
ABSTRACT
Hypervirulent K. pneumoniae variants (hvKP) have been increasingly reported worldwide, causing metastasis of severe infections such as liver abscesses and bacteremia. The capsular serotype K2 hvKP strains show diverse multi-locus sequence types (MLSTs), but with limited genetics and virulence information. In this study, we report a hypermucoviscous K. pneumoniae strain, RJF293, isolated from a human bloodstream sample in a Chinese hospital. It caused a metastatic infection and fatal septic shock in a critical patient. The microbiological features and genetic background were investigated with multiple approaches. The Strain RJF293 was determined to be multilocis sequence type (ST) 374 and serotype K2, displayed a median lethal dose (LD50) of 1.5 × 102 CFU in BALB/c mice and was as virulent as the ST23 K1 serotype hvKP strain NTUH-K2044 in a mouse lethality assay. Whole genome sequencing revealed that the RJF293 genome codes for 32 putative virulence factors and exhibits a unique presence/absence pattern in comparison to the other 105 completely sequenced K. pneumoniae genomes. Whole genome SNP-based phylogenetic analysis revealed that strain RJF293 formed a single clade, distant from those containing either ST66 or ST86 hvKP. Compared to the other sequenced hvKP chromosomes, RJF293 contains several strain-variable regions, including one prophage, one ICEKp1 family integrative and conjugative element and six large genomic islands. The sequencing of the first complete genome of an ST374 K2 hvKP clinical strain should reinforce our understanding of the epidemiology and virulence mechanisms of this bloodstream infection-causing hvKP with clinical significance.
Introduction
Klebsiella pneumoniae is a major opportunistic pathogen, primarily affecting severe and immunocompromised patients in intensive care units (ICUs), and causing various types of community and hospital-acquired infections [Citation1]. Hospital outbreaks of K. pneumoniae infection are often associated with multidrug resistance in the causative strains, in particular for sequence type 11 (ST11) and ST258 isolates determined by multi-locus sequence typing (MLST) [Citation2]. However, during the past three decades, hypervirulent variants of Klebsiella pneumoniae (hvKP) have been increasingly reported in the Asian Pacific Rim and Western countries, which cause severe infec-tions such as liver abscess, endophthalmitis, meningitis, osteomyelitis, and necrotizing fasciitis in healthy and ambulatory individuals [Citation3]. Recently, an epidemiological study showed that hvKP was identified in 31% of patients with K. pneumoniae bacteremia in Beijing Chao-Yang Hospital, China [Citation4].
In general, a positive string test (with the formation of a 5 mm mucoid string or greater) is employed to quickly examine ropy exopolysaccharide produced by K. pneumoniae, which is defined as hypermucoviscosity and used to facilitate distinction of hvKP from the classic K. pneumoniae (cKP) [Citation3]. However, new evidence has suggested that hypervirulence and hypermucoviscosity are two complementary but distinct phenotypes of K. pneumoniae [Citation5]. Notably, K. pneumoniae isolates negative by string test are also capable of producing an invasive infection. The string test is thus insufficient in determining the hypervirulence of a K. pneumoniae strain, and more hypervirulence-associated genes have to be explored [Citation5].
Isolates with serotypes K1 and K2 have been found to be the most common in hvKPs. Over 80% of hvKPs causing invasive pyogenic liver abscesses in Taiwan, South Korea, and mainland China were characterized to be of these two serotypes [Citation1,Citation6]. Serotype K1 has been determined to be closely associated with ST23 by MLST. In contrast, the K. pneumoniae strains with serotype K2 exhibit diverse MLST types (including ST65, ST66, ST86, ST374, ST375, ST380 and etc.), among which ST65 and ST86 are predominant and with higher virulence than other MLST types [Citation6,Citation7]. It thus requires more genetic parsing to get a deep understanding of the virulence of K2 K. pneumoniae compared to K1. To our knowledge, no complete genome sequences for ST374 and serotype K2 isolates are publicly available. Herein, we carried out a retrospective analysis of hvKP isolates collected over 19 months at a hospital setting in China. A hypermucoviscous clinical strain, RJF293, causing bacteremia metastatic infection was characterized as the sequence type ST374 and capsular serotype K2. The microbiological features and genome information (including the presence/absence pattern of virulence genes, mobile genetic elements, etc.) of this strain were characterized with multiple approaches, making up a supplement to the existing knowledge on K2 hvKP.
Materials and methods
Ethics and consent
This study was observational, using human bacteria samples for the ex vivo experiments and sampling patients' blood as part of routine work in hospital treatment, therefore verbal informed consent was obtained from all volunteers. The bacteria samples and data sheets were anonymized. This study protocol including the verbally informed consent procedure was approved by the ethics committee of the Ruijin Hospital affiliated with Shanghai Jiao Tong University, Shanghai, China (reference number: 201329). Protocols of mouse experiments have been approved by the same ethics committee.
Bacterial strains, plasmids, and growth media
During the study period of September 2014 to March 2016, K. pneumoniae isolates were collected from patients' samples at the Ruijin Hospital, Shanghai, China. K. pneumoniae identification was based on biochemical profiling by the VITEK2 compact system and 16S rRNA gene sequencing. The strains were cultivated in LB broth or on LB agar supplemented with the appropriate antibiotics. The detailed information of strains used in this study is listed in Supplementary Table S1.
String test for the hypermucoviscosity phenotype
The string test to determine the hypermucoviscosity phenotype was performed by touching a colony grown overnight on a blood agar plate at 37°C with a loop and pulling up. Strains exhibiting a mucoid string with length of 5 mm or longer were defined as hypermucoviscous [Citation3].
