. The ratios of different bacteria sampled from the co-cultures remained nearly the same with the exception of the co-cultures of S. aureus and K. oxytoca, where K. oxytoca grew 1.5 times faster than t111 and 1.7 times faster than t13595.
Genomic DNA extraction, genome sequencing and analysis
Libraries for whole genome sequencing were prepared from S. aureus t111, t13595, B. thuringiensis, and K. oxytoca DNA with the Nextera XT v2 kit, and run on Illumina next generation sequencing platforms (MiSeq) according to the manufacturers' instructions (Illumina, San Diego, CA, USA). Read data for the study isolates have been deposited in the National Center for Biotechnology Information (NCBI) under accession number SRP094393. The respective .fastq files were submitted for de novo sequence assembly using CLC Genomics Workbench v7.0.4 (CLC bio A/S, Aarhus, Denmark) after quality trimming (Qs > 28) with optimal word sizes based on the maximum N50 value. De novo assembled contigs of the S. aureus isolates were imported into the Seqsphere+ software version 1.0 (Ridom GmbH, Würzburg, Germany) for in silico multi-locus sequence typing (MLST) and spa typing as previously described [Citation76, Citation77]. Automatic genome annotation was done on the rapid annotation using subsystem technology (RAST) server 2.0 for all the genomes [Citation78, Citation79]. De novo assembled genomes of sequenced isolates were queried against the closest related reference genome of S. aureus strain N315 (GenBank: NC_007795.1), B. thuringiensis serovar konkukian str. 97–27 (GenBank: NC_017200), and K. oxytoca strain E718 (GenBank: NC_018106) using blastN in the WebACT comparison tool (http://www.webact.org/WebACT/prebuilt#) and analyzed in detail by the Artemis Comparison Tool (ACT) software [Citation80, Citation81]. Similarity matches were filtered based on their length (100 kb segments) and percentage similarity scores, and only the filtered hits with at least 80% sequence similarity were then displayed by ACT (e-value of 10.00000) and analyzed in detail.
Preparation of protein extracts
All samples were collected in duplicate according to the growth curves of the S. aureus isolates after 90 min of stationary growth. Cells were removed by centrifugation (10 min, 8000 x g, 4°C) from 2 mL culture aliquots and the supernatant was subsequently filtrated with a 0.22 µm filter (GE Healthcare Systems, Little Chalfont, United Kingdom). Proteins were precipitated with 10% w/v TCA on ice at 4°C overnight. The precipitates were harvested by centrifugation (20 min, 8000 × g, 4°C) and washed with ice-cold acetone. The protein pellets were dried at room temperature and stored at −20°C until further use. Total protein concentration was determined with the Pierce BCA protein quantification assay (ThermoFisher Scientific). Protein pellets were suspended in 50 mM ammonium bicarbonate buffer (Fluka, Buchs, Switzerland). As reducing agent, 10 mM dithiothreitol (Duchefa Biochemie, Haarlem, the Netherlands) was used for 30 min, and as alkylating agent, 10 mM iodoacetamide (Sigma-Aldrich, St. Louis, USA) was used for 30 min in the dark. Further, overnight incubation with 80 ng of trypsin (Promega, Madison, USA) at 37°C was followed by the addition of 0.1% trifluoroacetic acid (TFA, Sigma-Aldrich, St. Louis, USA) to end the digestive reaction. The samples were desalted by Zip-Tip purification (Millipore, Billerica, USA) as previously described by Dreisbach et al. [Citation82]. The final eluates were concentrated using a vacuum centrifuge (Eppendorf, Hamburg, Germany) and stored at 4°C until further use.
Mass spectrometry and data analysis
Tryptic peptides were separated by reversed phase liquid chromatography (LC). LC-MS/MS analyses were performed using a nanoACQUITY UPLC system (Waters, Milford, MA) coupled to an LTQ XL Orbitrap mass spectrometer (Thermo Fisher Scientific,Waltham, MA) creating an electro spray ionization through a Picotip Emitter (SilicaTip, FS360-20-10 Coating P200P, New Objective). Specifically, the peptides were loaded onto a trap column (Symmetry C18, 5 μm, 180 μm inner diameter × 20 mm, Waters). Elution was performed onto an analytical column (BEH130 C18, 1.7 μm, 100 μm inner diameter × 100 mm, Waters) by a binary gradient of buffer A (0,1% (v/v) acetic acid) and B (0,1% (v/v) acetic acid in acetonitrile) over a period of 80 min with a flow rate of 400 nL min−1. A stepped gradient was applied.
