
ABSTRACT
Current treatment for combatting Chagas disease, a life-threatening illness caused by the kinetoplastid protozoan parasite Trypanosoma cruzi is inadequate, and thus the discovery of new antiparasitic compounds is of prime importance. Previous studies identified the indirubins, a class of ATP kinase inhibitors, as potent growth inhibitors of the related kinetoplastid Leishmania. Herein, we evaluated the inhibitory activity of a series of 69 indirubin analogues screened against T. cruzi trypomastigotes and intracellular amastigotes. Seven indirubins were identified as potent T. cruzi inhibitors (low μΜ, nM range). Cell death analysis of specific compounds [3'oxime-6-bromoindirubin(6-BIO) analogues 10, 11 and 17, bearing a bulky extension on the oxime moiety and one 7 substituted analogue 32], as evaluated by electron microscopy and flow cytometry, showed a different mode of action between compound 32 compared to the three 6-BIO oxime- substituted indirubins, suggesting that indirubins may kill the parasite by different mechanisms dependent on their substitution. Moreover, the efficacy of four compounds that show the most potent anti-parasitic effect in both trypomastigotes and intracellular amastigotes (10, 11, 17, 32), was evaluated in a mouse model of T. cruzi infection. Compound 11 (3ʹpiperazine-6-BIO) displayed the best in vivo efficacy (1/6 mortality, 94.5% blood parasitaemia reduction, 12 dpi) at a dose five times reduced over the reference drug benznidazole (20 mg/kg vs100 mg/kg). We propose 3ʹpiperazine-6-BIO as a potential lead for the development of new treatments of Chagas disease.
Introduction
Chagas disease, or American trypanosomiasis, is endemic in 21 countries in the Central and South America and is considered the most deadly parasitic disease in Latin America, being worldwide estimated that 7–8 million people are infected with Trypanosoma cruzi, its causative agent [Citation1]. The disease is increasing in other continents due to population migration. There is no human vaccine against Chagas disease, and the current chemotherapy available is based on nifurtimox or benznidazole [Citation2]. These drugs show good efficacy when given during the acute phase, up to 2 to 6 months after infection. About 30% of the chronic cases will develop cardiovascular disease, while 10% will manifest neurological or/and gastrointestinal problems [Citation3]. As there are serious drawbacks of current treatments, especially their toxicity, new effective treatments for this disease are urgently needed.
Indirubin is a vivid red compound found in plants and molluscs. It is a minor ingredient of indigo dye derived from plants (Isatis spp., Indigofera spp., Polygonum spp.), and the infamous Tyrian purple prepared since antiquity from molluscs of the family Muricidae [Citation4]. In addition, indirubin as well as indigo are produced as a result of bacterial metabolism, a phenomenon that is evident in the clinical condition of Purple Urine Bag Syndrome (PUBS), where the urine of patients with a urinary tract infection turn purple [Citation5]. More importantly, indirubin was found to be the active constituent of the Traditional Chinese Medicine preparation Danggui Longhui Wan, used to treat leukemia [Citation4].
The basic indirubin scaffold can be modified to generate analogue compounds by different synthetic and therapeutic processes [Citation6]. Indirubins have shown to possess several biological properties, including anticancer [Citation7–Citation9], antiviral [Citation10,Citation11], as stem cell regulators [Citation12,Citation13], neuroprotective [Citation14,Citation15] and anti-inflammatory agent for psoriasis [Citation16] and endotoxemia [Citation17] by targeting different kinases such as glycogen synthase kinase 3 (GSK3), cyclin dependent kinases (CDKs) and dual-specificity tyrosine-regulated kinases (DYRKs) [Citation18–Citation21]. Additionally, they possess antiparasitic activity against the trypanosomatids Leishmania and Trypanosoma brucei, by inhibiting kinases, including Leishmania (L) glycogen synthase kinase 3 short (GSK-3s) [Citation22] and T. brucei (Tb) GSK-3s kinases [Citation23], respectively.
Since T. cruzi belongs to the same family of trypanosomatids, such as Leishmania and T. brucei [Citation24], in this study we performed a screening to identify novel T. cruzi inhibitors amongst 69 compounds of an in house indirubin collection (Table S1). The identified inhibitors were further studied to evaluate their mode of action by determining the mechanism of cell death using electron microscopy and FACS analysis. The results obtained highlighted the different mode of action between the 6- and 7- substituted indirubins. Finally, the efficacy of selected indirubins was evaluated in a murine model of T. cruzi infection.