PFGE, MLST, K-typing and detection of known virulence genes
The Pulsed-field Gel Electrophoresis (PFGE) protocol used was based on the PulseNet 1-day standardized PFGE protocol for Escherichia coli O157:H7, Salmonella, and Shigella [Citation8]. PFGE patterns were interpreted by using the criteria proposed by Tenover et al [Citation9]. A Salmonella serotype Braenderup strain (H9812) was used to normalize migration variation occurring across the gel and to determine sample band sizes accurately [Citation10].
MLST was also performed by amplifying and sequencing seven K. pneumoniae housekeeping genes, including gapA, infB, mdh, pgi, phoE, rpoB, and tonB, according to the protocol provided on the Pasteur MLST website [Citation11]. Serotype-specific genes for the K1 and K2 capsular serotypes were also amplified by PCR amplification of the cps gene cluster at the wzy and wzx loci as described previously [Citation12]. The presence of ten known K. pneumoniae virulence genes, including magA, rmpA, allS, mrkD, kfuBC, cf29a, fimH, uge, wab and ureA, was assessed by PCR [Citation13]. The PCR primers used are listed in Supplementary Table S2.
Antimicrobial susceptibility testing
The VITEK2 compact system (bioMérieux, Marcy l'Etoile, France) was used for antimicrobial susceptibility testing. Results were interpreted according to the interpretive standards of the Clinical and Laboratory Standards Institute [Citation14]. Carbapenem resistance was defined as resistance to imipenem using an MIC breakpoint of ≥ 4 μg/ml or ertapenem using an MIC breakpoint of ≥ 2 μg/ml following the revision of MIC breakpoints in 2014 [Citation15].
Mouse lethality assay
Evaluation of K. pneumoniae RJF293 was performed with the BALB/c mouse, as described previously [Citation16]. Five-week-old female BALB/c mice were used for the virulence assessment in the present study. Briefly, the overnight culture of the K. pneumoniae strains to be tested was inoculated into fresh LB broth at 1:100 and grown for 3 hours in a 37°C shaking incubator. Then the cells were harvested, washed once and resuspended in saline (0.9%) to OD600 of 1.0 (approximately 109 CFU/ml). Serial dilution was done to get the appropriate concentration, which was 102 CFU/ml, 103 CFU/ml, 104 CFU/ml and 105 CFU/ml for the determination of median lethal dosage (LD50), and 104 CFU /ml and 105 CFU/ml for the survival assay. One hundred microliters of the bacterial suspension was injected into the mice intraperitoneally. All mice were monitored daily for survival. For the survival analysis, 10 mice were involved in each group, with 5 per group used for the LD50 determination. The ST11 cKP strain HS11286 and the ST23 hvKP strain NTUH-K2044 were used for controls [Citation17,Citation18]. Kaplan-Meier survival estimation was performed with the R package ‘survival’.
DNA preparation, genome sequencing and annotation
The RJF293 isolate was grown overnight at 37°C to stationary phase in LB medium, and total DNA was isolated from harvested cells. The genomic DNA was extracted by using the CTAB/phenol-chloroform method that has been described previously with slight modification [Citation19]. The overnight culture of K. pneumoniae RJF293 was inoculated into 50 ml fresh LB broth at a ratio of 1:100. Bacteria were harvested by centrifuging when the OD600 of the subculture reached 0.8. The pellet was resuspended in 4 ml TE buffer by both vortexing and pipetting. The suspension was supplemented with 300 μl lysozyme (50 mg/ml) and placed into a 37°C water bath for 1 hour. Then SDS and proteinase K were added to a final concentration of 1% (m/v) and 0.5 mg/ml, respectively. The mixture was incubated at 65°C until completely lysed. One milliliter of NaCl solution (5 mol/L) and 1 ml CTAB/NaCl were added in order. The mixture was incubated at 65°C for 15 min, and then 7 ml phenol: chloroform was added (1:1). The tube was placed on a revolver for at least 1 hour. The supernatant containing DNA was separated by spinning. The application of phenol: chloroform was repeated once to eliminate protein as much as possible. Then, the DNA was precipitated with 0.6 volumes isopropanol and washed with 70% ethanol twice. The quality of the purified genomic DNA was assessed by both electrophoresis and NanoDrop™ 2000 fluorospectrometer (Thermo Fisher Scientific, MA, USA). The sequencing library was prepared using the Illumina Nextera XT DNA Library Prep kit as per the user instructions.
The genome sequence was first determined by using IlluminaMiseq short read sequencing (2 × 251 paired-end sequencing for a 400-bp library) and assembled with the Newbler Assembler, resulting in sequences with 124-fold coverage and 99 assembled contigs. Then, with the PacBio RSII sequencing platform, a 4-kb template was prepared, and the library was sequenced on one SMRT cell, yielding 96,326 direct reads with an average length of ∼4.7 kb. These long reads were assembled by using the PacBio HGAP3 workflow. Finally, the assembled contigs derived from the Miseq short reads were scaffolded with long PacBio reads, and then the resultant scaffolds were gap-filled with PBJelly [Citation20]. The genome assembly was improved by Pilon [Citation21]. Besides, the chromosomal replication origin (oriC) was predicted by OriFinder [Citation22]. The circular chromosome was subsequently confirmed by using PCR amplification and DNA sequencing of the upstream and downstream junctions of the oriC site.
The RJF293 genome sequence was annotated by the NCBI Prokaryote Genome Annotation Pipeline (PGAP) version 2.0 [Citation23]. Putative virulence factors were predicted by VRprofile with the BLASTp-based Ha-value >0.64 [Citation24], which collected 2454 virulence factors from the VFDB database [Citation25]. Unusual chromosomal regions with putative foreign origins were identified by VRprofile with default parameters, including prophage, integrative and conjugative element (ICE) and genomic islands [Citation25]. The type IV and VI secretion systems were also predicted by VRprofile. CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) arrays were identified by PILERCR [Citation26].
