
ABSTRACT
Bacterial lipooligosaccharide (LOS) is an important virulence-associated factor, and its sialylation largely confers its ability to mediate cell adhesion, invasion, inflammation, and immune evasion. Here, we investigated the function of the Haemophilus parasuis α-2,3-sialyltransferase gene, lsgB, which determines the terminal sialylation of LOS, by generating a lsgB deletion mutant as well as a complementation strain. Our data indicate a direct effect of lsgB on LOS sialylation and reveal important roles of lsgB in promoting the pathogenicity of H. parasuis, including adhesion to and invasion of porcine cells in vitro, bacterial load and survival in vivo, as well as a contribution to serum resistance. These observations highlight the function of lsgB in mediating LOS sialylation and more importantly its role in H. parasuis infection. These findings provide a more profound understanding of the pathogenic mechanism of this disease-causing bacterium.
Introduction
Haemophilus parasuis is a small, non-motile, pleomorphic rod-shaped, and nicotinamide adenine dinucleotide (NAD)-dependent bacterium in the Haemophilus genus of the family Pasteurellaceae [Citation1]. As a commensal pathogen, H. parasuis colonizes the upper respiratory tract of healthy pigs and forms a symbiotic relationship with the host [Citation1]. When the host suffers from environmental stresses or reduced immunity, it can spread to the lungs and cause pneumonia or even get into the blood circulation and invade multiple tissues to cause systemic Glässer’s disease, characterized by fibrinous polyserositis, polyarthritis and meningitis in pigs [Citation2–Citation4].
To date, 15 serotype strains of H. parasuis with diverse virulence have been described, but the virulence factors of this bacterium are not completely defined. Early studies described a neuraminidase activity located in the outer membrane of H. parasuis [Citation5,Citation6]. This indicates a new antigenic epitope of H. parasuis, but the detailed biological function of sialylation in this bacterium is unknown. Sialylation is formed by specific cellular sialyltransferase-mediated transfer of sialic acids to carbohydrate chains [Citation7]. The sialyltransferases catalyze the transfer of a sialic acid residue from a cytidine monophosphate (CMP)-activated donor substrate, typically cytidine monophosphate-N-acetylneuraminic acid (CMP-Neu5Ac), onto a carbohydrate receptor, which is usually a galactose residue, or N-acetyl residue (GalNAc) or other sialic acid [Citation8]. Sialylated lipooligosaccharide (LOS) is widely present in bacteria, including Neisseria gonorrhoeae, Campylobacter, Pasteurella and Luminescent Bacteria [Citation9–Citation12]. Sialylation is not only involved in the stability of protein structure, but also in extension of the half-life of glycoproteins; moreover, it is an important component in many biological processes, such as cell recognition [Citation13], leukocyte homing [Citation14], cell adhesion and invasion [Citation15] and cell signaling [Citation16]. The genes involved in sialic acid metabolism have been investigated in 21 H. parasuis strains from different clinical origins (including nasal and systemic isolates). These genes include the neuraminidase gene nanH, the CMPNeu5Ac synthetase, and the sialyltransferase genes neuA, siaB and lsgB [Citation17]. Commonly, the sialyltransferase has been classified into α-2,3-sialyltransferase, α-2,6-sialyltransferase and α-2,8-sialyltransferase [Citation18]. Based on their protein sequence homology, all the sialyltransferases identified to date could be divided into six glycosyltransferase (GT) families GT4, GT29, GT38, GT42, GT52 and GT80 according to the Carbohydrate-Active enZymes (CAZy) (http://www.cazy.org/) database [Citation19]. Noticeably, all sialyltransferases from eukaryotes and some viruses are grouped into GT29 families, while bacterial sialyltransferases are grouped into GT4, GT38, GT42, GT52 and GT80 families [Citation18]. The α-2,3-sialyltransferase encoded by H. parasuis lsgB gene has been grouped into GT38 family [Citation20]. This lsgB gene was predominantly present in the systemic H. parasuis isolates but was not present in any of the nasal isolates [Citation17], suggesting that lsgB-mediated sialylation is likely to affect bacterial pathogenicity. However, the influence of sialylation in H. parasuis pathogenicity is not clear and its mechanism of action remains to be determined.
In this study, we investigated the function of the α-2,3-sialyltransferase gene, lsgB, in H. parasuis serotype 5 virulent strain SH0165 by generating a lsgB deletion mutant and its complementation strain. We provide evidence that H. parasuis lsgB plays an important role in LOS terminal sialylation, bacterial pathogenicity, adhesion and invasion, and resistance against the complement alternative pathway-mediated bactericidal effect in the serum. Characterization of sialylation in H. parasuis expands understanding of bacterial infection and contributes to the elucidation of H. parasuis pathogenic mechanisms.
Results
H. parasuis strain SH0165 contains the conserved motifs of the sialyltransferase LsgB
To clarify the lsgB region available for deletion, we analyzed the lsgB gene locus and its flanking genes published in the SH0165 genomic sequence on the NCBI website. We found that lsgB has 10 bases that overlap with its downstream gene, HAPS-0043 (). Bacterial sialyltransferases usually contain two conserved motifs, the aspartic acid/glutamic acid-aspartic acid/glutamic acid-glycine motif (E/D-E/D-G), and the histidine-proline (H-P) motif, which are involved in substrate binding and catalytic activity [Citation21]. We compared LsgB protein sequences in multiple bacterial genera, including Mannheimia vargena, Pasteurella multocida, Actinobacillus suis, Haemophilus influenzae and H. parasuis, and found that the conserved LsgB motifs, marked with red triangles in , were present in H. parasuis SH0165, as well as in the other bacteria. Thus, an in-frame gene deletion was engineered in the region containing these conserved motifs.
Figure 1. Alignment of the LsgB protein among H. parasuis and other genera. (a) Simplified depiction of the lsgB gene and its flanking genes on the H. parasuis SH0165 chromosome. (b) Analysis of LsgB protein homology among different bacterial genera, including M. vargena, P. multocida, A. suis, H. influenzae and H. parasuis using the NCBI BLAST program with the default parameters. Conserved protein sequences are labeled in yellow, and the triangles indicate the conserved motifs of the LsgB sequence.

PCR identification of the ΔlsgB mutant and its complementation strain C-lsgB
The lsgB deletion mutant was constructed in wild-type SH0165 by natural transformation, and the complementation strain, C-lsgB, was generated by introducing the whole lsgB operon in the shuttle vector pSHK3-Gm as described in Materials and Methods. As shown in , two primer pairs, P9/P10 and P11/P12, were used to detect internal and intact lsgB fragments, respectively, and the primer pair P5/P6 was used to detect the kanamycin fragment. In the ΔlsgB mutant, P9/P10 cannot amplify the inner lsgB product, and the C-lsgB strain yielded the same band as the wild-type strain (513 bp) (). When amplified with P11/P12, the ΔlsgB mutant yielded a fragment (1167 bp) that was a little bigger than that from the wild-type (969 bp), indicating successful replacement of the internal lsgB fragment by the kanamycin cassette. As expected, amplification with P11/P12 in the C-lsgB strain generated two bands that were exactly the same as those from both wild-type and the ΔlsgB strains, indicating successful complementation of the mutant lsgB gene in the C-lsgB strain (). The kanamycin cassette (894 bp) was detected in both ΔlsgB and C-lsgB strains, but not in the wild-type strain (). Together, these results indicate successful construction of the lsgB deletion mutant and its complementation strain.
