
ABSTRACT
The increasing number of infections by species of Mucorales and their high mortality constitute an important concern for public health. This study aims to decipher the genetic basis of Mucor circinelloides pathogenicity, which displays virulence in a strain dependent manner. Assuming that genetic differences between strains may be linked to different pathotypes, we have conducted a study to explore genes responsible for virulence in M. circinelloides by whole genome sequencing of the avirulent strain NRRL3631 and comparison with the virulent strain CBS277.49. This genome analysis revealed 773 truncated, discontiguous and absent genes in the NRRL3631 strain. We also examined phenotypic traits resulting in reduced heat stress tolerance, chitosan content and lower susceptibility to toxic compounds (calcofluor white and sodium dodecyl sulphate) in the virulent strain, suggesting the influence of cell wall on pathogenesis. Based on these results, we focused on studying extracellular protein-coding genes by gene deletion and further pathotype characterization of mutants in murine models of pulmonary and systemic infection. Deletion of gene ID112092, which codes for a hypothetical extracellular protein of unknown function, resulted in significant reduction of virulence.
Although pathogenesis is a multifactorial process, these findings highlight the crucial role of surface and secreted proteins in M. circinelloides virulence and should promote further studies of other differential genes.
Introduction
Mucormycosis is an emerging opportunistic infection caused by fungi of the order Mucorales, which is becoming more frequent due to the increase in population with risk factors, such as immunosuppression, diabetes, blunt trauma and hematological malignancies. Mucor circinelloides is one of the most frequent species within Mucorales causing fatal mucormycosis. Mucorales infections are difficult to treat due to their fast dissemination within the host tissues and their low susceptibility to antifungal agents, which drives to worse outcomes than other common fungal diseases, such as aspergillosis [Citation1-3]. Experimental mucormycosis has demonstrated differences in virulence between strains, which points to individual factors responsible for pathogenesis. This phenomenon has been previously reported by Li et al [Citation4], who observed different pathogenicity among M. circinelloides strains in Galleria mellonella infections. The authors also observed phenotypical characteristics related to virulence, such as higher germination velocity, spore size and protein secretion.
In the present study we evaluated the virulence of two M. circinelloides f. lusitanicus strains by systemic and pulmonary infection murine models. One of the strains was previously identified as highly virulent (CBS277.49) and the other one as avirulent (NRRL3631) in moth wax infection [Citation4]. Once we verified the pathogenic capacity of both strains in the murine model, we carried out the whole genome sequencing of the avirulent strain (NRRL3631) and we conducted a genome comparison study with the virulent one (already sequenced). In this analysis we aim to summarize genetic differences between both M. circinelloides strains, directed for screening potential determinants of pathogenicity. Nevertheless, interpreting and understanding the role of genes in virulence is a difficult challenge, even for those encoding proteins with known function. Considering the strong association between extracellular proteins, cell wall structure and pathogenic potential [Citation5], we have addressed the study of genes coding for extracellular enzymes using gene deletion and replacement method. This approach has allowed us to examine the effect of gene lack on the virulence of the pathogenic strain. Overall, the results of the genetic screening carry out in the present study can provide a new route for understanding M. circinelloides pathogenesis and control of mucormycosis.
Results
CBS277.49 and NRRL3631 virulence in pulmonary and systemic murine models
Systemic and pulmonary infections with spores of CBS277.49 strain resulted on 100% mortality after challenge with 1×106 CFU/animal administered intravenously (i.v.), and 80% mortality after i.v inoculation with 1 × 105 CFU/animal or intranasally inoculation with 5 × 107 CFU/animal. However, infections performed with strain NRRL3631 resulted in 100% survival at any inocula and route of infection assayed ( and ).
Figure 1. Virulence of M. circinelloides CBS277.49 and NRRL3631 strains in neutropenic murine host. Survival of mice infected (A) intravenously (i.v.) with 1 × 106 and 1 × 105 CFU of fungal strains and (B) intranasally (i.n.) with 5 × 107 CFU of fungal strains show significant lower virulence of NRRL 3631 (#) (p < 0.05). (C) Fungal biomass quantification of liver, brain, kidney lung and spleen 3 days after i.v. infection with 1 × 106 CFU/animal of CBS277.49 and NRRL3631 using real-time PCR. Data represent µg of fungal biomass respect 1 g of mice tissue biomass. Significant differences are indicated (#). Bars indicate the standard deviation from independent experiments.
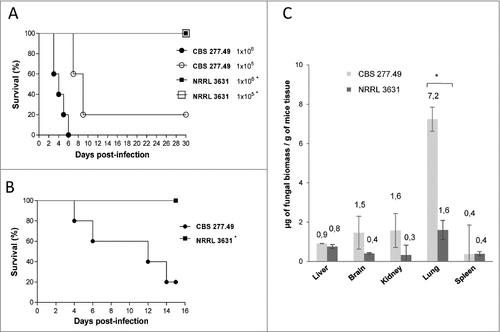
Fungal burden was quantified by real time PCR (qPCR) in relevant organs of mice infected i.v. with both wildtype strains (1 × 105 CFU / animal). Quantification of total genomic DNA (gDNA) and conversion to biomass values revealed greater amount of CBS277.49 in lung (7.2 µg of fungal biomass / g of mice tissue) in comparison to NRRL3631 strain (0.5 µg biomass / g of mice tissue) (p = 0.019) (). Lower amount of fungal biomass was detected in kidney, brain, liver and spleen with no significant differences among strains (p ≥ 0.33). Taken together, these mortality rate and fungal load results indicate higher infection capacity of CBS 277.49 in mice, being lung the preferred target organ.