For MS/MS analysis a full survey scan was recorded in the Orbitrap (m/z 300–2000) with a resolution of 30,000. The five most abundant precursor ions from each survey scan were consecutively isolated in the LTQ XL and fragmented via collision induced dissociation (CID). Precursors were dynamically excluded for 30 s and unassigned charge states as well as singly charged ions were rejected [Citation83]. Internal calibration was applied (lock mass 445.120025 m/z). Database searching was done with Sorcerer-SEQUEST 4 (Sage-N Research, Milpitas, USA). After extraction from the raw files, #.dta files were searched with Sequest against a concatenated target-pseudoreversed decoy database with a set of common laboratory contaminants. The target database was the Uniprot reference database (downloaded on September 2014) of B. thuringiensis (5815 protein hits), K. oxytoca (5663 protein hits), S. aureus t111 (2574 protein hits) and S. aureus t13595 (2625 protein hits). The #.out files were compiled with Scaffold 4 (version Scaffold_4.4.1, Proteome Software Inc., Portland, OR). Protein identification criteria were carried out as specified by Hempel et al., [Citation83]. but with the following XCorr values: ≥ 2.2, 3.3, 3.75 for doubly, triply and higher charged peptides. To allow protein abundance comparisons between samples, Scaffold MS/MS data were normalized by adjusting the sum of the selected quantitative values for all proteins in the list within each MS sample to a common value: the average of the sums of all MS samples present in the experiment. This was achieved by applying a scaling factor for each sample to each protein or protein group adjusting in this way the selected value to a normalized “Quantitative Value” [Citation84]. These values were exported from Scaffold and further curated in spreadsheets for analysis (Table S3). The MS raw data has been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD005596 [Username: [email protected] / Password: byH5iQGT]. Volcano plots used the normalized values for calculating the fold-changes in each protein among the monoculture samples versus the co-culture samples.
Bioinformatic analyses
Subcellular protein localization was predicted with TMHMM (version 2.0) [Citation85, Citation86], SignalP (version 4.1) [Citation87], LipoP (version 4.1) [Citation88], PSORTb (version 3.0) [Citation89], and ClusbSub-P (version 2.18.3) [Citation90]. Proteins with ambiguous predictions were manually curated by inspection of their individual sequences with InterProScan 5 (version 49.0) [Citation91]. For interpretation and visualization of biologically relevant protein functions, the gene ontology (GO) terms identified for the investigated exoproteomes were imported into the freely available online Revigo software (http://revigo.irb.hr/) [Citation92].
Biological and chemical safety
S. aureus and K. oxytoca are biosafety level 2 (BSL-2) microbiological agents and were accordingly handled following appropriate safety procedures. All experiments involving live bacteria and chemical manipulations of bacterial protein extracts were performed under appropriate containment conditions. All chemicals and reagents used in this study were handled according to the local guidelines for safe usage and protection of the environment.
Disclosure of potential conflicts of interest
The authors declare that they have no financial and non-financial competing interests in relation to the documented research.
Ethics
The present research has no particular ethical implications.
New_folder__3_.zip
Download Zip (819.8 KB)Acknowledgments
We thank Corinna Glasner, Andreas Otto, Tim Stobernack, Eleni Tsompanidou and Kai Zhou for helpful discussions.
Additional information
Funding
Related Research Data
References
- Kluytmans J, van Belkum A, Verbrugh H. Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin Microbiol Rev 1997;10:505–20.
- Heijer MD, den CD, van Bijnen MSc EM, PhD WJP, MD PMP, MD PHG, PhD PCAB, MD PFGS, PhD DEES, Team AS. Prevalence and resistance of commensal Staphylococcus aureus, including meticillin-resistant S aureus, in nine European countries: a cross-sectional study. Lancet Infect Dis 2013;13:409–15.