Figure 2. Schematic representation and PCR identification of the lsgB gene deletion and its complementation. (a) The primers for amplification of specific target fragments are labeled. Primer pair P9/P10 was used to detect the inner lsgB gene, P11/P12 was used to amplify the intact lsgB deletion region, and P5/P6 amplified the kanamycin cassette. (b) PCR identification of the inner-lsgB region (513 bp), the intact deletion region of lsgB (969 bp or 1167 bp), and the kanamycin resistance cassette (894 bp).
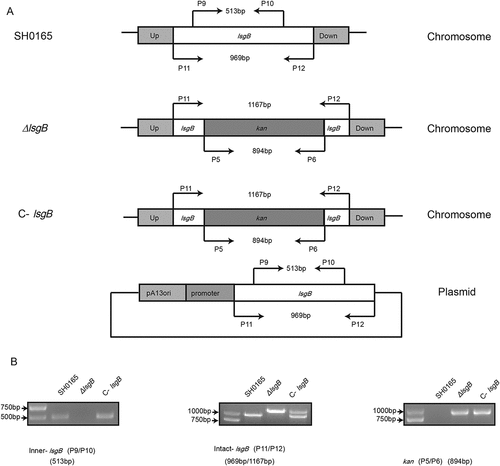
Deletion of the lsgB gene in H. parasuis leads to elongated rod shape morphology and terminal desialylation of LOS
We first compared the growth of wild-type SH0165, ΔlsgB mutant and C-lsgB strains. As shown in , the OD600 values of the ΔlsgB mutant and C-lsgB cultures increased slightly faster than that of wild-type SH0165, but the viable bacterial counts during 16 h of growth were not significantly different among strains. We next compared morphology of the bacteria by Gram-staining and scanning electron microscopy and found that deletion of lsgB led to an elongated rod shape, while the C-lsgB strain exhibited a similar morphology as the wild-type strain ().
Figure 3. Effects of lsgB deletion on bacterial growth, morphology, and LOS sialylation. (a-b) Comparison of growth among the wild-type SH0165, ΔlsgB mutant and C-lsgB strains. OD600 values of each strain during growth for 16 h were recorded every 2 h, and viable bacterial counts were calculated at 4 h intervals. (c) Both optical microscopy and scanning electron microscopy were used to observe the morphology of each strain. (d) Schematic depiction of the LOS structure and silver staining of LOS in the wild-type SH0165, ΔlsgB mutant and C-lsgB strains. (e-f) HPLC analysis of LOS sialic acid content in the SH0165, ΔlsgB and C-lsgB strains. The target peak was detected by fluorescence at appropriate time points based on the results from a standard substance (e), and the peak area ratios were compared (f).
lsgB determines the terminal sialylation of LOS (); therefore, we next extracted the LOS from each strain and compared potential differences via SDS-PAGE with silver staining. We observed a slight difference in the molecular weight of LOS, which was around 14.7 kDa in the ΔlsgB mutant, but slightly larger in both the wild-type and C-lsgB strains (around 15 kDa) (). This observation indicated differences in sialic acid content among these strains. Subsequently, HPLC was applied to quantitatively analyze the difference of sialic acid content. The results showed that total sialic acid content in the ΔlsgB mutant was significantly lower than that in the wild-type strain, while lsgB complementation could significantly increase the sialic acid content in the ΔlsgB mutant, although it was still much lower than the wild-type SH0165 (). These observations revealed an important role of lsgB in maintaining the sialylation of LOS, and deletion of lsgB led to a molecular weight reduction and the desialylation of LOS.
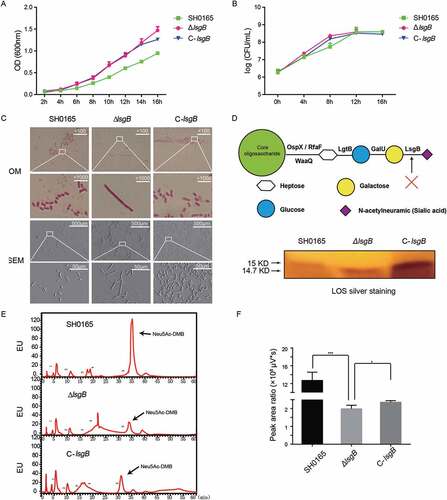
lsgb deletion in H. parasuis attenuates bacterial autoagglutination, pathogenicity, and in vivo colonization
Bacterial autoagglutination is a potential virulence-associated indicator in some Gram-negative bacteria [Citation22]; therefore, we next considered whether lsgB gene deletion affects bacterial virulence. In the autoagglutination test, the upper suspension of the static ΔlsgB culture rapidly decreased with time () and most bacteria had precipitated to the bottom of the culture after incubation for 6 h (), indicating significantly attenuated autoagglutination of the ΔlsgB mutant compared with the wild-type and C-lsgB strains.
Figure 4. H. parasuis lsgB deletion attenuates bacterial autoagglutination and pathogenicity, and colonization and survival in vivo. (a-b) OD600 values of the upper suspension of each strain were measured and compared every 30 min (a), and the culture suspension after 6 h of static incubation was photographed (b). (c) Virulence test of the wild-type SH0165, ΔlsgB mutant and C-lsgB strains by comparing survival of mice post infection. Each group contained 10 mice, and survival was monitored for 7 days. (d) Bacterial colonization in spleen and lung and survival in the blood were compared in an 8 h challenge assay. Bacterial counts were determined by serial dilution and plating and are expressed as logCFU/mL.
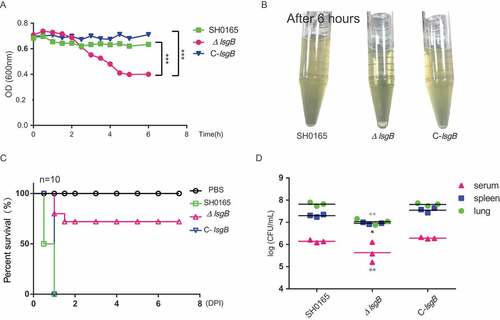
Moreover, these strains were intraperitoneally injected into mice and their in vivo pathogenicity was analyzed by monitoring the survival rates of the mice. After 12 h post challenge, only 50% of the mice challenged by the wild-type strain had survived, and all mice in both wild-type and the C-lsgB groups died within one day post infection. However, mice in the ΔlsgB challenge group had a survival rate of 75% at the end of the observation period. Mice receiving PBS injection were alive throughout the experiment (). We also compared the in vivo bacterial load of the wild-type SH0165, ΔlsgB mutant, and C-lsgB strains in mice in an 8 h infection assay. The bacterial load of the ΔlsgB mutant in the spleen and lung and its survival in serum was significantly reduced compared with that of the wild-type strain. The recovered C-lsgB strain exhibited similar level as the wild-type strain (). These results suggest that deletion of lsgB in H. parasuis largely attenuated its pathogenicity.