Small and large spores from CBS277.49 strain display different germination kinetics but same infectivity.
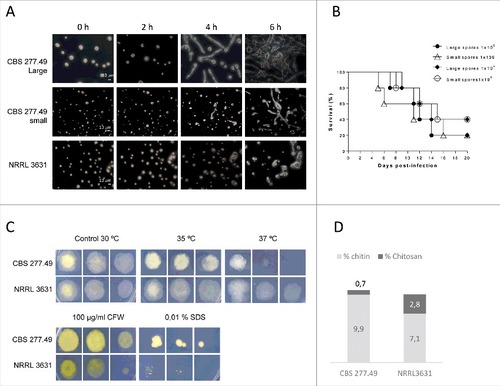
Microscopic observation revealed different germination rate between small (5 × 3.5 µm) and large (12.3 × 9 µm) sporangiospores from the pathogenic strain (CBS277.49). Large sporangiospores from CBS277.49 produced visible germ tubes after 3–4 h of incubation, while small ones showed a 2 hours delay (). The NRRL3631 strain, which only produces one-sized sporangiospores (5 × 3.5 µm), showed similar germination kinetics to CBS277.49 small sporangiospores i.e., around 6 hours post-incubation. Despite these differences, the systemic infection by small or large sporangiospores of the pathogenic strain resulted in similar virulence at both inocula (1 × 105 and 1 × 106 CFU/animal) (p ≥ 0.602) (). These results showed no correlation between sporangiospore size and pathogenic capacity, suggesting the existence of virulent factors inherent to CBS277.49 strain not related to spore dimensions and germination kinetics.
Figure 2. Fungal strains phenotypes. Small and large spores of CBS277.49 strain exhibit differences in (A) germination kinetics and (B) similar virulence capacity in murine model infection. (C) Fungal colonies from CBS277.49 and NRRL3631 M. circinelloides strains grown for 2–3 days at 30°C on SM plates containing sodium dodecyl sulphate (SDS) or calcofluor white (CFW) and 5 days at 35ºC and 37ºC, inoculating 103, 102 and 10 fresh spores. (D) Percentage of chitin and chitosan respect to initial mycelial dry weight.
Whole genome comparison between CBS277.49 and NRRL3631
Whole-genome resequencing of M. circinelloides NRRL3631 was performed by shotgun paired-end method using Illumina sequencing technology. The 91% of the reads were aligned to the reference genome, covering the 96% of the genome size (36.6 Gb) (). This search lead to the detection of single nucleotide polymorphisms (SNPs) and unmapped regions, including four scaffolds entirely unmapped of CBS277.49 (scaffolds 17, 19, 21 and 22) containing 17,493 Kb, 9,869 Kb, 7,638 Kb and 4,155 Kb, respectively (Suppl. Fig. S1). The uncovered gaps were examined and 543 genes were identified as unique genes in the virulent strain that were classified in functional categories based on KOG (). Among them, we identified a high proportion of protein coding genes with unknown functions that accounted for the 68% of total number of unique genes in CBS277.49. The genes that were exclusively found in the pathogenic strain with known functions are listed in . These CBS277.49-specific genes code for proteins involved in RNA processing, nuclear structure and chromatin dynamics, cell cycle control, signal-transduction pathways, post-translational modifications enzymes, vesicle formation and secretory pathway sorting, transport and metabolism, including enzymes involved in extracellular structures biogenesis, secondary metabolites biosynthesis and defense against oxidant compounds ().
Table 1. Assembly and mapping statistics of M. circinelloides NRRL3631 genome sequencing.
Figure 3. Genes identified in the genome comparison study clustered in functional categories based on KOG classification. (A) Absent genes and (B) discontiguous genes in NRRL3631 strain.
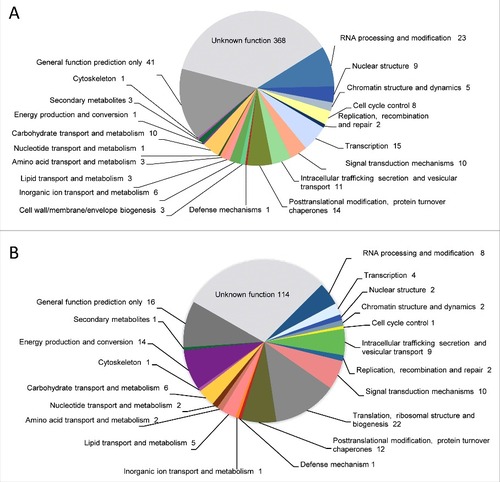
Table 2. Eukaryotic Orthologous Groups (KOG) classification of absent or discontiguos genes detected in NRRL3631 genome in comparison to CBS277.49. A total of 395 sequences assigned to KOG classifications within the principal 17 categories are shown.
To further analyze variation between both genomes, we conducted a pairwise comparison between NRRL3631 reads assembled into contigs and the reference genome. This analysis allows highlighting the variation in gene position, resulting in 230 discontiguous protein-coding sequences that were clustered by KOG classification (). Approximately the 50% of these discontiguous genes code for proteins with unknown function. In the functionally annotated genes we found many translation and ribosomal proteins, followed by post-translational modification, protein turnover, chaperones as well as energy production and conversion enzymes (). The total extracellular enzymes found in the analysis are listed in .