Results
Trypanocidal and cytotoxic activities
The trypanocidal activity of the 69 indirubins was initially evaluated against trypomastigotes (Y strain T. cruzi) through the determination of EC50 values. First, the collection of 69 indirubins (Table S1) was screened against the trypomastigote form of T. cruzi parasites at 10 μΜ and 1 μΜ. In the initial screening, only 7 compounds were found to cause at least 50% growth inhibition in T. cruzi parasites at both concentrations tested (data not shown) and subsequently their EC50 values was determined. Thus, 7 out of the 69 compounds showed a potent anti-T. cruzi activity, being able to kill trypomastigotes with EC50 values ranging from 0.25 to 1.3 μM (compounds 5, 10–13, 17 and 32, ). The standard drug benznidazole presented an EC50 of 11.3 µM. The 7 identified compounds active against T. cruzi trypomastigotes included six 3’substituted 6-BIO (5, 10–13 and 17) and one 7-substituted (32) indirubins. Compounds 5, 12 and 13 displayed high cytotoxicity against murine macrophages J774.1 (). In contrast, compounds 10, 11, 17 and 32, which had lower cytotoxicity against macrophages, were further investigated for their anti-trypanosomal activity against the intracellular amastigote form of the parasites. Importantly, the EC50 values for the amastigotes were also in the low micromolar/nanomolar range, between 0.22–1.51 μΜ and compounds displaying selectivity index (SI) values above 6.6 (; Figure S1).
Table 1. Cytotoxicity against murine macrophages J774.1 and anti-Trypanosoma cruzi activity against trypomastigote and amastigote forms of indirubins.
Investigating the mechanisms of action of indirubins
Transmission electron microscopy (TEM) was used to examine the ultrastructural alterations caused by the treatment with the indirubins 10, 11, 17 and 32. Thin sections of untreated trypomastigotes observed by TEM revealed normal appearance of organelles, intact plasma membrane and cytoplasm without alterations (). Treatment with 6-substituted indirubins 10, 11 and 17, however, caused loss of cytoplasm ()), resulted in pyknotic nuclei displaying DNA degradation (,)) and appearance of myelin figures ()). Similar alterations were observed after the treatment with the 7-substituted indirubin (32), which also caused a dilatation of Golgi cisternae ()).
Figure 1. Transmission electron microscopy of T. cruzi trypomastigotes incubated with indirubins for 24 h. (a) Untreated cells, scale bar: 1 μm. (B-F) After incubation with indirubins, we observed: (b) loss of cytoplasm after incubation with the EC50 concentration of the compound 10, scale bar: 0.5 μΜ, (c) dilatation of some Golgi cisternae after treatment with the EC50 concentration of the compound 32, scale bar: 0.2 μm, (d) DNA degradation with pyknotic nuclei after incubation with the EC50 concentration of the compound 11, scale bar: 0.5 μΜ, (e) DNA degradation with pyknotic nuclei after incubation with the EC50 concentration of the compound 17, scale bar: 0.2 μΜ and (f) appearance of myelin figures after incubation with the EC50 concentration of the compounds 10, 11, 17 and 32, scale bar: 0.5 μm. Black arrows indicate changes in the organelles.
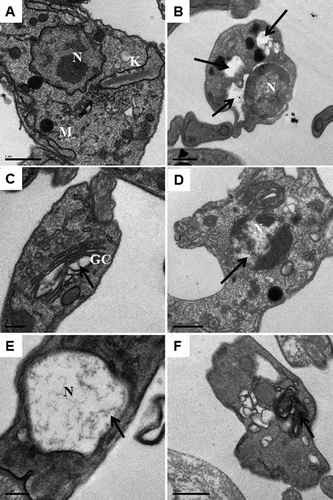
Scanning electron microscopy (SEM) was also employed to study the morphology of trypomastigotes treated or not with indirubins 10, 11, 17 and 32. Untreated trypomastigotes had the typical elongated shape of the parasite without visible alterations in the plasma membrane or in cell volume (). Trypomastigotes treated with 6-substituted indirubins 10, 11 and 17 and 7-substituted indirubin 32 displayed extensive loss of plasma membrane integrity. Representative images of trypomastigotes treated with the most potent compound herein, compound 11, at 0.25 and 0.5 µM for 24 h, are shown in ,), respectively.
Figure 2. Scanning electron microscopy of T. cruzi trypomastigotes incubated with indirubins for 24 h. Using SEM, we observed alterations in cell shape and loss of plasma membrane integrity after treatment with the EC50 and 2 x EC50 concentration of compound 11 (B and C respectively) and (A) untreated cells. White arrows indicate the alterations reported.