Genomic comparative analysis
Genome-wide single nucleotide polymorphism (SNP) calling and phylogenetic analysis was performed by using kSNP v3 [Citation27-29]. The 106 completely sequenced K. pneumoniae genomes, including RJF293 sequenced in this study, were downloaded from NCBI Genome (Supplementary Table S3). The phylogeny scheme was generated from the 429,267 kSNP3-detected SNP sites for all the chromosome sequences with k = 21, as determined by Kchooser [Citation27]. A parsimony tree was generated by kSNP3 based on an extended majority rule consensus of the equally most parsimonious trees from a sample of 100 trees [Citation27]. The tree was displayed with iTOL with midpoint rooting [Citation30].
Genome sequence comparisons between RJF293 and the other seven completely sequenced hvKP strains (Supplementary Table S4) were performed by mGenomeSubtractor [Citation31]. All the annotated RJF293 protein-coding genes (served as the query) were examined by mGenomeSubtractor-facilitated BLASTn searches, using an H-value cut-off for conserved genes, against the other hvKP genomes (served as subject). The H-value (0 ≤ H-value ≤ 1.0) reflects the degree of similarity in terms of the length of match and the degree of identity at the nucleotide level between the matching gene in the subject genome and the query gene examined [Citation31]. Klebsiella pneumoniae strains were defined as hvKP if they exhibited the hypermucoviscous phenotype and caused severe and metastatic infection [Citation3].
Detection of ICE and prophage excision
The excision of prophage or ICE from the chromosome was detected using PCR assays. Primers were picked upstream of the attL (P1) and downstream of the attR (P4) sites, with distance of 100–1000 bp (Supplementary Table S2). No amplicon can be obtained with this pair of primers if the ICE or prophage remains integrated in the chromosome, as the size of ICE or prophage is outside the range of DNA polymerase capability. The attB site after excision of the ICE or prophage is normally smaller than 100bp in size. When the ICE or prophage identified can excise from the chromosome, an amplicon with the expected size can be observed using PCR amplification (Supplementary Figure S1).
Nucleotide sequence accession numbers
The genome sequence of Klebsiella pneumoniae RJF293 has been submitted to GenBank under accession numbers CP014008 (RJF293 chromosome) and CP014009 (plasmid pRJF293).
Results
Collection and clinical characteristics of hypermucoviscous K. pneumoniae isolates
From September 2014 to March 2016, a total of 872 K. pneumoniae episodes were collected by the Clinical Microbiology Laboratory of the Ruijin Hospital. A total of 128 K. pneumoniae isolates (128/872, 14.67%) exhibited the hypermucoviscous phenotype (positive in string test), and the highest prevalence of 44.1% was detected in pus samples. The mean age of the patients was 62.2 ± 14.5 years. Ninety-one patients (91/128, 71.09%) were males, and 37 (37/128, 28.90%) were females. These clinical specimens were collected from respiratory samples (90/128, 70.3%), abscess samples (15/128, 11.7%), blood samples (5/128, 3.9%), abdominal drainage samples (5/128, 3.9%), urine samples (4/128, 3.1%), secretion samples (4/128, 3.1%), bile samples (3/128, 1.6%) and pleural effusion samples (2/128, 1.6%). The hypermucoviscous isolates occurred commonly in patients with diabetes mellitus (28.9%), biliary tract disease (27.3%), or cancer/immunosuppression (44.5%), and 27.3% of patients had a history of abdominal surgery. Of the hypermucoviscous K. pneumoniae infections, 25% led to sepsis, and 7% of patients developed septic shock and died during hospitalization.
Microbiological characteristics of K. pneumoniae isolates with ultra-long mucoid string
During the string test, 20 hypermucoviscous K. pneumoniae isolates exhibited an ultra-long viscous string (> 20 mm; RJF293 as representative in the Supplementary Figure S2). They included 4 isolates from blood samples, 9 from the respiratory tract, 4 from abscesses, 1 from abdominal drainage, and 2 from other samples (). PFGE results showed that these 20 isolates had 18 distinct patterns (Supplementary Figure S3A). For capsular serotypes, the K1 (n = 7) and K2 (n = 6) serotypes were predominant (). There were a total of 9 MLST types in these 20 isolates, including two new types (). The K2 serotype isolates belonged to three different sequence types, ST374, ST86 and ST375, whereas, 71.4% of K1 serotype isolates belonged to ST23.
Table 1. The capsular serotype, MLST type and distribution of ten virulence factor genes in the 20 hypermucoviscous K. pneumoniae isolates with ultra-long viscous string (>20 mm).
In order to determine whether the above-identified clones differ by their virulence potential, the presence of 10 reported genetic factors implicated in K. pneumoniae virulence was examined by PCR (). All the 20 hypermucoviscous K. pneumoniae isolates harbored the mrkD, fimH, uge, wabG and ureA genes. rmpA, the gene responsible for the hypermucoviscous phenotype was absent in one isolate. Nearly three-fourths of these isolates carried more than 7 virulence genes.
In addition, antimicrobial susceptibility testing showed that all 20 hypermucoviscous K. pneumoniae isolates were resistance to ampicillin but susceptible to the majority of the other antimicrobial agents tested, including cephalosporins, β-lactam/β-lactamase inhibitor and carbapenems (Supplementary Table S5). Namely, the percentage of strains susceptible to ampicillin/sulbactam, cefazolin, ceftazidime, ciprofloxacin, amikacin, and imipenem was 90%, 90%, 95%, 95%, 100% and 100%, respectively.
Clinical characteristics of hvKP isolate RJF293 with serotype K2 and ST374
Our attention was drawn to a fatal septic shock patient with a bloodstream infection caused by a hypermucoviscous K. pneumoniae strain. The patient was a 64-year-old male who was admitted to surgery at the Ruijin Hospital due to gastric stump cancer on March 2014. He received total gastrectomy, complicated with postoperative bleeding, and then underwent hemostatic laparotomy the next day. With high temperature and breathing difficulty, the patient developed septic shock due to the abdominal infection and was subsequently transferred to the ICU 20 days later and received abdominal drainage. Cefuroxime, meropenem and ceftazidime were administrated to this patient for anti-infection treatment, but unfortunately the infection persisted and this patient died 5 months after entrance into the ICU.