Table 3. Genes coding for extracellular enzymes identified in the genome comparison study among CBS277.49 and NRRL3631 genomes as absent, or discontiguous sequences in NRRL3631. aNumber of duplicated genes in CBS277.49 M. circinelloides genome based upon BLAST with E-value < E−50 and ≥ 70% identity. bDistribution of orthologues within the fungi species based upon BLAST with E-value < E−22 and ≥ 50% identity. Ac: Absidia glauca, Cc: Choanephora cucurbitarum, Cg: Colletotrichum gloeosporioides, Lc: Lichtheimia corymbifera Lr: Lichtheimia ramosa, Ma: Mucor ambiguos, Mc: Mucor circinelloides, Me: Morteriella elongata, Pb: Phycomyces blaskespeanus, Pp: Parasitella parasítica, Rm: Rhizopus microsporus, Ri: Rhizopus irregularis, Rd: Rhizopus delemar, Ro; Rhizopus oryzae, Um: Ustilago maydis.
Reads from NRRL3631 sequencing that did not map to the reference genome (2,183 reads and contigs) were aligned to NCBI's nucleotide database (nr/nt) using BLAST. The returned best hits showed significant similarity to mitochondrial proteins as cytochrome c oxidase, NADH dehydrogenase from other strains and species of Mucorales (M. circinelloides, Rhizopus oryzae or Rhizopus delemar), and some bacteria (data not shown).
Heat stress and cell wall-related phenotypes
As shown in , both strains were able to grow under heat stress conditions (35ºC and 37ºC), although the pathogenic strain exhibited slightly lower growth than NRLL3631 at both temperatures, being more evident the difference at 37°C.
Growth in presence of noxious compounds revealed higher sensitivity of NRRL3631 to SDS and CFW, which exhibited reduced colony size in comparison to CBS277.49 ().
Fungal cell wall extraction and quantification revealed that both strains contained similar amount of total N-acetylglucosamine polymers (chitin and chitosan), which represent approximately 10% of dry weight mycelial biomass. However, the chitosan fraction was lower in CBS277.49 (0.7%) than in NRRL3631 (2.5%) ().
Sequence analysis of ID112092 and ID108920 proteins of Mucor circinelloides
We focused the study on extracellular proteins as virulence factor candidates due to their potential effector [Citation6] or immunomodulating role on fungal pathogenesis [Citation7]. The screening was based on the following criteria: surface-exposed proteins, considering both cell wall anchored proteins and secreted peptides since many extracellular enzymes remain embedded in cell wall without predicted anchor domains; gene products with unknown function; genes with less than two paralogous and poorly conserved sequences in closer fungal species [Citation8-10]. From this first screening listed in , we next examined the deduced amino acid sequences of selected genes, focusing the study on ID112092 and ID108920. The deduced amino acid sequences of ID112092 and ID108920 were used to perform sequence and domain motif analysis. The results showed the presence of N-terminal signal peptide and several N- and O-glycosylation predicted sites along the sequences, which suggests that both genes code for highly glycosylated-secreted proteins (Suppl. Fig. S2 and S3). BLAST search against NCBI database showed that gene ID112092 is poorly conserved in other organisms being the closest match Mucor ambiguous (80.5% identity), followed by Absidia glauca (58.4% and 24.7% identity) and Parasitella parasitica (45.8% identity). All proteins, except the short protein SAM09789.1 from A. glauca, have a conserved Friend of Prmt1 motif (FoP), which corresponds to a novel chromatin target of protein arginine methyltransferase, involved in gene regulation and methylation. The study of the two orthologues genes identified in A. glauca genome: SAM09789.1 (hypothetical small-secreted protein) and SAL94833.1 (hypothetical intracellular protein with Fop domain) (http://fungi.ensembl.org/Absidia_glauca/Info/Index) is particularly interesting. Both genes are located in different scaffolds and display partial sequence similarity to N-terminal and C-terminal region of ID112092, respectively (Suppl. Fig. S2). These findings indicate that ID112092 is a unique gene, only present in M. circinelloides and M. ambiguous species, which contains an unusual combination of chromatin-targeting domain and a signal peptide for its extracellular secretion.
Unlike the previous gene, ID108920 is highly conserved from fungal to mammalian genomes and has a putative gamma-glutamyl transpeptidase activity. Sequence analysis revealed that all orthologues genes contain a transmembrane domain that compromises between 17–22 aminoacids, with the exception of M. circinelloides and M. ambiguous that show a signal peptide instead (Suppl. Fig. S3).
Target deletion of genes ID112092 and ID108920 encoding extracellular proteins
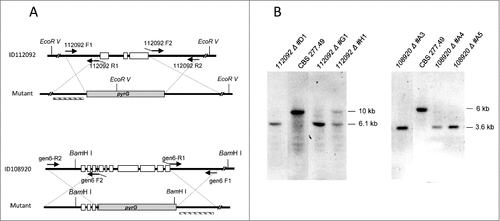
Target deletion of ID112092 and ID108920 was performed by replacement of the entire coding sequences by deletion vectors in MU402 strain (). Initial transformants were grown in selective medium and homologous recombination was confirmed by Southern blot analysis using appropriate restriction enzymes and probes. The wild type strain showed a 10 Kb EcoRV hybridizing band corresponding to ID112092 that was replaced by a 6.1 Kb band in the homologous integrative transformants #D1 and #G1, indicating deletion of the gene ID112092. Similarly, the 6 Kb BamHI fragment of wild type corresponding to the gene ID108920 was replaced by a 3.6 Kb band in the transformants #A3 and #A5, indicating homologous insertion of the disruption vector and therefore the deletion of ID108920 ().