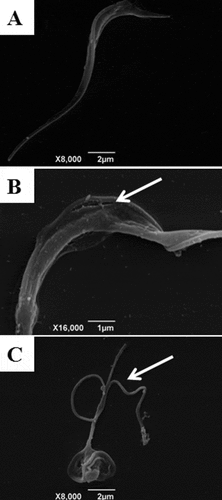
Next, to further understand the mechanism by which indirubins cause parasite death in T. cruzi, a double staining with annexin V and propidium iodide (PI) was performed in indirubin-treated parasites for 6 and 24 h followed by flow cytometry analysis. In untreated cultures, 97.6% of the cells were viable (annexin V-PI negative) (). Cultures treated with the EC50 concentration of 6-substituted indirubins showed an immediate increase in the number of double positive (apoptotic-like cells) and annexin V-negative, PI-positive (necrotic cells) populations after 6 h of treatment for indirubin 17 and after 24 h for indirubins 10, 11 and 17, without the appearance of early apoptotic-like parasites (annexin V positive-PI negative) (). These results indicate that 6-substituted indirubin treatment probably causes death by a necrotic process. On the other hand, treatment with the 7-substituted indirubin (32) caused an apoptotic-like cell death after 6 h of treatment, when we observed an increase of the early apoptotic population (12.2%), which then at 24 h progresses to late-apoptotic like and necrotic populations (27.6% and 16.2%, respectively) ().
Figure 3. Flow cytometry analysis of trypomastigotes treated with indirubins 10, 11, 17 and 32 at the EC50 concentration, upon propidium iodide (PI) and annexin-V staining. (A) Untreated trypomastigotes after 24 h of incubation; (B) trypomastigotes incubated with indirubins 10, 11, 17 and 32 for 6 and 24 h.
In vivo experiments
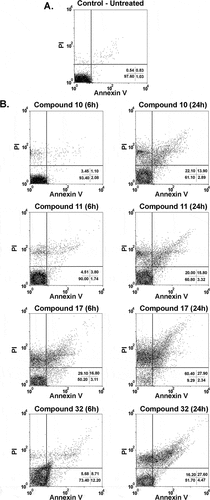
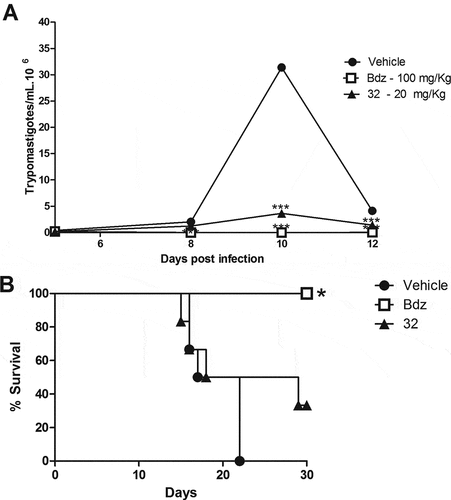
Next, the in vivo efficacy of the indirubins 10, 11, 17 and 32 was evaluated in a mouse model of acute Chagas disease. Indirubin doses of 10 or 20 mg/Kg were selected according to previous dosing regiments of other indirubin backbone analogues in mice [Citation25], and presented no signs of toxicity or mortality. More specifically, no mortality was observed during the treatment and for up to 3 months post treatment with indirubins tested at doses of 10 or 20 mg/Kg for 5 consecutive days. Mice exhibited normal behavior, good appetite and no weight loss or external signs of fatigue. Compound 11, that exhibited higher activity against macrophages (J774.1) in vitro, was further examined in vivo by evaluating the serum and urine levels of renal and hepatic function markers (Table S2), in which mice treated with this compound showed no signs of toxicity. Thus, the highest dose was selected (20 mg/kg) and administrated, as described in material and methods section. As shown in , all the 6-substituted indirubins 10, 11 and 17 tested significantly (P < 0.001) reduced blood parasitaemia (at a rate of 69.3%, 94.5% and 85%, respectively, after 12 days post infection – ) when compared to mice treated with an equal volume of vehicle (DMSO-PBS) ()). The 7- substituted indirubin, compound 32 was also shown to reduce parasitaemia but at a rate of 34.63% after 12 days post infection (, ). Treatment with benznidazole, the standard drug, at 100 mg/kg dose resulted in > 99% of inhibition of blood parasitaemia. Treatment with indirubins 11 and 17, in contrast to the indirubin 10 and 32, similar to the treatment with benznidazole, had a protective effect, reducing mortality ().
Table 2. Parasitemia and mortality in mice infected with Y Trypanosoma cruzi strain and treated daily with 20 mg/Kg of indirubins or 100 mg/Kg of benznidazole for 5 days.