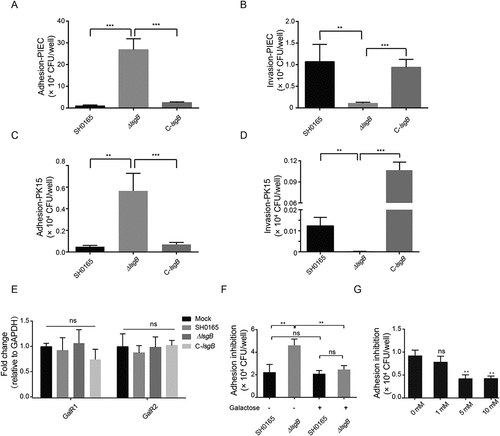
Desialylation in the ΔlsgB mutant enhances bacterial adhesion by exposure of its galactose residues
We have demonstrated in vivo attenuated pathogenicity of the ΔlsgB mutant compared to the wild-type and complementation strains. Therefore, we next investigated the in vitro adhesion and invasion abilities of these strains on PK15 and PIEC cells. As shown in , the invasion of the ΔlsgB mutant in both cell types was significantly lower than that of the wild-type and C-lsgB strains (), while unexpectedly, adhesion to both cell types by the ΔlsgB mutant was significantly higher than that of the wild-type and C-lsgB strains (). Since lsgB deletion led to LOS terminal desialylation as well as the galactose residues exposure, we next determined the transcriptional change of the cellular galactose receptors GalR1 and GalR2 in response to these strains. We did not observe the differential expression of these two receptors in response to the wild-type SH0165, ΔlsgB mutant, and the C-lsgB strain (), however, when cells were pretreated with galactose, the high level of adhesion by ΔlsgB mutant was significantly decreased, showing a similar level as that of the wild-type SH0165 (), indicating that addition of galactose might block the galactose receptors, thus preventing the LOS terminal galactose-mediated bacterial adhesion. Moreover, we showed dose-dependent blocking of ΔlsgB mutant adhesion by the addition of galactose (), further supporting our hypothesis that lsgB deletion in H. parasuis results in exposure of galactose, which promotes galactose-mediated bacterial adhesion to the cells. These observations indicate that lsgB deletion enhances galactose exposure and galactose-mediated adhesion to cells.
Figure 5. Effects of lsgB deletion in H. parasuis on bacterial adhesion to and invasion of porcine cells. (a-b) Adhesion to and invasion of PIECs by the wild-type SH0165, ΔlsgB mutant and C-lsgB strains. (c-d) Adhesion to and invasion of PK15 cells by the wild-type SH0165, ΔlsgB mutant and C-lsgB strains. (e) Real-time PCR detection of galactose binding receptor, GalR1 and GalR2, expression in response to the three strains. GAPDH was used as the endogenous control. (f) Elevated adhesion to cells by the ΔlsgB mutant was significantly blocked by the addition of galactose. (g) Galactose addition resulted in a dose-dependent inhibition of adhesion by the ΔlsgB mutant.
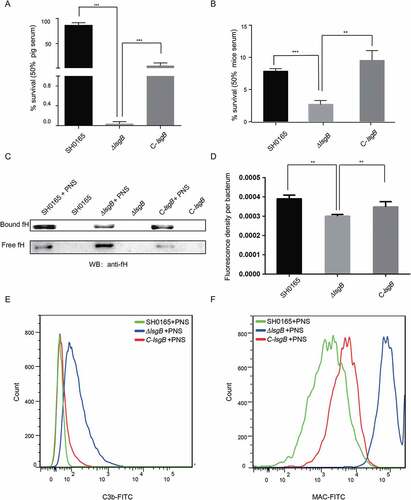
H. parasuis lsgB deletion attenuates resistance to complement system-mediated killing via decreased binding of the complement inhibitor fH and increased deposition of C3b and MAC
We observed in the mouse infection assay that the ΔlsgB mutant showed attenuated pathogenicity and decreased in vivo colonization ability, including reduced survival in serum (). We therefore questioned whether these attenuated abilities of the ΔlsgB mutant are associated with bacterial survival in the blood. Serum resistance assays with 50% porcine or mouse serum showed that the ΔlsgB mutant was much more sensitive to both sera, with the survival rate being significantly lower than those of the wild-type SH0165 and C-lsgB strains (). Moreover, these strains were incubated with or without porcine normal serum (PNS) for 1 h and their abilities of bind fH were investigated by western blotting and whole cell ELISA. After incubation, we observed that the amount of SH0165-bound fH was significantly higher than that of free fH in the supernatant. In contrast, the amount of ΔlsgB-bound fH was much lower than that of the free fH, and of SH0165-bound fH. The C-lsgB strain exhibited the same trend as the wild-type strain, showing a higher level of bound fH (). This result was further supported by the whole cell ELISA assay showing that bound-fH fluorescence density per bacterium in the ΔlsgB mutant was significantly lower than that in either the wild-type strain or the C-lsgB strain (). Complement-mediated killing relies on the deposition of C3b and formation of the MAC; therefore, we next used flow cytometry to investigate the surface deposition of these two components on the bacteria. Compared with the wild-type strain, the deposition of C3b on the ΔlsgB mutant was significantly increased after incubation with porcine normal serum for 30 min. The C-lsgB strain exhibited slightly more C3b deposition than the wild-type strain, but still less than the ΔlsgB mutant (). The deposition of MAC on these strains was similar to that of C3b, with a significantly increased deposition of MAC on the ΔlsgB mutant (). Together, these results reveal that lsgB deletion in H. parasuis leads to decreased fH binding and increased deposition of C3b and MAC on the bacteria, resulting in higher sensitivity to the serum complement system.
Figure 6. lsgB deletion increases bacterial sensitivity to serum complement-mediated killing. (a-b) Survival of the wild-type SH0165, ΔlsgB mutant and C-lsgB strains in response to 50% pig serum (a) and 50% mice serum (b). (c) Western blotting analyzing the recruitment of the complement inhibitor, fH, to each strain incubated with or without porcine normal serum (PNS). Both free fH (lower) and bound fH (upper) were all detected. (d) Whole cell ELISA was applied to analyze the bound fH on each strain. The results are presented as the average fluorescence density per bacterium. (e-f) Comparison of C3b or MAC deposition on the surface of the wild-type, ΔlsgB mutant and C-lsgB strains after incubation with PNS and flow cytometry.