Figure 4. Disruption of 112092 and 108920 genes. (A) Targeted replacement strategy using a vector generated by fusion PCR with the pyrG gene as selective marker. Black arrowheads indicate the primer pairs used for amplification of DNA fragments. Probes are indicated (dashed bar). (B) Southern analysis of gDNAs from M. circinelloides CBS277.49 and transformants. DNAs were digested with EcoR V and Sma I to detect 112092 and 108920 gene deletions, respectively.
Virulence of knockout mutants in genes coding for predicted extracellular proteins ID112092 and ID108920
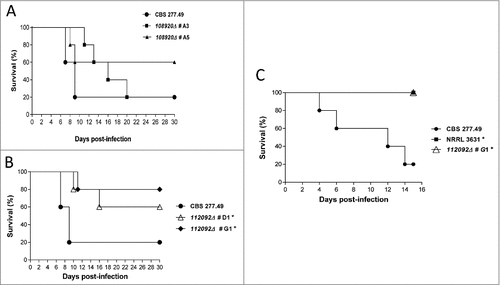
Virulence of knockout mutants was evaluated by experimental disseminated and pulmonary infections in mice. The null mutants in the gene ID108920 (hypothetical γ-glutamyltranspeptidase) (transformants #A3 and #A5) exhibited similar virulence to the wild-type strain (CBS277.49) in disseminated infections (p ≥ 0.081) (). In contrast, infection with spores of knockout mutants in the gene ID112092 (unknown function) (transformants #D1 and #G1) resulted in a significant reduction of mice mortality (p ≤ 0.043) (). Pulmonary infections performed with 112092∆ mutant (transformant #G1) resulted in high reduction of virulence with 100% survival in comparison to wild type strain (p = 0.017) ().
Figure 5. Virulence of knockout mutants in neutropenic mice infection. (A) and (B) Survival of mice infected intravenously (i.v.) with 1 × 105 CFU and (C) mice infected intranasally with 5 × 107 CFU of fungal strains. Data showed attenuated virulence of mutants in ID112092 and ID108920 genes. Significant differences are indicated (#).
Discussion
Only some members of Mucorales are able to cause disease in warm-blooded animals, including humans. These pathogen lineages have evolved by adaption to their environmental niches and acquisition of novel strategies, allowing them to cross-kingdom host jumps [Citation11]. However, some species of the Mucorales exhibit a variety of pathogenic potential traits in different strains, suggesting that small genetic changes can be responsible for pathogenicity. In fact, a previous study correlates virulence with spore size and germination velocity of M. circinelloides in G. mellonella infection, being the strains with faster germination and larger spores more pathogenic [Citation4]. This phenomenon has also been observed in other fungal species like Paracoccidioides brasiliensis and Cryptococcus neoformans [Citation12, Citation13]. However, the link between virulence and spore size is not so straightforward in M. circinelloides. In fact, our results point out that virulence of CBS277.49 is independent of sporangiospores size in both systemic and pulmonary infections. In addition, we must consider that NRRL3631 remains avirulent after pulmonary infection, despite small sporangiospores reach more easily alveoli and in consequence should favor infection [Citation14]; while the large CBS277.49 sporangiospores are highly pathogenic. The thermotolerance assay showed similar ability to grow at 35ºC and 37ºC in both strains, which is crucial to infect warm-blooded hosts. In addition, no alteration in growth capacity in nutritive or minimal media culture was observed in the avirulent strain, beside its delay in germination. Taking all together, our results suggest that other factors, not related to spore morphology or growth capacity, may be involved in the pathogenesis of M. circinelloides.
In an attempt to decipher novel virulence determinants responsible for the pathogenicity of the strain CBS277.49, the genomes comparison has provided strain-specific and discontiguous genes that merit to be further explored, with the aim to identify candidate virulence factors. We hypothesize that this genetic variation among strains may be the result of gene loss, as the same occurs in the obligate biotrophic human pathogen Pneumocystis jirovecii [Citation15], or horizontal gene transfer, as it happens in many other fungal lineages [Citation16, Citation17]. Furthermore, loss of genes can also be responsible of virulence acquisition, even though this hypothesis was discarded in M. circinelloides as we only detected NRRL3631 unique genes coding for mitochondrial enzymes.
Due to the overwhelming number of genetic differences found among strains, mainly related to RNA and DNA machinery, cell division, metabolism, secretion and transport, the big challenge lies on unravelling those genetic determinants involved in virulence. Taking advantage that phenotypic observations on M. circinelloides strains give evidence that thermotolerance, germination velocity and growth capacity do not correspond to virulence-associated traits, we discarded genes related to those phenotypes. On the other hand, cell wall differences among virulent and avirulent strain were considered relevant, since NRRL3631 displays higher sensitivity to CFW and SDS, toxic agents that bind to chitin and interfere with the phospholipid bilayer, respectively [Citation18]. Whereby we assume that cell wall permeability and structure might be altered in the pathogenic strain. Controversially, we detected less chitosan content in the cell wall of the virulent strain, a deacetylated form of chitin that has been extensively associated to virulence in other fungal pathogens, like C. neoformans [Citation19]. Recently, chitin and chitosan have been shown to exhibit different immune activation properties. Chitin seems to stimulate and activate Th2 immune response driving to less efficient protection against fungal infection, as observed O'Dea et al [Citation20]. in Aspergillus fumigatus strains with different chitin content. Whereas chitosan, but not chitin, has been shown to induce a strong IL-β response that contributes to the activation of inflammation, generally considered protective in fungal pathogens [Citation21, Citation22]. Considering these chitin and chitosan immune modulatory properties, it is possible that higher chitin and lower chitosan content on CBS277.49 cell wall are linked to its high virulence. Another hypothetical explanation to link lower amount of chitosan and virulence is that deacetylation of chitin to chitosan protects the fungal cell wall from being hydrolyzed by host chitinases, and consequently might contribute to lower levels of host innate immune response, as it occurs in plant infections [Citation23]. However, none of the genes identified in this genome analysis seem to be directly related to the lower chitosan content in the virulent strain.