Figure 4. Parasitemia and survival rate of BALB/c mice infected with T. cruzi and treated with 6-substituted indirubins. Female BALB/c mice were infected with 104 Y strain trypomastigotes. Five days after infection, mice (n = 6) were treated orally with indirubins 10, 11 and 17 (20 mg/Kg) or benznidazole (100 mg/kg) daily, during five consecutive days. (A) Parasitemia was monitored by counting the number of trypomastigotes in fresh blood samples. Values represent the mean± SEM of 6 mice per group. (B) Survival was monitored by 30 days after infection. Results are from one experiment of two performed. ** P < 0.01; *** P < 0.001 compared to untreated-infected group (vehicle).
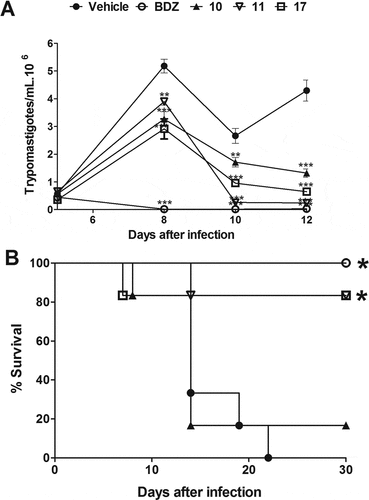
Figure 5. Parasitemia and survival rate of BALB/c mice infected with T. cruzi and treated with 7-substituted indirubin. Female BALB/c mice were infected with 104 Y strain trypomastigotes. Five days after infection, mice (n = 6) were treated orally with indirubin 32 (20 mg/Kg) or benznidazole (100 mg/kg) daily, during five consecutive days. (A) Parasitemia was monitored by counting the number of trypomastigotes in fresh blood samples. Values represent the mean± SEM of 6 mice per group. (B) Survival was monitored by 30 days after infection. Results are from one experiment of two performed. ** P < 0.01; *** P < 0.001 compared to untreated-infected group (vehicle).
Discussion
In previous studies, indirubins had been evaluated against a variety of diseases particularly in cancer and neurodegenerative disease therapy studies [Citation8,Citation26–Citation28] and have been shown to be potent inhibitors of mammalian kinases such as CDKs, GSK-3β, DYRKs and Aurora kinases [Citation4,Citation6,Citation18,Citation29]. Recently, indirubins have also drawn the attention of scientific groups working on antiparasitic drug discovery [Citation22,Citation23,Citation30,Citation31], highlighting their potential as chemotherapeutic agents against the kinetoplastid protozoans L. donovani [Citation22,Citation30] and T. brucei [Citation23]. Indirubins have been shown to target parasitic kinases, such as GSK-3 [Citation22,Citation23,Citation30], CRK3 (homologue of the mammalian CDK1) [Citation22,Citation30,Citation32] and casein kinase 1 (CK1) [Citation31].
The sequence homology of kinetoplastid kinases and the inhibitory action of indirubins on the activity of specific Leishmania and T. brucei kinases [Citation23] prompted us to further evaluate the antiparasitic activity of a 69-indirubin in-house collection against T. cruzi. Herein, our results showed that the indirubin compounds were effective inhibitors of the growth of T. cruzi parasites. Surprisingly, only 7 out of the 69 compounds showed a potent anti-T. cruzi activity, compared to 13 and 32 out of 69 that previously showed inhibitory activity against Leishmania and T. brucei, respectively [Citation22,Citation23,Citation30]. It is noteworthy that some of the most potent inhibitors of Leishmania and T. brucei parasites [Citation22,Citation23,Citation30], namely 6BIO and 5-Me-6BIO, did not exhibit any activity against the screening threshold of 10 μΜ in T. cruzi trypomastigotes. The latter observation could be due to: a) different intake of indirubins, b) different levels of expression of the proteins targeted by the compounds in the three parasites, c) different role of the protein targets between the three parasites, d) different affinity of the compounds in the ATP binding pocket of the protein-target or/and e) different protein targets in the three parasites. The identified indirubins that herein act as inhibitors of the T. cruzi trypomastigotes, however, are also potent inhibitors of Leishmania and T. brucei parasites and could serve as scaffolds for anti-kinetoplastid drug discovery.
In our study, four out of seven compounds (indirubins 10, 11, 17 and 32) were identified as inhibitors of both T. cruzi trypomastigotes and intracellular amastigotes, while having good selectivity against mammalian cells. Those compounds display lower EC50 values (EC50Try: 0.25–1.30 μΜ, EC50Ama: 0.22–1.51 μΜ) than the standard drug benznidazole (EC50 of 11.4 and 13.4 µM for trypomastigotes and intracellular amastigotes upon 24 h and 6 h of treatment, respectively) in vitro, which implies that they are potent inhibitors of the T. cruzi growth. In the literature, benznidazole is reported to inhibit T. cruzi amastigotes in vitro with EC50 values ranging from 1–15 μΜ, this variation being the result of the use of different host cell types, strains of parasites used and time of exposure to test compounds [Citation33–Citation35]. Compounds 10, 11 and 17 are 3’substituted-6BIO analogues and were identified as most potent inhibitors of Leishmania and T. brucei parasites, as well as of their parasitic kinase-targets (GKS-3s, CRK3) [Citation22,Citation23], while compound 32 is a 7-substituted indirubin. The 6-substituted compounds target T. cruzi trypomastigotes and amastigotes with EC50 values: 0.22–0.9 μΜ, while the active 7-substituted indirubin (compound 32) with an EC50 value 1.30 μΜ. As evidence exists that 3’substituted-6BIO analogues target more efficiently the CRKs and GSK-3 kinases in Leishmania, while 7-substituted are less potent inhibitors and are known inhibitors of human DYRKs and Aurora kinases [Citation18], it is possible that these selected indirubins act by inhibiting different kinases.