- DeLeo FR, Chambers HF. Reemergence of antibiotic-resistant Staphylococcus aureus in the genomics era. J Clin Invest 2009;119:2464–74.
- Lindsay JA, Holden MTG. Understanding the rise of the superbug: investigation of the evolution and genomic variation of Staphylococcus aureus. Funct Integr Genomics 2006;6:186–201.
- Bagnoli F, Bertholet S, Grandi G. Inferring Reasons for the Failure of Staphylococcus aureus Vaccines in Clinical Trials. Front Cell Infect Microbiol 2012;2:1–4.
- Proctor RA. Is there a future for a Staphylococcus aureus vaccine? Vaccine 2012;30:2921–7.
- Grundmann H, Schouls LM, Aanensen DM, Pluister GN, Tami A, Chlebowicz M, Glasner C, Sabat AJ, Weist K, Heuer O, et al. The dynamic changes of dominant clones of Staphylococcus aureus causing bloodstream infections in the European region: Results of a second structured survey. Eurosurveillance 2014;19:1–10.
- Morrison MA, Nadle J, Petit S, Greshman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, et al. Invasive Methicillin-Resistant Staphylococcus aureus infections in the United States. JAMA-J Am Med Assoc 2007;298:1763–71.
- van der Kooi-Pol MM, Duipmans JC, Jonkman MF, van Dijl JM. Host–pathogen interactions in epidermolysis bullosa patients colonized with Staphylococcus aureus. Int J Med Microbiol 2014;304:195–203.
- van der Kooi-Pol MM, de Vogel CP, Westerhout-Pluister GN, Veenstra-Kyuchukova YK, Duipmans JC, Glasner C, Buist G, Elsinga GS, Westra H, Bonarius HPJ, et al. High anti-staphylococcal antibody titers in patients with Epidermolysis Bullosa telate to long-term colonization with alternating types of Staphylococcus aureus. J Invest Dermatol 2013;133:847–50.
- Goerke C, Gressinger M, Endler K, Breitkopf C, Wardecki K, Stern M, Wolz C, Kahl BC. High phenotypic diversity in infecting but not in colonizing Staphylococcus aureus populations. Environ Microbiol 2007;9:3134–42.
- Mongkolrattanothai K, Gray BM, Mankin P, Stanfill AB, Pearl RH, Wallace LJ, Vegunta RK. Simultaneous carriage of multiple genotypes of Staphylococcus aureus in children. Journal of Medical Microbiology 2011;60:317–22.
- van der Kooi-Pol MM, Sadaghian Sadabad M, Duipmans JC, Sabat AJ, Stobernack T, Omansen TF, Westerhout-Pluister GN, Jonkman MF, Harmsen HJM, van Dijl JM. Topography of distinct Staphylococcus aureus types in chronic wounds of patients with epidermolysis bullosa. PLoS ONE 2013;8:e67272–6.
- Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, Nouwen JL. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis 2005;5:751–62.
- Kooi-Pol MM, Veenstra-Kyuchukova YK, Duipmans JC, Pluister GN, Schouls LM, Neeling AJ, Grundmann H, Jonkman MF, Dijl JM. High genetic diversity of Staphylococcus aureus strains colonizing patients with epidermolysis bullosa. Exp Dermatol 2012;21:463–6.
- Brandling-Bennett HA, Morel KD. Common wound colonizers in patients with epidermolysis bullosa. Pediatr Dermatol 2010;27:25–8.
- Sibbald MJJB, Ziebandt AK, Engelmann S, Hecker M, de Jong A, Harmsen HJM, Raangs GC, Stokroos I, Arends JP, Dubois JYF, et al. Mapping the pathways to staphylococcal pathogenesis by comparative secretomics. Microbiology and Molecular Biology Reviews 2006;70:755–88.
- Bravo A, Likitvivatanavong S, Gill SS, Soberón M. Bacillus thuringiensis: A story of a successful bioinsecticide. Insect Biochemistry and Molecular Biology 2011;41:423–31.