Being evident the cell wall alterations in CBS277.49 strain, and the lack of physiological basis to attribute its virulence to any growth property, we focused the screening of new virulence factors on extracellular proteins with unknown function or unknown biological role. Extracellular proteins have been crucial in Mucorales evolution, diversification and virulence [Citation24, Citation25]. In fact, many of these extracellular glycoproteins trigger inflammatory reactions, acting as pattern recognition receptors and affecting disease development [Citation26, Citation27]. Accordingly, extracellular enzymes uniquely present in the genome of the pathogenic strain or translocated to a different position in the avirulent one were considered the main candidates for virulence factors.
From this study, it is of particular interest the gene ID112092, found in a different locus in the NRRL3631 genome, with no paralogous and poorly conserved in other species. Curiously, this gene with unknown function contains a signal peptide and a chromatin associated FoP domain, which results controversial for a glycosylated-secreted protein. It displays close homology to A. glauca and P. parasitica sequences, both symbiotic Mucorales with reported horizontal gene transfer events [Citation28]. Specifically, A. glauca contains two different proteins and each one displays exclusive homology to N-terminal and to C-terminal regions of M. circinelloides sequence, respectively. In basis of these findings, we hypothesize that ID112092 may be a novel multidomain protein resulted from the evolutionary combination of two ancestral proteins. Whereas the functional features of ID112092 remain unknown, the knockout mutant displayed a drastic reduction of virulence in mice infection. Nevertheless, no phenotypical differences were observed in 112092∆ mutants in spore size or growth capacity in presence of toxic compounds (CFW, SDS) and under heat stress at 35–37ºC, comparing to the wild type strain CBS277.49 (data not shown).
In summary, our study gives evidence of different pathogenic potential of two wild type strains of M. circinelloides that display genetic differences, and provides candidate virulence factors related to cell wall surface and secreted proteins. These findings might be of general interest on the perspective of understanding pathogenicity of Mucorales, and open a new path to find novel target proteins and future attempts to develop efficient therapeutic strategies. Future studies on ID112092 gene are needed in order to understand its enzymatic activity and biological role. The functional characterization of this unique gene should provide greater detail of virulence properties of M. circinelloides in the context of pathogen-host interaction.
Material and methods
Strains and culture conditions
The wild type strains CBS277.49 and NRRL3631 of M. circinelloides f. lusitanicus were used in this study. Additionally, the leucine and uracil auxotroph MU402, derived from CBS277.49, was used as recipient strain for transforming experiments [Citation29].
To obtain fungal biomass, a sporangiospores suspension was poured into potato dextrose broth (PDB) and incubated at 30ºC under agitation at 150 rpm for 48 h. To obtain sporangiospores, strains were grown in yeast extract peptone glycerol agar (YPG) or potato dextrose agar (PDA, Pronadisa) at 30ºC for 5–7 days. Extraction of gDNA, was performed from cultures grown in PDB at 30ºC as previously described [Citation29]. For transformation experiments M. circinelloides was grown on MMC medium (1% casamino acids, 0.05% yeast nitrogen base without aminoacids and ammonium sulphate, 2% glucose) [Citation30].
Animal models
Four week-old male OF1 mice weighing 30 g (Charles River, Criffa S.A) were used. For the systemic infection mice were immunosuppressed 2 days prior the infection by intraperitoneal injection of 200 mg/kg body weight of cyclophosphamide (Genoxal®; Laboratories Funk S.A.) and once every 5 days thereafter. Mice were challenged intravenously (i.v.) via the lateral tail vein with 1 × 105 or 1 × 106 sporangiospores/animal in 0.2 mL of sterile saline.
Due to the heterogeneous size of sporangiospores from strain CBS277.49, in an additional experimental assay mice were infected i.v. with previously separated small and large sporangiospores both at 1 × 105 and 1 × 106 CFU/animal. Sporangiospores were fractioned by filtration through two Monodur filters (10 µm pore diameter) and consecutive centrifugations at 340g on two size range i.e., small (5 × 3.5 µm) and large (12.3 × 9 µm) sporangiospores.
For pulmonary infections mice were immunosuppressed by subcutaneous injection of 125 mg/kg of cortisone acetate 4 days prior to the infection and once every 2 days thereafter [Citation31]. Infection was performed in mice anesthetized with sevoflurane (Sevorane, Abbott lab.) by instillation of 0.03 mL of sterile saline containing 5 × 107 sporangiospores. All experimental groups of animals were checked twice daily for 20–30 days post-infection.
The murine models of systemic and pulmonary infection were used to evaluate the virulence of knockout mutants in comparison to wild type CBS277.49 and NRRL3631 strains.
Mice care procedures and experimental conditions were supervised and approved by the Universitat Rovira i Virgili Animal Welfare and Ethics Committee. Experiments were repeated three times with similar results. Data presented are from one representative experiment.