Differences in potency among the selected compounds were also correlated to differences in the mode of death. While all the 4 selected indirubins, namely compounds 10, 11, 17 and 32, caused the loss of the parasitic plasma membrane integrity and cytoplasm, DNA degradation with pyknotic nuclei and the appearance of myelin figures, only the 7-substituted indirubin 32 caused dilatation of Golgi cisternae. Thus, our results are suggestive of a supplementary or different mode of action or target of this compound against T. cruzi parasites.
Further investigation with the use of flow cytometry confirmed that parasitic death by 3’substituted-6BIO analogues, results in necrosis as parasites displayed rapid cell permeability and cytoplasmic swelling, associated frequently with necrotic parasitic death [Citation36,Citation37]. Similar results can be found when T. cruzi is treated with benznidazole [Citation38], whose action is strictly associated with oxidative stress induction in parasites [Citation39]. On the other hand, compound 32 causes an apoptotic-like cell death during the first 6 h of treatment, as there is an increase in the early apoptotic population (14.5%), which later on (24 h) gives rise to late-apoptotic like and necrotic populations (17.1% and 13.8%, respectively). What is worth mentioning is that the mode of death induced by 3’substituted-6BIO in T. cruzi is different from the mode of death caused by the same compounds against both Leishmania and T. brucei [Citation22,Citation23]. Conceivably, treatment of Leishmania and T. brucei by EC50 concentrations of 3’substituted-6BIO derivatives, causes apoptosis-like death, a term used here to describe the phenotype. The latter observation reinforces the suggestion that in T. cruzi there is a different target or mode of action.
Finally, through the in vivo test for the four compounds in the acute phase of the mouse model of T. cruzi infection, the 3ʹpiperazine-6BIO (compound 11) emerged as a promising scaffold for drug discovery against Chagas disease, as it reduced blood parasitaemia at a rate 94.5% after 12 days of infection and it had a protective effect on mortality rate (80% survival rate). It is interesting to note that although compounds 10 and 11 are the base and salt of the same structure, a large difference is observed in terms of the inducing mortality rate. The calculated logD for compound 10 [Citation20] is −0.87, suggesting that water solubility of this compound is probably high while lipophilicity is low, and therefore the permeability is expected to be reduced [Citation40]. In fact, treatment with 3ʹpiperazine-6BIO (compound 11) reduced blood parasitaemia (94.5%) and increase survival rate (80%) at a similar rate to benznidazole (100% survival rate), the current drug for Chagas disease, when tested in a dose of 5X lower than that of benznidazole. The benefits observed after the treatment with compound 11 in a short-term therapeutic scheme may be increased in future investigations where treatment for 20 consecutive days will be performed in the acute phase of the disease, instead of only 5 consecutive days. Moreover, compound 11 will be also tested against the chronic phase of Chagas disease in an animal model. As 3ʹpiperazine-6BIO (compound 11) is one of the most potent inhibitors against both Leishmania and T. brucei parasites, it may be used as a scaffold for future antikinetoplastid drug discovery. Combination of drugs is the new trend of combating parasitic diseases in order to prevent resistance of the therapy [Citation41,Citation42] and thus compound 11 could also serve in the future as a scaffold for designing a combinatorial therapy with benznidazole for treating Chagas disease. 3ʹpyrrolidine-6BIO (compound 17) also reduced blood parasitaemia (85%) and increase survival rate (80%) at a significant rate related to benznidazole (100% survival rate), when tested in a dose of 5X lower than that of benznidazole, which implies that the insertion of a ring (piperazine or pyrrolidine) at position 3ʹ of 6BIO could lead to more effective inhibition of T. cruzi parasites. The latter observation is in accordance to literature, where the insertion of a ring (piperazine or pyrrolidine) at position 3ʹ of 6BIO led to increased activity against Leishmania parasites in vitro by inhibiting more specifically leishmanial Glycogen Synthase Kinase-3short (LGSK3s) [Citation22]. Compound 32, a 7-substituted indirubin, presented a low inhibition of blood parasitaemia after 12 days of infection and it had a weak protective effect on mortality rate.