Routine microbiological cultures of sputum, drainage and blood samples were collected from this patient. Four K. pneumoniae isolates were identified, including RJA726 (not included in the previous 20 hypermucoviscous strains due to its isolation period) isolated from the abdominal drainage in April, RJF293 and RJF294 from the blood in September, and RJA898 from the sputum in September. All four K. pneumoniae isolates were characterized as hypermucoviscous by string test (RJF293 as example, Supplementary Figure S2). These 4 isolates had the same PFGE pattern, capsular serotype (K2), MLST type (ST374) and antimicrobial susceptibility testing results (Supplementary FigureS3B, Table1 and Supplementary Table S5). This evidence suggested toward the metastasis of the same hypermucoviscous K. pneumoniae strain in this patient. RJF293 collected from the bloodstream was selected as the representative for further virulence assays, genome sequencing, and comparative analysis.
Mouse lethality assay exhibited hypervirulence of RJF293
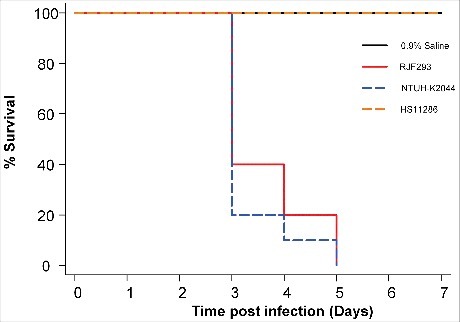
The susceptibility of BALB/c mice to the K. pneumoniae RJF293 strain was examined to study its pathogenicity. Five-week-old mice in healthy condition were infected with a serial dilution of bacteria, and the survival was recorded every 24 hours, within 7 days post-infection (). RJF293 showed an LD50 of 1.5 × 102 CFU in the BALB/c mice (calculated by Reed-Muench method). For the survival analysis, 103 CFU of different strains as well as only saline were injected into the mice intraperitoneally. The Kaplan-Meier survival estimate revealed that RJF293 showed virulence similar to that of hvKP strain NTUH-K2044 (p = 0.461, by log-rank test). The ST11 cKP strain HS11286 did not cause mortality within the observation duration. These results showed that the ST374 serotype K2 strain RJF293 was equivalently virulent to the well-documented ST23 serotype K1 hvKP strain NTUH-K2044 in the mouse infection model.
Figure 1. Kaplan-Meier survival curves for K. pneumoniae RJF293 infected mice. Mice were infected with 103 CFU of different K. pneumoniae strains intraperitoneally. The previously reported hvKP strain NTUH-K2044 (ST23, K1 serotype), cKP strain HS11286 (ST11, KL103 serotype) and saline were applied as the controls. RJF293 showed virulence not statistically significant from that of NTUH-K2044 (p > 0.6, by log-rank test). No death of mice in the HS11286 or saline groups was observed during seven days.
RJF293 genome analysis
Since no K2 ST374 K. pneumoniae strain had yet been completely sequenced, we decided to explore the virulence factors of RJF293 via WGS. Our analysis showed that the RJF293 genome consists of a circular chromosome of 5,226,330 base pairs with average GC content of 57.5%, and one circular plasmid with 224,263 bases with average GC content of 50.1%. The RJF293 chromosome contains 4,995 annotated protein-coding sequences (CDSs) (Supplementary Table S4). The size of the plasmid was confirmed using S1-nuclease treatment followed by PFGE (Supplementary Figure S4).
The RJF293 strain exhibits a distinct genetic background from those of the other sequenced K2 and K1 serotype hvKP strains, as supported by both phylogenetic analysis and genome alignments. Whole genome SNP-based phylogenetic analysis showed that the ST374 RJF293 was failed to be grouped with any other K2 serotype hvKP strains (ST66 or ST86), also distant from the clade grouped by the ST23 hvKP strains (). In addition, when all 4,995 CDSs of RJF293 were analyzed for the presence of homologs against seven completely sequenced hvKP chromosomes (Supplementary Figure S5), only 88% (4,412/4,995) of homologous CDSs were found across all eight hvKP genomes being compared (BLASTn-based H-value > 0.81) [Citation31].
Figure 2. Putative virulence genes (gene clusters) detected among the 106 completely sequenced K. pneumoniae genomes (Supplementary Table S3). (A) A parsimony tree generated from 429,267 SNPs using kSNP3 for the 106 completely sequenced K. pneumoniae chromosomes and displayed by iTOL with midpoint rooting. The hvKP and cKP isolates listed in Supplementary Table S4 are highlighted by color (red or orange, hvKP; blue, cKP). (B) The virulence genes predicted in the RJF293 genome are listed as an example in Supplementary Table S5. The presence of genes in the chromosome and/or plasmid is indicated in different gray scales.

A total of 69 putative virulence genes were detected on the RJF293 chromosome (Supplementary Table S6), including gene clusters coding for enterobactin synthesis (fep, ent), pilus (yag), type 3 fimbriae (mrk), lipopolysaccharide synthesis (lpx) and iron ABC transporter (kfu). These RJF293 virulence genes show a unique presence/absence pattern (). Compared with the ST23 K1 serotype hvKP strains, the two-component regulatory system KvgAS is found to be present in only RJF293 (K2, ST374) and another five genomes of K. pneumoniae K2 strains. Besides, 8 virulence genes (clusters), including glxKR, ybbWY, allABCDRS, ylbEF, hyi, arcC, fdrA, KP1_1364 and KP1_1371, were detected only in the five ST23 K1 serotype hvKP strains and another strain (YH43 with a novel sequence type and absence of capsular serotype information). The cps gene clusters of K1 and K2 K. pneumoniae strains have conserved regions at the 5′ and 3′ extremities, while the variable region is located between wzb and wcaJ (Supplementary Figure S6). RJF293 showed 100% identity to the previously reported K2 strain CG43 (ST86) in the cps gene cluster region, though with different MLST patterns [Citation32]. The LPS cluster of RJF293 was characterized as O1, which is the most prevalent serotype in K. pneumoniae strains, and an IS102 element was located upstream of the RJF293 LPS cluster (Supplementary Figure S7) [Citation33].