Discussion
H. parasuis is recognized worldwide as one of the most important bacterial pathogens in the pig industry and is the leading cause of mortality during porcine weaning and nursery stages [Citation23]. As an important opportunistic pathogen, it normally colonizes the upper respiratory tract of healthy pigs without causing any clinical symptoms as well as inducing the production of specific antibodies [Citation24]. However, the disease is always easily to be observed under certain stress conditions especially when piglets are experiencing the immunocompromised status. For examples, maternal antibody reduction is a critical factor that allows the development of H. parasuis infection. Sow-reared piglets are usually protected against early infection of H. parasuis via acquisition of the maternal antibody [Citation25], whereas the colostrums-deprived piglets are extremely susceptible to the systemic infection [Citation26]. Selective pressure from the antibiotics sometimes induces the disease by the emergence of antimicrobial-resistance strains and the diversity of the isolates, thereby facilitating the distribution of H. parasuis [Citation27]. Additionally, things will get worse in piglets co-infected with some other respiratory pathogens or immunosuppressive pathogens, for example, swine influenza virus could aggravate the lesions to the lung and thus facilitates microbial replication in the lung [Citation28]. Despite of these external causes, H. parasuis also utilizes its internal factors for initial colonization, probably via adhesion to and invasion of host epithelial cells [Citation29–Citation31], disrupting mucosal immunity by degradation of IgA [Citation32], resisting against phagocytosis by macrophage as well as bactericidal effect by the complement system [Citation33–Citation35]. Therefore, identification and characterization of the critical virulence factors that participating in these important phenotypes should provide better understanding of H. parasuis pathogenicity. However, only a few virulence factors in this bacterium have been truly reported to date, and the knowledge regarding the pathogenesis of H. parasuis still needs expanding.
Sialic acid, a carbohydrate residue at the terminus of LPS in most Gram-negative bacteria, is one of the first molecules to directly interact with host cells during bacteria-host interactions [Citation36]. Many pathogenic bacteria, such as H. influenzae, N. gonorrhoeae, and N. meningitides, can decorate their LOS by sialylation, either by incorporating Neu5Ac into their LOS, cell membrane or capsule. Alternatively, some of the Escherichia coli strains and N. meningitidis produce capsular polysaccharides containing Neu5Ac [Citation37]. The mechanism of LOS sialylation in these microbes differs. For example, N. gonorrhoeae cannot synthesize Neu5Ac but utilizes a surface α-2, 3-sialylatransferase to transfer Neu5Ac from blood circulatory CMP-Neu5Ac to the terminal galactose residue of LOS [Citation38,Citation39]. In H. parasuis, the lsgB gene product, α-2,3-sialyltransferase, sialylates the terminal galactose of LOS by using 5ʹ-cytidinemonophospho-N-acetylneuraminic acid or other donors [Citation20]. Moreover, H. influenzae sialylates its LOS by a precursor scavenging mechanism [Citation40], and an analogous strategy has also been reported in H. ducreyi [Citation41] and H. somnus [Citation42]. These observations indicate that LOS sialylation is an important modification in members of the Haemophilus genus.
To reveal the biological function of sialic acid in LOS modification in H. parasuis, we constructed an α-2,3-sialyltransferase lsgB gene deletion mutant, and its complementation strain. The basic characteristics of these strains were compared, including growth, morphology, and LOS sialylation. The viable bacterial counts among these strains remained the same during 16 h of culture, but the OD600 values of the ΔlsgB mutant and C-lsgB strains increased faster than that of the wild-type strain. We assumed that this difference might result from an elongated shape of the ΔlsgB bacteria, which was demonstrated by optical microscopy and SEM. However, this assumption does not apply to the complementation strain because the rod-shape of these cells was not elongated, indicating that the lsgB gene complementation might not fully revert all wild-type phenotypes. On one hand, the alteration of some phenotypes resulting from gene deletion might be irreversible; and on the other hand, the higher abundance of the lsgB gene in the C-lsgB strain might account for inconsistencies between the wild-type and complementation strains. Moreover, we investigated specific differences in LOS from these three strains and found that lsgB deletion in H. parasuis caused a molecular weight reduction of LOS, and correspondingly LOS sialylation in the ΔlsgB mutant was significantly lower than in the wild-type and C-lsgB strains. This reveals that the lsgB gene is essential for LOS sialylation in H. parasuis.
Self-autoagglutination is an indirect indicator of bacterial colonization and virulence in vivo [Citation22,Citation43]. Here, we observed that the autoagglutination ability of the ΔlsgB mutant was significantly decreased, and we also demonstrated a significant attenuation of in vivo pathogenicity, as well as reduced colonization and survival of the ΔlsgB mutant, compared to the wild-type and complementation strains. To further dissect these differences, we subsequently investigated in vitro adhesion and invasion abilities on two types of porcine cell, PIEC and PK15 cells. LOS is important in host cell adhesion and invasion, which is an important mechanism for H. parasuis infection [Citation30]. Also, the lsgB gene is reported to be present predominantly in systemic clinical isolates but not in nasal isolates [Citation17], indicating a potential relationship in H. parasuis strains between the lsgB gene and adhesion or invasion. The ΔlsgB mutant exhibited significantly attenuated invasion of both PIEC and PK15 cells; however, unexpectedly; we observed a prominent and significant increase in the adhesion of the ΔlsgB mutant to both cell types. Deletion of lsgB led to the exposure of the penultimate galactose of LOS; therefore, we tested whether these cell types have differences in the expression of the galactose binding domains or receptors, such as the galactose binding lectin domain (GalR) of the adhesion G protein-coupled receptor [Citation44,Citation45], but the results did not suggest any difference on the expression of GalR1 or GalR2 in response to these strains. However notably, our adhesion inhibition assays showed that addition of galactose significantly blocked adhesion of the ΔlsgB mutant in a dose-dependent manner, while adhesion of the wild-type strain was completely unaffected, revealing that it is the terminal galactose residue, which is exposed after the removal of sialic acid by lsgB deletion (see ), that mediates the high level of adhesion.
We also compared the serum survival of the wild-type, ΔlsgB mutant, and C-lsgB strains, and found that deletion of lsgB, which results in desialylation of LOS in H. parasuis, significantly attenuated bacterial resistance against serum-mediated killing. Bacterial serum resistance is also recognized to be an important virulence indicator by assisting with bacterial survival in host blood. A previous study has also reported the contribution of LOS sialylation to serum resistance in N. gonorrhoeae [Citation46]. This sialylation of N. gonorrhoeae LOS converts serum-sensitive strains to serum resistant strains and requires the exogenous addition of CMP-Neu5Ac and has been termed “unstable serum resistance” [Citation47]. Several hypotheses concerning the mechanism of this “unstable serum resistance” have been raised, such as decreased binding of IgG, or increased binding of host complement alternative pathway inhibitor, fH [Citation48]. Here, we observed that the binding of fH by ΔlsgB mutant was significantly less than that by both wild-type SH0165 and C-lsgB strains, indicating that the H. parasuis lsgB gene can facilitate bacterial binding of fH in response to serum challenge. Together with the attenuated inhibition of the host complement system, we also observed more deposition of C3b and MAC on the surface of the ΔlsgB mutant. C3b and MAC are key players in the activation of complement-mediated killing; therefore, the desialylation of LOS, because of lsgB deletion, resulted in a higher activation of the host complement system, and an enhanced bactericidal effect. There is an additional possibility that sialylated LOS masks epitopes that are usually recognized by pentameric IgM and that restrict the recognition of C1q and the following activation of the classical pathway in the complement system [Citation49]. Whether LOS desialylation following lsgB deletion partially facilitates the engagement of IgM with certain surface epitopes in H. parasuis, thus boosting the classical complement pathway-mediated killing, should be further investigated.