Altogether, these findings reinforce that indirubins are potent and selective trypanocidal agents, especially 3’substituted-6BIO analogues. Therefore, the screening for structurally-related indirubins derivative for Chagas disease treatment is an attractive line of drug development.
Materials and methods
Drugs
Indirubin derivatives (Table S1) were synthesized from isatin, as previously described [Citation19,Citation20,Citation43,Citation44]. Purity of the tested compounds was > 98%, and determined by means of HPLC-DAD. Benznidazole (LAFEPE) was used as a reference anti-T. cruzi drug. All compounds were dissolved in DMSO (Sigma-Aldrich; Catalog number: W387520) and diluted in cell culture medium for use in the assays. The final concentration of DMSO was less than 1% in all in vitro experiments and less than 20% in all in vivo experiments.
Parasites
The T. cruzi Y strain (DTU II) [Citation45] was used in this study. Tissue culture trypomastigotes were obtained from the supernatants of 5 to 6-day-old infected LLC-MK2 cells maintained in Dulbecco’s modified Eagle medium (DMEM; Catalog number: 11,965–084) supplemented with 10% fetal bovine serum (FBS; GIBCO) and 50 µg/mL of gentamycin (ThermoFisher Scientific, Catalog number: 15,750,060) at 37°C in a 5% humidified CO2 atmosphere. For in vivo analysis, bloodstream trypomastigotes used were obtained from BALB/c mice previously infected intraperitoneally with 10,000 Y trypomastigotes, and then bled after 8–12 days when they reached the peak of parasitaemia.
Animals
Female BALB/c mice (18–22 g) were provided by the animal breeding facility and maintained in sterilized cages under a controlled environment, receiving water ad libitum and a balanced diet for rodent at Instituto Gonçalo Moniz (Fundação Oswaldo Cruz, Bahia, Brazil).
Assessment of cytotoxicity to mammalian cells
Mouse macrophages (J774.1 cell line) were seeded into 96-well plates at a cell number 1 × 104 cells/well in DMEM medium supplemented with 10 % of FBS and incubated for 24 h at 37°C and 5% CO2. Samples were then added at eight concentrations (ranging from 0.078 to 10 µM) in triplicate, and incubated for 72 h, followed by the addition of 20 µL/well of Alamar Blue (Invitrogen; Catalog number: DAL1025). Colorimetric readings were performed 24 h later at 570 and 600 nm. CC50 values were calculated using data-points gathered from three independent experiments. Benznidazole was tested in concentrations ranging from 0.78 to 100 µM.
Cytotoxicity for trypomastigotes
Trypomastigotes (4x105 cells/well) were seeded into 96-well plates and the indirubins were added at eight concentrations ranging from 0.08 to 10 µM, in triplicate. Plates were incubated for 24 h at 37ºC and 5% of CO2. Aliquots of each well were collected and the number of viable parasites, based on parasite integrity and motility, was assessed in a Neubauer chamber and compared to untreated cultures. Three independent experiments were performed.
In vitro infection assay
Peritoneal exudate macrophages were obtained by washing, with cold PBS, the peritoneal cavity of BALB/c mice 4–5 days after injection of 3% thioglycolate (Sigma-Aldrich) in saline (1.5 mL per mice). Then, cells were plated at a cell density of 2 × 105 cells/well in 24-well plates with sterile coverslips on the bottom in RPMI supplemented with 10% FBS and incubated for 24 h at 37° C and 5% CO2. Cells were then infected with trypomastigotes at a ratio of 10 parasites per macrophage for 2 h. Free trypomastigotes were removed by successive washes using saline solution. Cultures were incubated in complete medium alone or with the compounds under investigation in different concentrations for 6 h. The medium was replaced with fresh medium and the plate was incubated for 3 days. Cells were fixed in absolute alcohol and the percentage of infected macrophages and the mean number of amastigotes/100 macrophages was determined by manual counting after hematoxylin and eosin staining in an optical microscope (Olympus). The percentage of infected macrophages and the relative number of amastigotes per macrophage was determined by counting 100 cells per slide.