- Gillis A, Mahillon J. Phages preying on Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis: past, present and future. Viruses 2014;6:2623–72.
- Gilbert RJ, Stringer MF, Peace TC. The survival and growth of Bacillus cereus in boiled and fried rice in relation to outbreaks of food poisoning. J Hyg (Lond) 1974;73:433–44.
- Jensen GB, Hansen BM, Eilenberg J, Mahillon J. The hidden lifestyles of Bacillus cereus and relatives. Environ Microbiol 2003;5:631–40.
- Ghelardi E, Celandroni F, Salvetti S, Fiscarelli E, Senesi S. Bacillus thuringiensis pulmonary infection: critical role for bacterial membrane-damaging toxins and host neutrophils. Microbes Infect 2007;9:591–8.
- Hernandez E, Ramisse F, Gros P, Cavallo J-D. Super-infection by Bacillus thuringiensis H34 or 3a3b can lead to death in mice infected with the influenza A virus. FEMS Immunol Med Mic 2000;29(3):177–81.
- Podschun R, Ullmann U. Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity. Clin Microbiol Rev 1998;11:589.
- Yinnon AM, Butnaru A, Raveh D, Jerassy Z, Rudensky R. Klebsiella bacteraemia: community versus nosocomial infection. Q J Med 1996;89:993–941.
- Bodey GP, Elting LS, Rodríguez S, Hernández M. Klebsiella bacteremia. Cancer 1989;64:2368–76.
- Ronald A. The etiology of urinary tract infection: Traditional and emerging pathogens. Dis Mon 2003;49:71–82.
- Vergnano S, Sharland M, Kazembe P, Mwansambo C, Heath PT. Neonatal sepsis: an international perspective. Arch Dis Child-Fetal 2005;90:F220–4.
- Gonzalez DJ, Haste NM, Hollands A, Fleming TC, Hamby M, Pogliano K, Nizet V, Dorrestein PC. Microbial competition between Bacillus subtilis and Staphylococcus aureus monitored by imaging mass spectrometry. Microbiology 2011;157:2485–92.
- Mäder U, Nicolas P, Depke M, Pané-Farré J, Debarbouille M, van der Kooi-Pol MM, Guérin C, Dérozier S, Hiron A, Jarmer H, et al. Staphylococcus aureus Transcriptome Architecture: From Laboratory to Infection-Mimicking Conditions. PLoS Genet 2016;12:e1005962–32.
- Lycklama a, Nijeholt JA, Driessen AJM. The bacterial Sec-translocase: structure and mechanism. Philosophical Transactions of the Royal Society B: Biological Sciences 2012;367:1016–28.
- Sibbald MJJB, Winter T, van der Kooi-Pol MM, Buist G, Tsompanidou E, Bosma T, Schafer T, Ohlsen K, Hecker M, Antelmann H, et al. Synthetic Effects of secG and secY2 Mutations on Exoproteome Biogenesis in Staphylococcus aureus. J Bacteriol 2010;192:3788–800.
- Ziebandt A-K, Kusch H, Degner M, Jaglitz S, Sibbald MJJB, Arends JP, Chlebowicz MA, Albrecht D, Pantuček R, Doškar J, et al. Proteomics uncovers extreme heterogeneity in the Staphylococcus aureus exoproteome due to genomic plasticity and variant gene regulation. Proteomics 2010;10:1634–44.
- Pocsfalvi G, Cacace G, Cuccurullo M, Serluca G, Sorrentino A, Schlosser G, Blaiotta G, Malorni A. Proteomic analysis of exoproteins expressed by enterotoxigenic Staphylococcus aureus strains. Proteomics 2008;8:2462–76.
- Burlak C, Hammer CH, Robinson M-A, Whitney AR, McGavin MJ, Kreiswirth BN, DeLeo FR. Global analysis of community-associated methicillin-resistant Staphylococcus aureus exoproteins reveals molecules produced in vitro and during infection. Cell Microbiol 2007;9:1172–90.