In summary, we characterized the function of the lsgB gene in H. parasuis by generating its deletion mutant and the complementation strain. We provide evidence supporting the involvement of lsgB in LOS terminal sialylation, bacterial autoagglutination, and pathogenicity in vivo and in vitro. Strikingly, we found the lsgB deletion significantly increased bacterial adhesion to host cells by enhancing exposure of the galactose residue and its interaction with cellular galactose binding domains. Furthermore, the ΔlsgB mutant attenuated its binding with fH and recruited the deposition of C3b and MAC, thereby activating complement-mediated elimination of the bacteria. Characterization of lsgB-mediated LOS sialylation in H. parasuis should deepen our understanding of lsgB gene function and help to reveal the pathogenic mechanism of infection by this bacterium.
Materials and methods
Bacterial strains, plasmids and cell culture conditions
The bacterial strains and plasmids used in this study are listed in . All plasmids were propagated in Escherichia coli DH5α growing in Luria-Bertani medium (Oxoid, Hampshire, UK) at 37°C. H. parasuis clinical isolate SH0165 and its derivatives were cultured on tryptic soy agar (TSA) or in tryptic soy broth (TSB) (Difco Laboratories, Michigan, USA) supplemented with 10 μg· mL−1 NAD (Sigma-Aldrich, Missouri, USA) and 5% (v/v) inactivated bovine serum at 37°C (TSA/V/S, TSB/V/S, respectively). For selection of the plasmid-containing strains, the culture medium was supplemented with kanamycin at 50 μg· mL−1 (Kan50) or gentamicin at 20 μg· mL−1 (Gen20). Porcine kidney cells (PK15) and porcine iliac artery endothelial cells (PIEC) were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA, USA) with 10% heat-inactivated fetal bovine serum in a 37°C incubator under 5% CO2 until monolayer confluence.
Table 1. Bacterial strains and plasmids used in this study.
Alignment of LsgB protein sequences
Based on available sequence information of H. parasuis SH0165, the lsgB gene encodes an α-2,3-sialyltransferase, which belongs to glycosyltransferase family 52 and modifies bacterial LOS by sialylation. Bacterial sialyltransferases in different CAZy families (GT-38 and GT-52) and pfam family 05855 usually contain two short motifs (D/E-D/E-G and HP) [Citation21]. The LsgB protein sequence of H. parasuis SH0165 was therefore compared with sequences in other congenetic Gram-negative bacteria, such as M. vargena, P. multocida, A. suis, and H. influenzae, to analyze protein homology and to identify possible conserved motifs using the National Center for Biotechnology Information (NCBI) Basic Local Alignment Search Tool (BLAST) program with the default settings [Citation50].
Construction of an lsgB deletion mutant and the complementation strain
Oligonucleotide primers used to engineer an lsgB deletion mutant and its complementation strain are listed in . The 546-bp upstream and 527-bp downstream fragments flanking lsgB were amplified from the SH0165 genome using primer pairs P1/P2 and P3/P4, respectively. Both sets of primers contained a 9-bp core DNA Uptake Signal Sequence (USS) of 5ʹ-ACCGCTTGT-3ʹ [Citation51]. The kanamycin resistance cassette (894 bp) was amplified from pSHK3 with primers P5/P6. These three fragments were then fused by overlapping extension PCR using primers P1/P4. The resulting product was cloned into pK18mobsacB at BamHI and PstI sites to obtain the recombinant plasmid, pK18-lsgB-UKD. This was introduced into H. parasuis strain SH0165 by natural transformation as previously described with slight modifications [Citation52]. Briefly, 20 μL of cAMP (8 mM) was added to 20 μL of recipient bacterial suspension in logarithmic phase (at OD600 of 1.0) and incubated for 10 min. 1 μg of the recombinant plasmid was added into this bacterial suspension and incubated for another 10 min, which was subsequently spotted on a TSA/V/S plate for 5 h at 37℃. Bacterial cells were finally collected and transferred to the TSA/V/S/Kan50 plate and incubated at 37℃ for 48 h. All the natural transformants growing on TSA/V/S/Kan50 were PCR identified with primer pairs P5/P6, P9/P10, and P11/P12. For complementation, the lsgB gene operon including the promoter and terminator regions was amplified and cloned into the E. coil – H. parasuis shuttle vector pSHK3-Gm [Citation2] to generate the complementation plasmid, pSHK3-C-lsgB. This plasmid was then introduced into the ΔlsgB mutant by electroporation under the condition of 2.5kV voltage, 5ms pulse time and 2mm electrode gap to obtain the complementation strain C-lsgB. The bacterial suspension was grown in TSB/V/S at 37°C for 5h with circular agitation and then spread onto TSA/V/S/Kan50/Gen20 plates at 37°C for 48 h. All electroporation transformants were PCR identified with the primers mentioned above ( and ).
Table 2. Sequence of the PCR primers used in this study.
Growth analysis of H. parasuis wild-type, ΔlsgB mutant and C-lsgB strains
To compare the growth of wild-type SH0165, ΔlsgB mutant, and C-lsgB strains, overnight cultures of each strain were diluted in TSB/V/S and adjusted to the optical density at 600 nm (OD600) of 1.0. The cultures were then inoculated at 1:1000 into fresh medium and incubated at 37°C with circular agitation (180 rpm· min−1) for 16 h. The OD600 of the cultures was measured at 2 h intervals and colony forming units (CFUs) were determined by serial dilution and plating at 6 h intervals. Data collection at each time point was performed three times independently.
Autoagglutination test
The autoagglutination ability of H. parasuis wild-type, ΔlsgB mutant and C-lsgB strains was evaluated as previously described with slight modifications [Citation22]. Briefly, overnight cultures of each strain were inoculated into 5mL of TSB/V/S medium and cultured at 37°C for 10 h. Bacterial cells were collected and diluted in fresh medium to OD600 0.7, and the mixture was subsequently statically incubated at 25°C. The OD600 values of the upper suspensions were measured every 30 min during 6 h of observation.
Lipooligosaccharide analysis
LOS from the wild-type SH0165, ΔlsgB mutant and C-lsgB strains was extracted using the phenol-water method as previously described with slight modifications [Citation53]. Briefly, overnight bacterial cells were collected and resuspended in sterile deionized water at a ratio of 10% (g/ml). An equivalent amount of 90% phenol solution was added and the mixture was incubated in a water-bath with agitation at 68°C for 30 min and was then precipitated overnight at 4°C. This operation was repeated 4 times and the supernatant recovered was the LOS crude extract. Next, this crude extract was dialyzed to remove the phenol, and the aqueous phase was further precipitated with 70% ethanol. This precipitate was finally dissolved in the sterile deionized water and used as the LOS refined extracts. LOS preparations were then separated via sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE; 9% stacking gel and 17% separating gel) and visualized via silver staining.