One likely intact prophage was identified within the 40-kb region (GI5) specific to the RJF293 chromosome. The corresponding excision form of this prophage was identified by a PCR assay (Supplementary Figure S8) and the sequence of attB site after excision was determined to be TTGAACAT. This prophage coded for the prophage core component proteins, including the coat, plate, portal, tail, and tail fiber proteins and integrases. Interestingly, it also contains a 3-kb variable region coding for seven RJF293-specific CDSs with unknown function. Six other large unusual GI-like regions (>10 kb) were detected on the RJF293 chromosome ( and Supplementary Figure S5), which contained G+C contents lower than the genomic G+C content. For example, the 20-kb region GI8 includes a kvgAS locus (RJF2_RS21335-RJF2_RS21340) and three genes coding for fimbrial (or chaperone) proteins.
Table 2. Large genomic island-like regions (>10 kb) identified on the K. pneumoniae RJF293 chromosome.
The type VI secretion system (T6SS) has been shown to be present in several Gram-negative species [Citation34]. There are three different type VI secretion system loci on the RJF293 chromosome (T6SS-1, RJF2_RS10835-10935; T6SS-2, RJF2_RS15665-15720; T6SS-3, RJF2_RS18290-18405), among which T6SS-3 was located on the 41-kb island GI7. This T6SS locus is present in five out of the 105 other completely sequenced K. pneumoniae strains, including CAV1042, Kp_Goe_121641, Kp_Goe_71070, 234-12, and Kpn555. Its cognate effector (RJF2_RS18365) contains a type I dockerin repeat domain, which is the binding partner of the cohesin domain, and the cohesin-dockerin interaction is the crucial interaction for complex formation in the cellulosome. The finding of multiple T6SS loci is consistent to previous studies [Citation35,Citation36]. and it still remains to be uncovered in the further study whether the three T6SS loci of RJF293 can function independently or not.
The RJF293 chromosomally borne ICE (GI6), called ICEKpnRJF293, is inserted into a tRNAAsn locus and contains a virulence factor gene cluster involved in the synthesis, regulation, and transport of the siderophore yersiniabactin (Supplementary Figure S9). This 59-kb ICE also codes for a conjugation transfer-associated type IV secretion system. The PCR assay showed that this ICE was able to site-specifically excise from the 3′-end of the tRNAAsn gene on the RJF293 chromosome (Supplementary Figure S10). ICEKpnRJF293 is highly syntenic to ICEKp1 in the liver abscess-causing hvKP strain NTUH-K2044 (ST23, K1 serotype) but lacks a virulence-associated region with an iroBCDN gene cluster responsible for salmochelin biosynthesis (Supplementary Figure S9) [Citation37]. Notably, ICEKpnRJF293 contains a 10-kb strain-unique region in the tRNA-distal end, which codes for a restriction-modification system, an ABC transporter, a hypothetical protein and two transposases (Supplementary Figure S9).
Two CRISPR arrays were also detected: one is located from 2,991,230 to 2,991,753bp and contains 8 spacers; another is from 3,001,570 to 3,001,903bp and contains 5 spacers. The seven CRISPR-associated protein genes (type I-E) are located between the two CRISPR arrays, forming a sandwich structure like the experimentally verified system in K. pneumoniae NTUH-K2044 [Citation38]. The DRs of the CRISPR arrays are conserved between RJF293 and NTUH-K2044, but the spacer sequences vary, revealing the difference in habitats of these two strains. In addition, the chromosome contains 12 annotated insertion sequence (IS) elements belonging to five families, i.e., IS5 (n = 5), IS3 (n = 4), IS1 (n = 1), IS481 (n = 1) and Tn3 (n = 1).
RJF293 carries a virulence plasmid, pRJF293
The 224-kb plasmid pRJF293 exhibits high sequence similarity to the reported 219-kb virulence plasmid pLVPK (NCBI accession no. NC_005249) in the hvKP strain CG43 (ST86, K2 serotype) (Supplementary Figure S11) [Citation39]. It codes for 11 putative virulence genes (Supplementary Table S6), including the mucoid phenotype regulator genes rmpA (RJF2_RS26130) and rmpA2 (RJF2_RS26605), the iroBCDN gene clusters involved in salmochelin biosynthesis (RJF2_RS26145– RJF2_RS26160), and the iucABCD/iutA gene cluster related to aerobactin biosynthesis (RJF2_RS26550- RJF2_RS26570). These virulence determinants are encoded on pRJF293 or similar virulence plasmids reported in K. pneumoniae (120-230 kb in size), which has been identified as restricted to hvKP isolates, including RJF293, CG43, KCTC2242, RJF999, ED23, Kp52.145 and NTUH-K2044 (). We also annotated 14 IS elements in pRJF293. They belong to seven families, IS5 (n = 5), IS1 (n = 3), IS3 (n = 2), Tn3 (n = 2), IS21 (n = 1), and IS66 (n = 1). Additionally, compared to plasmid pLVPK of the ST86 K2 serotype hvKP strain CG43, pRJF293 carries a novel 5.7-kb transposon (region 184,481..190,190 bp) [Citation39]. This transposon is composed of two IS102 elements at the ends, which are related to the IS903 group of the IS5 family. It also contains five annotated CDSs coding for putative relaxase and five putative proteins that may confer DNA conjugative transfer. This pRJF293-carrying transposon is only present in the virulence plasmid of hvKP isolate RJF999 (ST23, K1 serotype, isolated in the same hospital as RJF293, GenBank accession no. CP014010.1), but absent from the other 104 completely sequenced K. pneumoniae genomes.