- Wang W, Jeffery CJ. An analysis of surface proteomics results reveals novel candidates for intracellular/surface moonlighting proteins in bacteria. Molecular BioSystems 2016;12:1420–31.
- Dreisbach A, van Dijl JM, Buist G. The cell surface proteome of Staphylococcus aureus. Proteomics 2011;11:3154–68.
- Otto A, van Dijl JM, Hecker M, Becher D. The Staphylococcus aureus proteome. Int J Med Microbiol 2014;304:110–20.
- Escutia MR, Val G, Palacín A, Geukens N, Anné J, Mellado RP. Compensatory effect of the minor Streptomyces lividans type I signal peptidases on the SipY major signal peptidase deficiency as determined by extracellular proteome analysis. Proteomics 2006;6:4137–46.
- Hansmeier N, Chao T-C, Kalinowski J, Pühler A, Tauch A. Mapping and comprehensive analysis of the extracellular and cell surface proteome of the human pathogen Corynebacterium diphtheriae. Proteomics 2006;6:2465–76.
- Voigt B, Antelmann H, Albrecht D, Ehrenreich A, Maurer K-H, Evers S, Gottschalk G, van Dijl JM, Schweder T, Hecker M. Cell physiology and protein secretion of Bacillus licheniformis compared to Bacillus subtilis. J Mol Microbiol Biotechnol 2009;16:53–68.
- Tjalsma H, Antelmann H, Jongbloed JDH, Braun PG, Darmon E, Dorenbos R, Dubois J-YF, Westers H, Zanen G, Quax WJ, et al. Proteomics of protein secretion by bacillus subtilis: separating the “secrets” of the secretome. Microbiology and Molecular Biology Reviews 2004;68:207–33.
- Stobernack T, Glasner C, Junker S, Gabarrini G, de Smit M, de Jong A, Otto A, Becher D, van Winkelhoff AJ, van Dijl JM. Extracellular Proteome and Citrullinome of the Oral Pathogen Porphyromonas gingivalis. J Proteome Res 2016;15:4532–43.
- Krishnappa L, Dreisbach A, Otto A, Goosens VJ, Cranenburgh RM, Harwood CR, Becher D, van Dijl JM. Extracytoplasmic proteases determining the cleavage and release of secreted proteins, lipoproteins, and membrane proteins in Bacillus subtilis. J Proteome Res 2013;12:4101–10.
- Bendtsen JD, Kiemer L, Fausbøll A, Brunak S. Non-classical protein secretion in bacteria. BMC Microbiol 2005;5:58–13.
- Henderson B, Martin A. Bacterial moonlighting proteins and bacterial virulence. Current Topics in Microbiology and Immunology 2013;358:155–213.
- Götz F, Yu W, Dube L, Prax M, Ebner P. Excretion of cytosolic proteins (ECP) in bacteria. Int J Med Microbiol 2015;305:230–7.
- Ebner P, Rinker J, Götz F. Excretion of cytoplasmic proteins in Staphylococcus is most likely not due to cell lysis. Current Genetics 2016;62:19–23.
- Wang G, Xia Y, Song X, Ai L. Common non-classically secreted bacterial proteins with experimental evidence. Curr Microbiol 2015;72:102–11.
- Ebner P, Rinker J, Nguyen MT, Popella P, Nega M, Luqman A, Schittek B, Di Marco M, Stevanovic S, Götz F. Excreted cytoplasmic proteins contribute to pathogenicity in Staphylococcus aureus. Infect Immun 2016;84:1672–81.
- Antelmann H, Tjalsma H, Voigt B, Ohlmeier S, Bron S, van Dijl JM, Hecker M. A proteomic view on genome-based signal peptide predictions. Genome Research 2001;11:1484–502.
- Westers H, Dorenbos R, Dijl JM, Kabel J, Flanagan T, Devine KM, Jude F, Séror SJ, Beekman AC, Darmon E, et al. Genome engineering reveals large dispensable regions in Bacillus subtilis. Molecular Biology and Evolution 2003;20:2076–90.