Fungal burden quantification
Mice infected i.v. with 1 × 105 and 1 × 106 sporangiospores of both wild type strains were euthanatized 6 and 3 days post-infection respectively by CO2 anoxia. Spleen, lungs, liver, brain and kidneys were aseptically removed and weighed for fungal burden quantification by qPCR. Organs were ground up on liquid nitrogen and gDNA was extracted as previously described [Citation32]. Specific primers of M. circinelloides chitin synthase gene (ID153118) and of mice β2-microglobulin gene (ID12010) were used (). Samples analysis were carried out in triplicate in 15 µl PCR reactions containing 180 ng of test sample gDNA using SybrGreen kit (Fast SYBR® Green Master Mix -ABI) in a StepOne™ Real-Time PCR System (ABI). gDNA from non-infected mice was used as negative control. Relative amount of fungal and mice gDNA was quantified on the basis of their standard curves, elaborated with known fungal DNA concentrations (0.01 ng – 10 ng) and mice (1 ng – 200 ng) and their corresponding amplification cycle threshold (Ct). The amount of gDNA calculated for each test sample was translated into mg of biomass using the DNA: biomass ratio estimated for each organism (127 µg gDNA/ 1mg fungal biomass and 100 µg gDNA/ 1 mg mice tissue).
Table 4. Oligonucleotides used in this study. Italics and lower case indicate nucleotide sequences added for fusion purposes. Positions are referred to the start codon, (+) downstream or (-) upstream of ATG. Orientation is indicated, (s) sense, (as) antisense.
We determined the average number of nuclei per hyphae by DAPI staining [Citation33] and microscopic observation. The differences among strains were negligible, displaying similar number of nuclei (20 nuclei/ 100 µm) and amount of gDNA (127 µg/g) (Suppl. Fig. S4 and Suppl. Table S1). Experiments were repeated three times with similar results.
Whole-genome sequencing, assembly and comparative genomics
The whole genome of the strain NRRL3631 was sequenced using Illumina technology platform (High seq) (Macrogen, Korea). A Truseq DNA library with 200 bp insert fragments was constructed and Paired-End sequenced producing 56 million sequencing reads with a read length of 100 bp. Adaptors were removed with Trimmomatic [Citation34] and quality checked with FastqC [Citation35]. De novo assembly was performed using SOAPdenovo2 software [Citation36] (kmer size of 49). Reads with quality less than 30 over 70% of the read were filtered using NGS QC Toolkit [Citation37]. Final assembly was generated using ABACAS and IMAGE (PAGIT) [Citation38], using Mucor circinelloides CBS277.49 (GenBank assembly accession GCA_001638945.1) as reference. ABACAS was run twice, with an intermediate IMAGE step to close additional gaps. For running IMAGE, contigs of less than 500 bp were removed, kmer parameter was set to 51 and reads with quality less than 30 over 70% of the read were filtered using NGS QC Toolkit [Citation37]. ABACAS was run with nucmer and sensitive settings. Whole-genome sequencing reads from the strain NRRL3631 were aligned to the reference genome (http://genome.jgi.doe.gov/vista_embed/?organism = Mucci2) using Burrow-Wheeler Aligner (BWA) software, version 0.7.13 [Citation39] using bwa mem with the default parameters and the alignment results were visualized using the program Tablet [Citation40]. Regions with no read aligned were examined and considered unique regions in the CBS277.49 genome. Variant calling to identify single nucleotide polymorphisms (SNPs) were conducted using Samtools 1.3 calling method, using mpileup function with default parameters [Citation41]. Unmapped reads were assembled and analyzed by Blast search against NCBI database and sequences with best hits E ≤ e-48 were identified. Pairwise genome comparison with ACT, Artemis Comparison Tool [Citation42], was performed aligning the NRRL3631 contigs against the reference genome (CBS277.49).
Germination and sensitivity assays
For the germination kinetics study, 10 µl containing 5 × 104 spores were incubated at 30ºC on 1% agarose and 0.5% casamino acids media (Difco Laboratories). Spore growth was monitored by optical microscopy every hour over a period of 6 hours. Due to the variable size of spores produced by CBS277.49, growth kinetics was determined separately from small (5 × 3 µm) and large sporangiospores (13 × 9 µm) and from sporangiospores of NRRL3631, which show homogeneous size (5 × 3 µm), following the procedure for sporangiospores fractioning indicated in animal model section.
For cell wall-associated sensitivity assays, 10 µl containing 103, 102 and 10 fresh sporangiospores were spotted onto plates of Arne synthetic medium (SM) supplemented with either 100 µg/mL calcofluor white (CFW) or 0.01% sodium dodecyl sulphate (SDS), and incubated at 30°C for 2 days. For heat stress assay, SM plates inoculated with the same procedure previously described were incubated at 35°C and 37ºC for 5 days.
Cell wall fractioning and chitin and chitosan purification
For chitin and chitosan quantification, mycelia from both wild-type strains of M. circinelloides were grown in PDB for 24 h, filtrated, washed with distilled water and freeze-dried. The dry mycelia (3 g) was grounded on a IKA A11 grinder and treated with 0.5 M NaOH at 90ºC with shaking during 2 h, as previously described [Citation43]. The cell walls fraction obtained was collected by 10.000g centrifugation and dialyzed against distilled water (14 kDa cut-off). Subsequently, the water insoluble fraction was collected by 10.000g centrifugation and subjected to chitosan-chitin extraction by 1M HCl treatment following the previously described protocol [Citation44]. The resulting soluble (chitosan) and insoluble (chitin) fractions in HCl were collected by 10.000g centrifugation, dialyzed against distilled water (14 kDa cut-off), freeze-dried and weighted.