Ultrastructural studies
Trypomastigotes at a cell density of 1 × 107 cells/mL in 24 well-plates were treated with indirubins at EC50 value, twice EC50 value or not for 24 h. Parasites were then fixed with 2% formaldehyde and 2.5% glutaraldehyde in sodium cacodylate buffer (0.1 M, pH 7.2) for 1 h at room temperature. After fixation, parasites were washed 3 times with sodium cacodylate buffer (0.1 M, pH 7.2), and post-fixed with a 1.0 % solution of osmium tetroxide containing 0.8% potassium ferrocyanide (Sigma-Aldrich; Catalog number: P3289) for 1 h. Cells were subsequently dehydrated in increasing concentrations of acetone (30, 50, 70, 90 and 100%) for 10 min at each step and embedded in polybed resin (PolyScience; Catalog number: 00552–500). Ultrathin sections on copper grids were contrasted with uranyl acetate and lead citrate and observed under a ZEISS 109 transmission electron microscope. For scanning electron microscopy, trypomastigotes treated and fixed in the same conditions were washed in 0.1 M cacodylate buffer, and allowed to adhere in coverslips pre-coated with poly-L-lysine (Sigma-Aldrich; Catalog number: P4707). Cells were then post-fixed with a solution of osmium tetroxide containing 0.8% of potassium ferrocyanide for 30 min and dehydrated in crescent concentrations of ethanol (30, 50, 70, 90 and 100%). The samples were dried until the critical point, metallized with gold and analyzed in a JEOL JSM-6390LV scanning electron microscope. Two independent experiments were performed.
Propidium iodide and annexin v staining
T. cruzi Y strain trypomastigotes (1x107) were incubated for 6 and 24 h at 37 °C in the absence or presence of indirubins (EC50 values). After incubation, the parasites were labeled for propidium iodide (PI) and annexin V using the annexin V-FITC apoptosis detection kit (Sigma-Aldrich; Catalog number: APOAF-20TST), according to the manufacturer’s instructions. Acquisition was performed using a BD FACS Calibur flow cytometer (Becton Dickinson) and data were analyzed in BD CellQuest software. A total of 10,000 events were acquired in the region previously established as that corresponding to trypomastigotes forms of T. cruzi. Three independent experiments were performed.
Infection in mice
Female BALB/c mice (18–22 g) were infected with bloodstream trypomastigotes by intraperitoneal inoculation of 104 parasites in 100 µL of saline solution and then mice were randomly divided in groups (six animals per group). Before starting the treatment on the fifth day post infection, the infection was confirmed in all animals where on average 0.4 × 106 parasites per ml of blood were found. Thus, after day 5 of infection, treatment with 20 mg/kg weight of indirubins 10, 11, 17 and 32 were given orally for five consecutive days. For the control group, benznidazole was given orally at dose of 100 mg/kg weight. Saline containing 20% DMSO was used as a vehicle and administrated on untreated and infected group. Infection was monitored daily by counting the number of motile parasites in 5 µL of fresh blood sample drawn from the lateral tail veins, as recommended by standard protocol [Citation46]. Survival of mice was monitored for 30 days after treatment.
Statistical analyses
To determine the cytotoxicity concentration 50% of J774.1 (CC50) and the effective concentration 50% (EC50) of the trypomastigotes and amastigotes forms of T. cruzi, we used nonlinear regression. The selectivity index (SI) was defined as the ratio of CC50 by EC50 (trypomastigotes). The one-way ANOVA followed by Bonferroni’s multiple comparison test was used to determine the statistical significance of the group comparisons in the in vitro infection and in vivo studies. Results were considered statistically significant when P < 0.05. All analyzes were performed using Graph Pad Prism version 5.01
Ethics statement
All in vivo experiments were carried out in accordance with the recommendations of Ethical Issues Guidelines and were approved by the local Animal Ethics Committee (Approved number: L-IGM-016/2013)
Supplemental Material
Download Zip (736.8 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental material for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Hotez PJ, Bottazzi ME, Franco-Paredes C, et al. The neglected tropical diseases of latin America and the caribbean: a review of disease burden and distribution and a roadmap for control and elimination. PLoS Negl Trop Dis. 2008;2:e300.
- Coura JR, Junqueira AC, Fernandes O, et al. Emerging chagas disease in amazonian Brazil. Trends Parasitol. 2002;18:171–176.
- WHO. Chagas disease (American trypanosomiasis). 2017.
- Vougogiannopoulou K, Skaltsounis AL. From tyrian purple to kinase modulators: naturally halogenated indirubins and synthetic analogues. Planta medica. 2012;78:1515–1528.
- Kalsi DS, Ward J, Lee R, et al. Purple urine bag syndrome: a rare spot diagnosis. Dis Markers. 2017;2017:9131872.
- Gaboriaud-Kolar N, Vougogiannopoulou K, Skaltsounis AL. Indirubin derivatives: a patent review (2010 - present). Expert Opin Ther Pat. 2015;25:583–593.
- Zhou A, Yan L, Lai F, et al. Design, synthesis and biological evaluation of novel indolin-2-ones as potent anticancer compounds. Bioorg Med Chem Lett. 2017;27:3326–3331.