- Lottenberg R, Broder CC, Boyle MDP, Kain SJ, Schroeder BL, Curtiss R III. Cloning, sequence analysis, and expression in Escherichia coli of a streptococcal plasmin receptor. J Bacteriol 1992;174:5204–310.
- Pancholi V, Fischetti VA. A Major Surface Protein on Group A streptococci is a glyceraldehyde-3-phosphate-dehydrogenase with multiple binding activity. J Exp Med 2015;176:415–26.
- Kenny B, Finlay BB. Protein secretion by enteropathogenic Escherichia coli is essential. PNAS 1995;92:7991–5.
- Bergmann S, Wild D, Diekmann O, Frank R, Bracht D, Chhatwal GS, Hammerschmidt S. Identification of a novel plasmin(ogen)-binding motif in surface displayed α-enolase of Streptococcus pneumoniae. Mol Microbiol 2003;49:411–23.
- Hara H, Ohta H, Inoue T. Cell Surface-Associated Enolase in Actinobacillus actinomycetemcomitans. Microbio Immunol 2000;44:349–56.
- Shams F, Oldfield NJ, Wooldridge KG, Turner DPJ. Fructose-1,6-bisphosphate aldolase (FBA)–a conserved glycolytic enzyme with virulence functions in bacteria: “ill met by moonlight”. Biochm Soc Trans 2014;42:1792–5.
- Dallo SF, Kannan TR, Blaylock MW, Baseman JB. Elongation factor Tu and E1 beta subunit of pyruvate dehydrogenase complex act as fibronectin binding proteins in Mycoplasma pneumoniae. Mol Microbiol 2002;46:1041–51.
- Granato D, Bergonzelli GE, Pridmore RD, Marvin L, Rouvet M, Corthésy-Theulaz IE. Cell surface-associated elongation factor Tu mediates the attachment of Lactobacillus johnsonii NCC533 (La1) to human intestinal cells and mucins. Infect Immun 2004;72:2160–9.
- Liu H, Cheng Z, Song W, Wu W, Zhou Z. Immunoproteomic to analysis the pathogenicity factors in leukopenia caused by Klebsiella pneumonia bacteremia. PLoS ONE 2014;9:e110011–5.
- Pancholi V, Chhatwal GS. Housekeeping enzymes as virulence factors for pathogens. Int J Med Microbiol 2003;293:391–401.
- Mekonnen SA, Medina LMP, Glasner C, Tsompanidou E, de Jong A, Grasso S, Schaffer M, Mäder U, Larsen AR, Gumpert H, et al. Signatures of cytoplasmic proteins in the exoproteome distinguish community- and hospital-associated methicillin-resistant Staphylococcus aureus USA300 lineages. Virulence 2017;0:1–17.
- Christie-Oleza JA, Scanlan DJ, Armengaud J. “You produce while I clean up,” a strategy revealed by exoproteomics during Synechococcus-Roseobacter interactions. Proteomics 2015;15:3454–62.
- Amissah NA, Glasner C, Ablordey A, Tetteh CS, Kotey NK, Prah I, van der Werf TS, Rossen JW, van Dijl JM, Stienstra Y. Genetic diversity of Staphylococcus aureus in buruli ulcer. PLoS Negl Trop Dis 2015;9:e0003421–17.
- Prunier AL, Leclercq R. Role of mutS and mutL genes in hypermutability and recombination in Staphylococcus aureus. J Bacteriol 2005;187:3455–64.
- Wang S, Wu C, Shen J, Wu Y, Wang Y. Hypermutable Staphylococcus aureus strains present at high frequency in subclinical bovine mastitis isolates are associated with the development of antibiotic resistance. Veterinary Microbiology 2013;165:410–5.
- Canfield GS, Schwingel JM, Foley MH, Vore KL, Boonanantanasarn K, Gill AL, Sutton MD, Gill SR. Evolution in Fast Forward: a Potential Role for Mutators in Accelerating Staphylococcus aureus Pathoadaptation. J Bacteriol 2013;195:615–28.
- Schaaff F, Reipert A, Bierbaum G. An Elevated Mutation Frequency Favors Development of Vancomycin Resistance in Staphylococcus aureus. Antimicrobial Agents and Chemotherapy 2002;46:3540–8.