Nuclei acid manipulation and cloning
Total gDNA was extracted from M. circinelloides mycelium according to previously reported protocol [Citation45]. The DNA quality and quantity was determined by running aliquots in RedSafe-stained agarose gels and by spectrophotometric analysis. DNA and protein sequences were obtained from genome database (http://genome.jgi.doe.gov/vista_embed/?organism = Mucci2). Specific primers to amplify target genes construct disruption vectors and probes are indicated in . Cloning experiments were performed using pBluescript and Escherichia coli strain DH5α [Citation46].
Target gene knockout and fungal transformation
PCR reactions to construct deletion vectors were performed with Herculase II Fusion DNA Polymerase (Agilent Technologies, Santa Clara, CA) using specific primers listed in . Briefly, the 5′ and 3′ flanking sequences of the target gene were obtained by PCR amplification of CBS277.49 gDNA using specific primers with 5′ tails (30 bp) complementary to pyrG, and the resulting 1 Kb fragments were fused to the selective marker pyrG gene. The whole replacement fragment was PCR amplified using primers of the flanking regions and cloned into the vector pBluescript by blunt end ligation at Sma I site ().
Transformation was carried out on protoplasts as previously described [Citation47] and the homologous recombination events were confirmed by Southern analysis of gDNA using the indicated probes and restriction enzymes () as previously described [Citation48] using the non-isotopic digoxigenin labeling kit (Roche Applied Science).
Statistical analysis
The Mantel-Cox method was used to assess statistical significance of differences in survival among groups. Fungal burden data were expressed as mean ± standard deviation, and differences among organs were tested by two-tailed t-test. Data were analyzed using Graph Pad Prism software and the values P < 0.05 were considered statistically significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
143529_supp.zip
Download Zip (4.5 MB)Acknowledgments
The authors gratefully acknowledge Dr. F. Martin and Dr. C. Murat for their helpful advice on bioinformatics tools and the use of the facilities at INRA-Nancy research center, and Rosa María Ruiz-Vázquez and Santiago Torres-Martínez from the Universidad de Murcia for their advice in the development of this work.
Additional information
Funding
Related Research Data
References
- Torres-Narbona M, Guinea J, Martinez-Alarcon J, et al. Impact of zygomycosis on microbiology workload: a survey study in Spain. J Clin Microbiol. 2007;45(6):2051−3.
- Kontoyiannis DP, Lewis RE. How I treat mucormycosis. Blood. 2011;118(5):1216−24.
- Katragkou A, Walsh TJ, Roilides E. Why is mucormycosis more difficult to cure than more common mycoses? Clin Microbiol Infect. 2014;20(Suppl 6):74−81.
- Li CH, Cervantes M, Springer DJ, et al. Sporangiospore size dimorphism is linked to virulence of Mucor circinelloides. PLoS Pathog. 2011;7(6):e1002086.
- Arana DM, Prieto D, Roman E, et al. The role of the cell wall in fungal pathogenesis. Microbial Biotechnology. 2009;2(3):308−20.
- Stergiopoulos I, de Wit PJ. Fungal effector proteins. Ann Rev Phytopath. 2009;47:233−63.
- Kurokawa CS, Sugizaki MF, Peracoli MT. Virulence factors in fungi of systemic mycoses. Rev Inst Med Trop Sao Paulo. 1998;40(3):125−35.
- De Groot PW, Ram AF, Klis FM. Features and functions of covalently linked proteins in fungal cell walls. Fungal Genet Biol. 2005;42(8):657−75.
- Castillo L, Calvo E, Martinez AI, et al. A study of the Candida albicans cell wall proteome. Proteomics. 2008;8(18):3871−81.
- Lopez-Fernandez L, Roncero MI, Prieto A, et al. Comparative proteomic analyses reveal that Gnt2-mediated N-glycosylation affects cell wall glycans and protein content in Fusarium oxysporum. J Proteom. 2015;128:189−202.
- Bliska JB, Casadevall A. Intracellular pathogenic bacteria and fungi–a case of convergent evolution? Nature Rev. 2009;7(2):165−71.
- Seider K, Heyken A, Luttich A, et al. Interaction of pathogenic yeasts with phagocytes: survival, persistence and escape. Curr Opinion Microbiol. 2010;13(4):392−400.
- Okagaki LH, Strain AK, Nielsen JN, et al. Cryptococcal cell morphology affects host cell interactions and pathogenicity. PLoS Path. 2010;6(6):e1000953.
- Yamamoto N, Bibby K, Qian J, et al. Particle-size distributions and seasonal diversity of allergenic and pathogenic fungi in outdoor air. The ISME journal. 2012;6(10):1801−11.
- Cisse OH, Pagni M, Hauser PM. Comparative genomics suggests that the human pathogenic fungus Pneumocystis jirovecii acquired obligate biotrophy through gene loss. Gen Biol Evol. 2014;6(8):1938−48.
- Kellner M, Burmester A, Wostemeyer A, et al. Transfer of genetic information from the mycoparasite Parasitella parasitica to its host Absidia glauca. Current Genetics. 1993;23(4):334−7.
- Khaldi N, Collemare J, Lebrun MH, et al. Evidence for horizontal transfer of a secondary metabolite gene cluster between fungi. Genome Biol. 2008;9(1):R18.