Sialic acid extraction and high-performance liquid chromatography (HPLC)
Sialic acid content was quantitatively determined using a Sialic Acid Fluorescence Labeling kit according to the manufacturer’s instructions (Takara, Japan). Briefly, sialic acid from the wild-type, ΔlsgB mutant and C-lsgB strain was extracted with 50 mM HCl at 80°C for 3 hours, and 50 μL of cell-free culture supernatant was then collected by centrifugation and mixed with 200 μL 1,2-diamino-4,5-methyleneoxybenzene (DMB) at 50°C for 2.5 hours. Samples were next applied to a PALPAK Type R column which had been washed with 100% methanol and equilibrated with 30% methanol and a flow rate of 0.9 mL/min. Chromatographic analysis was performed on a Waters 2695 series (USA) instrument and the peak was detected by fluorescence (Ex.373nm, Em.488nm) at appropriate time points according to the standard substance.
Gram staining and scanning electron microscopy
Morphological observation of the wild-type, ΔlsgB mutant and C-lsgB strains was performed via Gram staining and scanning electron microscopy (SEM). Gram staining of each strain was performed following standard procedures, and SEM was conducted according to our previously described methods [Citation2].
Serum resistance assay
The porcine serum used consisted of a pool of sera collected from eleven healthy piglets (5–6 months old) that were free of Glässer’s disease and negative to H. parasuis antibodies. Mouse serum was collected from twenty specific pathogen free grade BALB/c mice and pooled. Sera were sterilized using a 0.22 μm filter and aliquots were stored at −80°C until use. Some aliquots were heat-treated at 56°C for 30 min to inactivate the complement components. The serum resistance assay was performed following an established procedure with brief modification [Citation52]. Briefly, 100 μL of bacterial suspension in logarithmic phase (approximately 2 × 108 CFU· mL−1) was mixed with either 100 μL of the normal serum (NS) or the heat-treated serum (HS) to obtain a final concentration of 50% serum. The mixture was maintained at 37°C with slight circular agitation (20 rpm· min−1) for 1 h. After incubation, the samples were serially diluted 10-fold and spotted onto plates and incubated at 37°C for 24 h and bacterial colonies counted. Results are presented as the percent survival calculated by dividing the number of bacteria treated with normal serum (NS) by the number of bacteria treated with heat-inactivated serum (HS). The assay was performed in triplicate.
In vitro adhesion and invasion assays
Both PK15 and PIEC cells were used to test the adhesion and invasion abilities of the wild-type, ΔlsgB mutant, and C-lsgB strains as previously described [Citation29,Citation31,Citation54]. For the adhesion assay, bacteria were grown overnight in 5 mL TSB/V/S at 37°C with agitation. Bacterial cells were collected, washed and resuspended in fresh cell culture medium, and added at 2 × 107 CFUs/well onto confluent cell monolayers grown in 24-well plates. The plates were incubated for up to 2 h at 37°C to allow bacterial adhesion. Cells were then washed five times with PBS to eliminate unbound bacteria and treated with 200 μL of 0.25% trypsin/EDTA. Thereafter, 800 μL of prechilled-deionized water was added to lyse the cells, and bacterial counts were determined by plating at appropriate dilutions. In adhesion inhibition tests, the cells were pretreated with D-galactose prior to the addition of bacteria.
For the invasion assay, bacteria were added to cells as described above. Subsequently, cells were washed three times to remove unbound bacteria and then incubated in medium containing 100 U· mL−1 penicillin and 10 μg· mL−1 streptomycin sulfate for a further 1 h to kill extracellular bacteria. Cells were washed and lysed as described above. The released intracellular bacteria were quantified by plating at appropriate dilutions. Each assay was conducted in triplicate.
Western blotting, whole cell ELISA, and real-time PCR
Serum factor H (fH) binding to H. parasuis was detected by western blotting and by whole cell ELISA. For Western blotting, 4 × 108 CFU bacterial cells resuspended in 180 μL of Hanks’ Balanced Salt Solution (HBSS, Mediatech, Inc., MD, USA) were incubated with 20 μL of normal porcine serum at 37°C for 1 h with moderate agitation. The mixtures were then added to HBSS buffer containing 20% sucrose and centrifuged at 10,000 × g for 3 min to separate the upper free fH from the lower bacteria-bound fH. The pellets were resuspended in 1% SDS and boiling-lysed for immunoblotting analysis with 0.45μm PVDF membrane. Blots were incubated with polyclonal anti-fH antibody (Abcam, 1:500) for 3–5 h at room temperature, and with horseradish peroxidase (HRP)-conjugated anti-goat secondary antibody (Abclonal, 1:5000). Blots were visualized using enhanced chemiluminescence solution (Bio-Rad).
For whole cell ELISA, the assay was performed as previously described with slight modifications [Citation55]. Briefly, 108 CFU of bacteria were incubated with 20 μL of normal porcine serum (60 min at 37°C) as described above for western blotting. Incubation was stopped by washing three times with chilled-HBSS containing 5 mM PMSF in a refrigerated microcentrifuge. The bacterial pellets were resuspended in 200 ml of the same buffer, and 50 μL of each sample post-incubation was added per well of a 96-well U-bottomed polystyrene microtiter plate and coated overnight at 4°C. The plates were then washed with PBS containing 0.05% Tween-20. Primary anti-fH antibody was diluted in PBS as abovementioned in Western blotting (1:500) and incubated at room temperature for 2h, and secondary Cy3-labeled anti-rabbit IgG was diluted in PBS/0.05% Tween-20 (Beyotime Biotechnology, 1:5000) and incubated for another 1h. To normalize the measurement, the result was presented as the average fluorescence of each single bacterium.
For real-time PCR, cells were challenged with the different strains for 2 h as mentioned above. Cells were immediately washed with chilled-PBS, and total RNAs were extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. cDNAs were synthesized using a PrimeScript™ RT reagent kit with gDNA Eraser (TaKaRa, Japan) according to the manufacturer’s instructions. Real-time PCR was performed with a ViiA™ 7 Real-Time PCR System using Power SYBR Green PCR Master Mix (Applied Biosystems, USA). GAPDH was amplified as an endogenous control and results were normalized to GAPDH and calculated by the 2−ΔΔCT method. The assays were repeated three times to confirm the results. Primers used for real-time PCR are listed in .