- O'Neill AJ, Chopra I, Bierbaum G, Reipert A, Schaaff F. Lack of Evidence for Involvement of Hypermutability in Emergence of Vancomycin-Intermediate Staphylococcus aureus. Antimicrobial Agents and Chemotherapy 2003;47:1484–5.
- Trong HN, Prunier AL, Leclercq R. Hypermutable and Fluoroquinolone-Resistant Clinical Isolates of Staphylococcus aureus. Antimicrobial Agents and Chemotherapy 2005;49:2098–101.
- Hamilton WD. The genetical evolution of social behaviour. I. J Theoret Biol 1964;7:1–16.
- West SA, Diggle SP, Buckling A, Gardner A, Griffin AS. The Social Lives of Microbes. Annu Rev Ecol Evol Syst 2007;38:53–77.
- Kruse CR, Nuutila K, Lee CCY, Kiwanuka E, Singh M, Caterson EJ, Eriksson E, Sørensen JA. The external microenvironment of healing skin wounds. Wound Rep and Reg 2015;23:456–64.
- Veloo ACM, Elgersma PE, Friedrich AW, Nagy E, van Winkelhoff AJ. The influence of incubation time, sample preparation and exposure to oxygen on the quality of the MALDI-TOF MS spectrum of anaerobic bacteria. Clinical Microbiology and Infection 2014;20:O1091–7.
- Leopold SR, Goering RV, Witten A, Harmsen D, Mellmann A. Bacterial whole-genome sequencing revisited: portable, scalable, and standardized analysis for typing and detection of virulence and antibiotic resistance genes. J Clin Microbiol 2014;52:2365–70.
- Harmsen D, Claus H, Witte W, Rothganger J, Claus H, Turnwald D, Vogel U. Typing of Methicillin-Resistant Staphylococcus aureus in a University Hospital Setting by Using Novel Software for spa Repeat Determination and Database Management. J Clin Microbiol 2003;41:5442–8.
- Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Research 2013;42:D206–14.
- Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 2008;9:75–15.
- Carver TJ, Rutherford KM, Berriman M, Rajandream M-A, Barrell BG, Parkhill J. ACT: the Artemis Comparison Tool. Bioinformatics 2005;21:3422–3.
- Rutherford K, Parkhill J, Crook J, Hornsnell T, Rice P, Rajandream M-A, Barrell B. Artemis: sequence visualization and annotation. Bioinformatics 2000;16:944–5.
- Dreisbach A, Hempel K, Buist G, Hecker M, Becher D, van Dijl JM. Profiling the surfacome of Staphylococcus aureus. Proteomics 2010;10:3082–96.
- Hempel K, Pané-Farré J, Otto A, Sievers S, Hecker M, Becher D. Quantitative cell surface proteome profiling for SigB-dependent protein expression in the human pathogen Staphylococcus aureus via biotinylation approach. J Proteome Res 2010;9:1579–90.
- Proteome Software, Inc. Scaffold Version 4.0 User's Manual [Internet]. 2014. Available from: http://www.proteomesoftware.com/
- Krogh A, Larsson B, Heijne von G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. Journal of Molecular Biology 2001;305:567–80.
- Sonnhammer EL, Heijne von G, Krogh A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc Int Conf Intell Syst Mol Biol 1998;6:175–82.
- Petersen TN, Brunak S, Heijne von G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 2011;8:785–6.
- Juncker AS, Willenbrock H, Heijne von G, Brunak S, Nielsen H, Krogh A. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci 2003;12:1652–62.
- Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Sahinalp SC, Ester M, Foster LJ, et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 2010;26:1608–15.
- Paramasivam N, Linke D. ClubSub-P: Cluster-Based Subcellular Localization Prediction for Gram-Negative Bacteria and Archaea. Front Microbiol 2011;2:218.
- Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics 2014;30:1236–40.
- Supek F, Bošnjak M, Škunca N, Šmuc T. REVIGO Summarizes and Visualizes Long Lists of Gene Ontology Terms. PLoS ONE 2011;6:e21800–9.