Discussion
Recently hypervirulent K. pneumoniae variants have been emerging as a cause of severe hospital-acquired invasive infections in critical patients [Citation1]. Increasing numbers of hvKP isolates being collected from blood samples of ICU patients in our study and in other clinical settings was reported [Citation4,Citation13]. The identification of multiple MLST allelic types and capsular serotypes reflects the genetic diversity of hypermucoviscous K. pneumoniae isolates. Among the 79 known capsular serotypes of K. pneumoniae, K1 and K2 have been shown to be the most prevalent in hvKP, and their association with invasive infection has been corroborated using murine models [Citation6,Citation40,Citation41]. In practice, K2 hvKP shows high clinical significance due to its high pathogenicity and metastatic infection capability. In comparison to K1 hvKP, K2 hvKP shows high MLST diversity, calling for more genomic data for understanding its pathogenesis [Citation6].
In our study, the K2 serotype ST374 strain RJF293 was collected from a human blood sample in an ICU patient. RJF293 exhibited three typical features of hvKP, showing the hypermucoviscous phenotype (viscous string of >20 mm in length) and causing severe and metastatic infection [Citation3]. The mouse lethality assay also confirmed that RJF293 was as virulent as the ST23 K1 serotype strain NTUH-K2044. We examined the K. pneumoniae isolates collected in our ICU department. However, no isolates from other patients showed the same MLST or PFGE pattern as RJF293.
The existence of 47 virulence-associated genes (gene clusters) in the 106 K. pneumoniae strains with a complete genome sequence was determined in our study. Strains with serotype K1 and K2 showed a remarkable difference in the carriage of these gene/gene clusters, and the K1 strains carried significantly more of these genes (gene clusters) than the K2 strains. allABCDR, arcC, fdrA, gcl, glxKR, hyi, KP1_1364, KP1_1371, ybbWY, ylbEF, fcl, and gmd are present in only serotype K1 K. pneumoniae genomes. These genes have been reported to be contributive to bacterial virulence. For example, the allantoinase gene cluster allABCDR involved in allantoin metabolism has been shown to contribute to virulence in mice after gastrointestinal inoculation [Citation42]. By mouse lethality assay, K. pneumoniae RJF293 was determined to have the same level of virulence as the K1 K. pneumoniae NTUH-K2044. This revealed that having more virulence-associated genes (gene clusters) is not equal to a higher level of virulence. The contribution of each known virulence determinant needs to be assessed quantitatively.
Different from the virulence determinants stated above, most of the genes/gene clusters located on plasmids, ICEs and other mobile genetic elements are shared by both K1 and K2 K. pneumoniae. These include the well-documented hypermucoviscous phenotype regulators rmpA and rmpA2, the aerobactin synthesis cluster iucABCD, the salmochelin synthesis cluster iroBCDN etc. The rmpA and rmpA2 genes have both been shown to activate capsule production, exhibiting the hypermucoviscous phenotype [Citation43,Citation44]. Siderophore systems are considered integral to bacterial virulence, allowing bacteria to scavenge for iron from host transport proteins, thereby enhancing the ability to survive and replicate within the host [Citation45]. The aerobactin has been shown to be the predominant siderophore in the hypervirulent K. pneumoniae [Citation46]. The research conducted by Russo et al demonstrated that the contributions of different siderophores for K. pneumoniae growth and survival in human ascites fluid were not equivalent. Aerobactin showed relatively higher biological activity than yersiniabactin or salmochelin [Citation47]. These genes (gene clusters) are usually carried by a virulence plasmid belonging to the IncHI1B group or an ICE belonging to the ICEKp1 family. The virulence plasmid (in this study, pRJF293) is normally 120 kb to 230 kb in size. It is majorly carried by K. pneumoniae strains with capsular serotype K1 or K2. However, very recently, the virulence plasmid was found in a K47 ST11 K. pneumoniae strain in China [Citation48]. It was characterized as greatly contributive to the ST11 hvKP and might be a sign of the dissemination of virulence plasmids in the future. The ICEKp1 family is another group of mobile genetic elements shared by K1 and K2 K. pneumoniae strains. The cargo genes (gene clusters) on this ICE can code for yersiniabactin, salmochelin and colibactin. In fact, the ICE is widely distributed in K. pneumoniae (47 out of 106) with more capsular serotypes and MLST types, for example K. pneumoniae AATZP (K64, ST147) [Citation49]. The virulence plasmid together with the ICE make up a platform for the intra- and even inter-species dissemination of virulence determinants.
In summary, we investigated the genetic background and microbiological features of a hypermucoviscous clinical isolate (RJF293) of K. pneumoniae collected from human blood. The metastatic infection case, string test, and mouse lethality assay confirmed the hypervirulence of this ST374 K2 serotype strain. Thus, the RJF293 genomics study reported here represents the first complete genome sequence of an ST374 K. pneumoniae strain with the K2 serotype, making a valuable addition to the growing list of diverse hvKP genomes for the scientific research community worldwide. Our results suggest that the ST374 K2 serotype hvKP case should not be underestimated and its clinical impact would be particularly challenging.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
1421894_supp.pdf
Download PDF (5.8 MB)Acknowledgments
The authors are grateful to the anonymous referees for many helpful comments and suggestions.