Flow cytometry analysis
Flow cytometry was applied to compare the deposition of C3b or membrane attack complex (MAC) on wild-type, ΔlsgB mutant, and C-lsgB strains. In brief, 5 × 108 CFUs of log-phase bacteria were incubated with 250 μL of 50% pig serum at 37°C for 30 min followed by extensive washes and resuspension. For detection of the MAC deposition, bacterial cells were incubated with primary antibody against MAC (Abcam, Cambridge, Massachusetts) at room temperature for 30 min, and then incubated with FITC-conjugated secondary antibody. For detection of the C3b deposition, bacterial suspension was incubated directly with FITC-conjugated anti-C3 antibody (Biolegend, California, USA) without secondary antibody incubation. Bacterial surface deposition of C3b or MAC was analyzed on a FACS Calibur Flow Cytometer (Becton Dickinson, Franklin Lakes, NJ, USA).
Virulence assays
Animal infection assays were conducted to compare the pathogenicity and the in vivo bacterial load of these strains. All procedures and handling techniques conformed to the guidelines established by the China Regulations for the Administration of Affairs Concerning Experimental Animals (1988) and Regulations for the Administration of Affairs Concerning Experimental Animals in Hubei (2005) (Project No.00263863 and Animal Welfare Assurance No.170211). Female BALB/c mice (4 weeks old) were purchased from China Three Gorges University (Hubei, China, Quality Certifcate No.42000600020031). All efforts were made to provide the ethical treatment and minimize suffering of animals employed in this study.
Mice were randomly divided into four groups of 10, and intraperitoneally injected with the different strains at a dose of 1 × 109 CFU per mouse. Clinical symptoms post infection was carefully monitored for 7 days of observation, and survival was recorded every day. In another assay, at 8 h post infection, blood was taken for serum isolation, and mice were then euthanized and perfused. The spleen and lung were harvested, weighed, homogenized, and plated to determine bacterial colony counts.
Statistical analysis
GraphPad Prism software ver. 6.0 was used for graph plotting and statistical analysis. The data were evaluated by analysis of variance (Two-way ANOVA, Dunnett’s multiple comparisons test) and Student’s t-test. A P value of < 0.05 was considered to be statistically significant (*), while p < 0.01 was considered extremely significant (**).
Abbreviations
| LOS | = | lipooligosaccharide |
| NAD | = | nicotinamide adenine dinucleotide |
| CMP | = | Cytidine monophosphate |
| MP-Neu5Ac | = | Cytidine monophosphate-N-Acetylneuraminic acid |
| TSA | = | tryptic soy agar |
| TSB | = | tryptic soy broth |
| PK15 | = | porcine kidney cells |
| PIEC | = | porcine iliac artery endothelial cells |
| OD600 | = | optical density at 600 nm |
| CFUs | = | colony forming units |
| HPLC | = | high performance liquid chromatography |
| SEM | = | scanning electron microscopy |
| DMB | = | 1,2-diamino-4,5-methyleneoxybenzene |
| NS | = | normal serum |
| HS | = | heat-treated serum |
| fH | = | factor H |
| HBSS | = | Hanks’ Balanced Salt Solution |
| MAC | = | membrane attack complex |
| GalR | = | galactose binding lectin domain |
| Kan | = | kanamycin |
| Gm | = | gentamicin |
Acknowledgments
We thank Jeremy Allen, PhD, from Liwen Bianji, Edanz Group China for editing and improving the language.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Oliveira S, Pijoan C. Haemophilus parasuis: new trends on diagnosis, epidemiology and control. Vet Microbiol. 2004;99:1–12.
- Huang J, Wang X, Cao Q, et al. ClpP participates in stress tolerance and negatively regulates biofilm formation in Haemophilus parasuis. Vet Microbiol. 2016;182:141–149.
- Cai X, Chen H, Blackall PJ, et al. Serological characterization of Haemophilus parasuis isolates from China. Vet Microbiol. 2005;111:231–236.
- Rubies X, Kielstein P, Costa L, et al. Prevalence of Haemophilus parasuis serovars isolated in Spain from 1993 to 1997. Vet Microbiol. 1999;66:245–248.
- Lichtensteiger CA, Vimr ER. Purification and renaturation of membrane neuraminidase from Haemophilus parasuis. Vet Microbiol. 2003;93:79–87.
- Lichtensteiger CA, Vimr ER. Neuraminidase (sialidase) activity of Haemophilus parasuis. FEMS Microbiol Lett. 1997;152:269–274.
- Honke K, Taniguchi N. Sulfotransferases and sulfated oligosaccharides. Med Res Rev. 2002;22:637–654.
- Harduin-Lepers A, Vallejo-Ruiz V, Krzewinski-Recchi MA, et al. The human sialyltransferase family. Biochimie. 2001;83:727–737.
- Brockhausen I. Crossroads between bacterial and mammalian glycosyltransferases. Front Immunol. 2014;5:492.
- Houliston RS, Vinogradov E, Dzieciatkowska M, et al. Lipooligosaccharide of Campylobacter jejuni: similarity with multiple types of mammalian glycans beyond gangliosides. J Biol Chem. 2011;286:12361–12370.
- Whitfield C, Trent MS. Biosynthesis and export of bacterial lipopolysaccharides. Annu Rev Biochem. 2014;83:99–128.
- Yuki N, Odaka M. Ganglioside mimicry as a cause of Guillain-Barre syndrome. Curr Opin Neurol. 2005;18:557–561.
- Varki A. Glycan-based interactions involving vertebrate sialic-acid-recognizing proteins. Nature. 2007;446:1023–1029.
- Rosen SD. Ligands for L-selectin: homing, inflammation, and beyond. Annu Rev Immunol. 2004;22:129–156.
- Stencel-Baerenwald JE, Reiss K, Reiter DM, et al. The sweet spot: defining virus-sialic acid interactions. Nat Rev Microbiol. 2014;12:739–749.
- Lopez PH, Schnaar RL. Gangliosides in cell recognition and membrane protein regulation. Curr Opin Struct Biol. 2009;19:549–557.
- Martinez-Moliner V, Soler-Llorens P, Moleres J, et al. Distribution of genes involved in sialic acid utilization in strains of Haemophilus parasuis. Microbiology. 2012;158:2117–2124.
- Li Y, Chen X. Sialic acid metabolism and sialyltransferases: natural functions and applications. App Microbiol Biotechnol. 2012;94:887–905.
- Cantarel BL, Coutinho PM, Rancurel C, et al. The carbohydrate-active enzymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 2009;37:D233–D238.
- Xu Z, Yue M, Zhou R, et al. Genomic characterization of Haemophilus parasuis SH0165, a highly virulent strain of serovar 5 prevalent in China. PloS One. 2011;6:e19631.
- Freiberger F, Claus H, Gunzel A, et al. Biochemical characterization of a Neisseria meningitidis polysialyltransferase reveals novel functional motifs in bacterial sialyltransferases. Mol Microbiol. 2007;65:1258–1275.
- Labandeira-Rey M, Brautigam CA, Hansen EJ. Characterization of the CpxRA regulon in Haemophilus ducreyi. Infect Immun. 2010;78:4779–4791.
- Macedo N, Rovira A, Torremorell M. Haemophilus parasuis: infection, immunity and enrofloxacin. Vet Res. 2015;46:128.