- de Groot PW, Ruiz C, Vazquez de Aldana CR, et al. A genomic approach for the identification and classification of genes involved in cell wall formation and its regulation in Saccharomyces cerevisiae. Comp Func Genomics. 2001;2(3):124−42.
- Baker LG, Specht CA, Donlin MJ, et al. Chitosan, the deacetylated form of chitin, is necessary for cell wall integrity in Cryptococcus neoformans. Eukaryotic Cell. 2007;6(5):855−67.
- O'Dea EM, Amarsaikhan N, Li H, et al. Eosinophils are recruited in response to chitin exposure and enhance Th2-mediated immune pathology in Aspergillus fumigatus infection. Infect Immunity. 2014;82(8):3199−205.
- Elieh Ali Komi D, Sharma L, Dela Cruz CS. Chitin and its effects on inflammatory and immune responses. Clinic Rev Allergy Immunol. 2017;[Epub ahead of print];http://doi.org/10.1007/s12016-017-8600-0.
- Bueter CL, Lee CK, Wang JP, et al. Spectrum and mechanisms of inflammasome activation by chitosan. J Immunol. 2014;192(12):5943−51.
- Sanchez-Vallet A, Mesters JR, Thomma BP. The battle for chitin recognition in plant-microbe interactions. FEMS Microbiol Rev. 2014;39(2):171−83.
- Schwartze VU, Winter S, Shelest E, et al. Gene expansion shapes genome architecture in the human pathogen Lichtheimia corymbifera: an evolutionary genomics analysis in the ancient terrestrial mucorales (Mucoromycotina). PLoS Genet. 2014;10(8):e1004496.
- Corrochano LM, Kuo A, Marcet-Houben M, et al. Expansion of signal transduction pathways in fungi by extensive genome duplication. Curr Biol. 2016;26(12):1577−84.
- Bueter CL, Specht CA, Levitz SM. Innate sensing of chitin and chitosan. PLoS Pathog. 2013;9(1):e1003080.
- Gao S, Li Y, Gao J, et al. Genome sequence and virulence variation-related transcriptome profiles of Curvularia lunata, an important maize pathogenic fungus. BMC Genomics. 2014;15:627.
- Wöstemeyer J, Ellenberger S, Schulz E, et al. Fusion parasitism between Parasitella parasitica and its host Absidia glauca: A system between sexuality and parasitism. Endocyt Cell Res. 2016;27(3):24–32.
- Heeswijck R, Roncero M. High frequency transformation of Mucor with recombinant plasmid DNA. Calsberg Res Commun. 1984;49:691−702.
- Nicolas FE, de Haro JP, Torres-Martinez S, et al. Mutants defective in a Mucor circinelloides dicer-like gene are not compromised in siRNA silencing but display developmental defects. Fungal Genet Biol. 2007;44(6):504−16.
- Dixon DM, Polak A, Walsh TJ. Fungus dose-dependent primary pulmonary aspergillosis in immunosuppressed mice. Infect Immun. 1989;57(5):1452−6.
- Fernandez-Molina JV, Abad-Diaz-de-Cerio A, Sueiro-Olivares M, et al. Rapid and specific detection of section Fumigati and Aspergillus fumigatus in human samples using a new multiplex real-time PCR. Diagn Microbiol Infect Dis. 2014;80(2):111−8.
- Lopez-Fernandez L, Ruiz-Roldan C, Pareja-Jaime Y, et al. The Fusarium oxysporum gnt2, encoding a putative N-acetylglucosamine transferase, is involved in cell wall architecture and virulence. PloS One. 2013;8(12):e84690.
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114−20.
- Leggett RM, Ramirez-Gonzalez RH, Clavijo BJ, et al. Sequencing quality assessment tools to enable data-driven informatics for high throughput genomics. Front Genet. 2013;4:288.
- Luo R, Liu B, Xie Y, et al. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience. 2012;1(1):18.
- Patel RK, Jain M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PloS One. 2012;7(2):e30619.
- Swain MT, Tsai IJ, Assefa SA, et al. A post-assembly genome-improvement toolkit (PAGIT) to obtain annotated genomes from contigs. Nature Protocols. 2012;7(7):1260−84.
- Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25(14):1754−60.
- Milne I, Bayer M, Cardle L, et al. Tablet–next generation sequence assembly visualization. Bioinformatics. 2009;26(3):401−2.
- Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAM tools. Bioinformatics. 2009;25(16):2078−9.
- Carver T, Harris SR, Berriman M, et al. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics. 2012;28(4):464−9.
- Zamani A, Edebo L, Sjostrom B, et al. Extraction and precipitation of chitosan from cell wall of zygomycetes fungi by dilute sulfuric acid. Biomacromolecules. 2007;8(12):3786−90.
- Amorim RV, Souza W, Fukushima K, et al. Faster chitosan production by Mucoralean strains in submerged culture. Braz J Microbiol. 2001;32:20−3.
- Torres AM, Weeden NF, Martin A. Linkage among isozyme, RFLP and RAPD markers in Vicia faba. Theor Appl Genet. 1993;85(8):937−45.
- Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol. 1983;166(4):557−80.
- Gutierrez A, Lopez-Garcia S, Garre V. High reliability transformation of the basal fungus Mucor circinelloides by electroporation. J Microbiol Methods. 2011;84(3):442−6.
- Di Pietro A, Roncero MIG. Cloning, Expression, and Role in Pathogenicity of pg1 Encoding the Major Extracellular Endopolygalacturonase of the Vascular Wilt Pathogen Fusarium oxysporum. MPMI. 1997;11(2):91−8.