Additional information
Funding
References
- Siu LK, Yeh KM, Lin JC, et al. Klebsiella pneumoniae liver abscess: a new invasive syndrome. Lancet Infect Dis. 2012;12:881–7. doi:10.1016/S1473-3099(12)70205-0
- Qi Y, Wei Z, Ji S, et al. ST11, the dominant clone of KPC-producing Klebsiella pneumoniae in China. J Antimicrob Chemother. 2011;66:307–12. doi:10.1093/jac/dkq431
- Shon AS, Bajwa RP, Russo TA. Hypervirulent (hypermucoviscous) Klebsiella pneumoniae: a new and dangerous breed. Virulence. 2013;4:107–18. doi:10.4161/viru.22718
- Liu YM, Li BB, Zhang YY, et al. Clinical and molecular characteristics of emerging hypervirulent Klebsiella pneumoniae bloodstream infections in mainland China. Antimicrob Agents Chemother. 2014;58:5379–85. doi:10.1128/AAC.02523-14
- Catalan-Najera JC, Garza-Ramos U, Barrios-Camacho H. Hypervirulence and hypermucoviscosity: Two different but complementary Klebsiella spp. phenotypes? Virulence. 20178:1111–23.
- Struve C, Roe CC, Stegger M, et al. Mapping the Evolution of Hypervirulent Klebsiella pneumoniae. MBio. 2015;6:e00630. doi:10.1128/mBio.00630-15
- Lin JC, Koh TH, Lee N, et al. Genotypes and virulence in serotype K2 Klebsiella pneumoniae from liver abscess and non-infectious carriers in Hong Kong, Singapore and Taiwan. Gut Pathog. 2014;6:21. doi:10.1186/1757-4749-6-21
- Ribot EM, Fair MA, Gautom R, et al. Standardization of pulsed-field gel electrophoresis protocols for the subtyping of Escherichia coli O157:H7, Salmonella, and Shigella for PulseNet. Foodborne Pathog Dis. 2006;3:59–67. doi:10.1089/fpd.2006.3.59
- Tenover FC, Arbeit RD, Goering RV, et al. Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J Clin Microbiol. 1995;33:2233–9.
- Hunter SB, Vauterin P, Lambert-Fair MA, et al. Establishment of a universal size standard strain for use with the PulseNet standardized pulsed-field gel electrophoresis protocols: converting the national databases to the new size standard. J Clin Microbiol. 2005;43:1045–50. doi:10.1128/JCM.43.3.1045-1050.2005
- Diancourt L, Passet V, Verhoef J, et al. Multilocus sequence typing of Klebsiella pneumoniae nosocomial isolates. J Clin Microbiol. 2005;43:4178–82. doi:10.1128/JCM.43.8.4178-4182.2005
- Fang CT, Lai SY, Yi WC, et al. Klebsiella pneumoniae genotype K1: an emerging pathogen that causes septic ocular or central nervous system complications from pyogenic liver abscess. Clin Infect Dis. 2007;45:284–93. doi:10.1086/519262
- Wu H, Li D, Zhou H, et al. Bacteremia and other body site infection caused by hypervirulent and classic Klebsiella pneumoniae. Microb Pathog. 2017;104:254–62. doi:10.1016/j.micpath.2017.01.049
- Wayne. Clinical and Laboratory Standards Institute. Performance standards for antimicrobial susceptibility testing-twenty fourth edition: approved standard M100eS24. CLSI. 2014;
- Facility Guidance for Control of Carbapenem- Resistant Enterobacteriaceae (CRE) November 2015 Update. CDC. 2015;1–19.
- Arvanitis M, Li G, Li DD, et al. A Conformationally constrained cyclic acyldepsipeptide is highly effective in mice infected with methicillin-susceptible and -resistant Staphylococcus aureus. PloS One. 2016;11:e0153912. doi:10.1371/journal.pone.0153912
- Bi D, Jiang X, Sheng ZK, et al. Mapping the resistance-associated mobilome of a carbapenem-resistant Klebsiella pneumoniae strain reveals insights into factors shaping these regions and facilitates generation of a ‘resistance-disarmed’ model organism. J Antimicrob Chemother. 2015;70:2770–4. doi:10.1093/jac/dkv204
- Wu KM, Li LH, Yan JJ, et al. Genome sequencing and comparative analysis of Klebsiella pneumoniae NTUH-K2044, a strain causing liver abscess and meningitis. J Bacteriol. 2009;191:4492–501. doi:10.1128/JB.00315-09
- Wilson K. Preparation of genomic DNA from bacteria. Curr Protoc Mol Biol. 2001; Chapter 2:Unit 2:4.
- English AC, Richards S, Han Y, et al. Mind the gap: upgrading genomes with Pacific Biosciences RS long-read sequencing technology. PloS One. 2012;7:e47768. doi:10.1371/journal.pone.0047768
- Walker BJ, Abeel T, Shea T, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PloS One. 2014;9:e112963. doi:10.1371/journal.pone.0112963
- Luo H, Zhang CT, Gao F. Ori-Finder 2, an integrated tool to predict replication origins in the archaeal genomes. Front Microbiol. 2014;5:482. doi:10.3389/fmicb.2014.00482
- Angiuoli SV, Gussman A, Klimke W, et al. Toward an online repository of Standard Operating Procedures (SOPs) for (meta)genomic annotation. Omics. 2008;12:137–41. doi:10.1089/omi.2008.0017
- Li J, Tai C, Deng Z, et al. VRprofile: gene-cluster-detection-based profiling of virulence and antibiotic resistance traits encoded within genome sequences of pathogenic bacteria. Brief Bioinform. 2017;bbw141.