- Vahle JL, Haynes JS, Andrews JJ. Experimental reproduction of Haemophilus parasuis infection in swine: clinical, bacteriological, and morphologic findings. J Vet Diagn Invest. 1995;7:476–480.
- Pomorska-Mol M, Markowska-Daniel I, Rachubik J, et al. Effect of maternal antibodies and pig age on the antibody response after vaccination against Glässers disease. Vet Res Commun. 2011;35:337–343.
- Oliveira S, Pijoan C, Morrison R. Evaluation of Haemophilus parasuis control in the nursery using vaccination and controlled exposure. J Swine Health Prod. 2004;12:123–128.
- Olvera A, Cerda-Cuellar M, Nofrarias M, et al. Dynamics of Haemophilus parasuis genotypes in a farm recovered from an outbreak of Glässer’s disease. Vet Microbiol. 2007;123:230–237.
- Pomorska-Mol M, Dors A, Kwit K, et al. Coinfection modulates inflammatory responses, clinical outcome and pathogen load of H1N1 swine influenza virus and Haemophilus parasuis infections in pigs. BMC Vet Res. 2017;13:376.
- Bouchet B, Vanier G, Jacques M, et al. Interactions of Haemophilus parasuis and its LOS with porcine brain microvascular endothelial cells. Vet Res. 2008;39:42.
- Bouchet B, Vanier G, Jacques M, et al. Studies on the interactions of Haemophilus parasuis with porcine epithelial tracheal cells: limited role of LOS in apoptosis and pro-inflammatory cytokine release. Microb Pathog. 2009;46:108–113.
- Frandoloso R, Martinez-Martinez S, Gutierrez-Martin CB, et al. Haemophilus parasuis serovar 5 Nagasaki strain adheres and invades PK-15 cells. Vet Microbiol. 2012;154:347–352.
- Mullins MA, Register KB, Bayles DO, et al. Haemophilus parasuis exhibits IgA protease activity but lacks homologs of the IgA protease genes of Haemophilus influenzae. Vet Microbiol. 2011;153:407–412.
- Cerda-Cuellar M, Aragon V. Serum-resistance in Haemophilus parasuis is associated with systemic disease in swine. Vet J. 2008;175:384–389.
- Olvera A, Ballester M, Nofrarias M, et al. Differences in phagocytosis susceptibility in Haemophilus parasuis strains. Vet Res. 2009;40:24.
- Costa-Hurtado M, Ballester M, Galofre-Mila N, et al. VtaA8 and VtaA9 from Haemophilus parasuis delay phagocytosis by alveolar macrophages. Vet Res. 2012;43:57.
- Angata T, Varki A. Chemical diversity in the sialic acids and related alpha-keto acids: an evolutionary perspective. Chem Rev. 2002;102:439–469.
- Hood DW, Cox AD, Gilbert M, et al. Identification of a lipopolysaccharide alpha-2,3-sialyltransferase from Haemophilus influenzae. Mol Microbiol. 2001;39:341–350.
- Gao L, Linden L, Parsons NJ, et al. Uptake of metabolites by gonococci grown with lactate in a medium containing glucose: evidence for a surface location of the sialyltransferase. Microb Pathog. 2000;28:257–266.
- Mandrell RE, Lesse AJ, Sugai JV, et al. In vitro and in vivo modification of Neisseria gonorrhoeae lipooligosaccharide epitope structure by sialylation. J Exp Med. 1990;171:1649–1664.
- Vimr E, Lichtensteiger C. To sialylate, or not to sialylate: that is the question. Trends Microbiol. 2002;10:254–257.
- Schilling B, Goon S, Samuels NM, et al. Biosynthesis of sialylated lipooligosaccharides in Haemophilus ducreyi is dependent on exogenous sialic acid and not mannosamine. Incorporation studies using N-acylmannosamine analogues, N-glycolylneuraminic acid, and 13C-labeled N-acetylneuraminic acid. Biochemistry. 2001;40:12666–12677.
- Inzana TJ, Glindemann G, Cox AD, et al. Incorporation of N-acetylneuraminic acid into Haemophilus somnus lipooligosaccharide (LOS): enhancement of resistance to serum and reduction of LOS antibody binding. Infect Immun. 2002;70:4870–4879.
- Janda JM, Oshiro LS, Abbott SL, et al. Virulence markers of mesophilic aeromonads: association of the autoagglutination phenomenon with mouse pathogenicity and the presence of a peripheral cell-associated layer. Infect Immun. 1987;55:3070–3077.
- Hamann J, Aust G, Arac D, et al. International union of basic and clinical pharmacology. XCIV. Adhesion G protein-coupled receptors. Pharmacol Rev. 2015;67:338–367.
- Uenishi H, Eguchi-Ogawa T, Shinkai H, et al. PEDE (Pig EST Data Explorer) has been expanded into Pig Expression Data Explorer, including 10 147 porcine full-length cDNA sequences. Nucleic Acids Res. 2007;35:D650–D653.
- Parsons NJ, Patel PV, Tan EL, et al. Cytidine 5ʹ-monophospho-N-acetyl neuraminic acid and a low molecular weight factor from human blood cells induce lipopolysaccharide alteration in gonococci when conferring resistance to killing by human serum. Microb Pathog. 1988;5:303–309.
- Lewis LA, Gulati S, Burrowes E, et al. alpha-2,3-sialyltransferase expression level impacts the kinetics of lipooligosaccharide sialylation, complement resistance, and the ability of Neisseria gonorrhoeae to colonize the murine genital tract. MBio. 2015;6:e02465-1.
- Ram S, McQuillen DP, Gulati S, et al. Binding of complement factor H to loop 5 of porin protein 1A: a molecular mechanism of serum resistance of nonsialylated Neisseria gonorrhoeae. J Exp Med. 1998;188:671–680.
- Zaleski A, Scheffler NK, Densen P, et al. Lipooligosaccharide P(k) (Galalpha1-4Galbeta1-4Glc) epitope of moraxella catarrhalis is a factor in resistance to bactericidal activity mediated by normal human serum. Infect Immun. 2000;68:5261–5268.
- Altschul SF, Madden TL, Schaffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402.
- Zhang B, Feng S, Xu C, et al. Serum resistance in Haemophilus parasuis SC096 strain requires outer membrane protein P2 expression. FEMS Microbiol Lett. 2012;326:109–115.
- Zhang B, He Y, Xu C, et al. Cytolethal distending toxin (CDT) of the Haemophilus parasuis SC096 strain contributes to serum resistance and adherence to and invasion of PK-15 and PUVEC cells. Vet Microbiol. 2012;157:237–242.
- Hitchcock PJ, Brown TM. Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J Bacteriol. 1983;154:269–277.
- Frandoloso R, Pivato M, Martinez-Martinez S, et al. Differences in Haemophilus parasuis adherence to and invasion of AOC-45 porcine aorta endothelial cells. BMC Vet Res. 2013;9:207.
- Ram S, Sharma AK, Simpson SD, et al. A novel sialic acid binding site on factor H mediates serum resistance of sialylated Neisseria gonorrhoeae. J Exp Med. 1998;187:743–752.