- Chen L, Zheng D, Liu B, et al. VFDB 2016: hierarchical and refined dataset for big data analysis–10 years on. Nucleic Acids Res. 2016;44:D694–7. doi:10.1093/nar/gkv1239
- Edgar RC. PILER-CR: fast and accurate identification of CRISPR repeats. BMC Bioinformatics. 2007;8:18. doi:10.1186/1471-2105-8-18
- Gardner SN, Slezak T, Hall BG. kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics (Oxford, England). 2015;31:2877–8. doi:10.1093/bioinformatics/btv271
- Wright MS, Perez F, Brinkac L, et al. Population structure of KPC-producing Klebsiella pneumoniae isolates from midwestern U.S. hospitals. Antimicrob Agents Chemother. 2014;58:4961–5. doi:10.1128/AAC.00125-14
- Wright MS, Suzuki Y, Jones MB, et al. Genomic and transcriptomic analyses of colistin-resistant clinical isolates of Klebsiella pneumoniae reveal multiple pathways of resistance. Antimicrob Agents Chemother. 2015;59:536–43. doi:10.1128/AAC.04037-14
- Letunic I, Bork P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016;44:W242–5. doi:10.1093/nar/gkw290
- Shao Y, He X, Harrison EM, et al. mGenomeSubtractor: a web-based tool for parallel in silico subtractive hybridization analysis of multiple bacterial genomes. Nucleic Acids Res. 2010;38:W194–200. doi:10.1093/nar/gkq326
- Ho JY, Lin TL, Li CY, et al. Functions of some capsular polysaccharide biosynthetic genes in Klebsiella pneumoniae NTUH K-2044. PloS One. 2011;6:e2.1664. doi:10.1371/journal.pone.0021664
- Follador R, Heinz E, Wyres KL, et al. The diversity of Klebsiella pneumoniae surface polysaccharides. Microb Genom. 2016;2:e000073. doi:10.1099/mgen.0.000073
- Cianfanelli FR, Monlezun L, Coulthurst SJ. Aim, Load, Fire: The Type VI Secretion System, a Bacterial Nanoweapon. Trends Microbiol. 2016;24:51–62. doi:10.1016/j.tim.2015.10.005
- Liu L, Ye M, Li X, et al. Identification and Characterization of an Antibacterial Type VI Secretion System in the Carbapenem-Resistant Strain Klebsiella pneumoniae HS11286. Front Cell Infect Microbiol. 2017;7:442. doi:10.3389/fcimb.2017.00442
- Sarris PF, Zoumadakis C, Panopoulos NJ, et al. Distribution of the putative type VI secretion system core genes in Klebsiella spp. Infect, Genet Evol. 2011;11:157–66. doi:10.1016/j.meegid.2010.09.006
- Lin TL, Lee CZ, Hsieh PF, et al. Characterization of integrative and conjugative element ICEKp1-associated genomic heterogeneity in a Klebsiella pneumoniae strain isolated from a primary liver abscess. J Bacteriol. 2008;190:515–26. doi:10.1128/JB.01219-07
- Lin TL, Pan YJ, Hsieh PF, et al. Imipenem represses CRISPR-Cas interference of DNA acquisition through H-NS stimulation in Klebsiella pneumoniae. Sci Rep. 2016;6:31644. doi:10.1038/srep31644
- Chen YT, Chang HY, Lai YC, et al. Sequencing and analysis of the large virulence plasmid pLVPK of Klebsiella pneumoniae CG43. Gene. 2004;337:189–98. doi:10.1016/j.gene.2004.05.008
- Hsu CR, Lin TL, Chen YC, et al. The role of Klebsiella pneumoniae rmpA in capsular polysaccharide synthesis and virulence revisited. Microbiology. 2011;157:3446–57. doi:10.1099/mic.0.050336-0
- Gomez-Simmonds A, Uhlemann AC. Clinical Implications of Genomic Adaptation and Evolution of Carbapenem-Resistant Klebsiella pneumoniae. J Infect Dis. 2017;215:S18–s27. doi:10.1093/infdis/jiw378
- Chou HC, Lee CZ, Ma LC, et al. Isolation of a chromosomal region of Klebsiella pneumoniae associated with allantoin metabolism and liver infection. Infect Immun. 2004;72:3783–92. doi:10.1128/IAI.72.7.3783-3792.2004
- Yu WL, Lee MF, Tang HJ, et al. Low prevalence of rmpA and high tendency of rmpA mutation correspond to low virulence of extended spectrum beta-lactamase-producing Klebsiella pneumoniae isolates. Virulence. 2015;6:162–72. doi:10.1080/21505594.2015.1016703
- Cheng HY, Chen YS, Wu CY, et al. RmpA regulation of capsular polysaccharide biosynthesis in Klebsiella pneumoniae CG43. J Bacteriol. 2010;192:3144–58. doi:10.1128/JB.00031-10
- Russo TA, Shon AS, Beanan JM, et al. Hypervirulent K. pneumoniae secretes more and more active iron-acquisition molecules than “classical” K. pneumoniae thereby enhancing its virulence. PloS One. 2011;6:e26734. doi:10.1371/journal.pone.0026734
- Russo TA, Olson R, Macdonald U, et al. Aerobactin mediates virulence and accounts for increased siderophore production under iron-limiting conditions by hypervirulent (hypermucoviscous) Klebsiella pneumoniae. Infect Immun. 2014;82:2356–67. doi:10.1128/IAI.01667-13
- Russo TA, Olson R, MacDonald U, et al. Aerobactin, but not yersiniabactin, salmochelin, or enterobactin, enables the growth/survival of hypervirulent (hypermucoviscous) Klebsiella pneumoniae ex vivo and in vivo. Infect Immun. 2015;83:3325–33. doi:10.1128/IAI.00430-15
- Gu D, Dong N, Zheng Z, et al. A fatal outbreak of ST11 carbapenem-resistant hypervirulent Klebsiella pneumoniae in a Chinese hospital: a molecular epidemiological study. Lancet Infect Dis. 2017;3099:1–10.
- Lam MMC, Wick RR, Wyres KL, et al. Frequent emergence of pathogenic lineages of Klebsiella pneumoniae via mobilisation of yersiniabactin and colibactin. BioRxiv. 2017. doi: 10.1101/098178