- Cheng X, Merz KH, Vatter S, et al. Identification of a water-soluble indirubin derivative as potent inhibitor of insulin-like growth factor 1 receptor through structural modification of the parent natural molecule. J Med Chem. 2017;60:4949–4962.
- Choi SJ, Lee JE, Jeong SY, et al. 5,5ʹ-substituted indirubin-3ʹ-oxime derivatives as potent cyclin-dependent kinase inhibitors with anticancer activity. J Med Chem. 2010;53:3696–3706.
- Jie C, Luo Z, Chen H, et al. Indirubin, a bisindole alkaloid from Isatis indigotica, reduces H1N1 susceptibility in stressed mice by regulating MAVS signaling. Oncotarget. 2017;8:105615–105629.
- Medina-Moreno S, Dowling TC, Zapata JC, et al. Targeting of CDK9 with indirubin 3ʹ-monoxime safely and durably reduces HIV viremia in chronically infected humanized mice. PloS one. 2017;12:e0183425.
- Clough BH, Zeitouni S, Krause U, et al. Rapid osteogenic enhancement of stem cells in human bone marrow using a glycogen-synthease-kinase-3-beta inhibitor improves osteogenic efficacy in vitro and in vivo. Stem Cells Transl Med. 2018;7(4):342–353.
- Sato N, Meijer L, Skaltsounis L, et al. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat Med. 2004;10:55–63.
- Chen L, Huang C, Shentu J, et al. Indirubin derivative 7-bromoindirubin-3-oxime (7bio) attenuates abeta oligomer-induced cognitive impairments in mice. Front Mol Neurosci. 2017;10:393.
- Toledo EM, Colombres M, Inestrosa NC. Wnt signaling in neuroprotection and stem cell differentiation. Prog Neurobiol. 2008;86:281–296.
- Sekhon S, Koo J. Indirubin: a novel topical agent in the treatment of psoriasis. Br J Dermatol. 2018;178:21.
- Qi T, Li H, Li S. Indirubin improves antioxidant and anti-inflammatory functions in lipopolysaccharide-challenged mice. Oncotarget. 2017;8:36658–36663.
- Myrianthopoulos V, Kritsanida M, Gaboriaud-Kolar N, et al. Novel inverse binding mode of indirubin derivatives yields improved selectivity for DYRK kinases. ACS Med Chem Lett. 2013;4:22–26.
- Polychronopoulos P, Magiatis P, Skaltsounis AL, et al. Structural basis for the synthesis of indirubins as potent and selective inhibitors of glycogen synthase kinase-3 and cyclin-dependent kinases. J Med Chem. 2004;47:935–946.
- Vougogiannopoulou K, Ferandin Y, Bettayeb K, et al. Soluble 3ʹ,6-substituted indirubins with enhanced selectivity toward glycogen synthase kinase −3 alter circadian period. J Med Chem. 2008;51:6421–6431.
- Hoessel R, Leclerc S, Endicott JA, et al. Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat Cell Biol. 1999;1:60–67.
- Efstathiou A, Gaboriaud-Kolar N, Smirlis D, et al. An inhibitor-driven study for enhancing the selectivity of indirubin derivatives towards leishmanial glycogen synthase kinase-3 over leishmanial cdc2-related protein kinase 3. Parasit Vectors. 2014;7:234.
- Efstathiou AG-KN, Myrianthopoulos V, Vougogiannopoulou K, et al. Indirubin analogues inhibit typanosoma brucei glycogen synthase kinase 3 short and T. brucei growth. submitted. 2018.
- Stuart K, Brun R, Croft S, et al. Kinetoplastids: related protozoan pathogens, different diseases. J Clin Invest. 2008;118:1301–1310.
- Heredia A, Natesan S, Le NM, et al. Indirubin 3ʹ-monoxime, from a Chinese traditional herbal formula, suppresses viremia in humanized mice infected with multidrug-resistant HIV. AIDS Res Hum Retroviruses. 2014;30:403–406.
- Cheng X, Merz KH. The role of indirubins in inflammation and associated tumorigenesis. Adv Exp Med Biol. 2016;929:269–290.
- Gaboriaud-Kolar N, Myrianthopoulos V, Vougogiannopoulou K, et al. Natural-based indirubins display potent cytotoxicity toward wild-type and t315i-resistant leukemia cell lines. J Natur Prod. 2016;79:2464–2471.
- Broecker-Preuss M, Becher-Boveleth N, Gall S, et al. Induction of atypical cell death in thyroid carcinoma cells by the indirubin derivative 7-bromoindirubin-3ʹ-oxime (7BIO). Cancer Cell Inter. 2015;15:97.
- Liu L, Kritsanida M, Magiatis P, et al. A novel 7-bromoindirubin with potent anticancer activity suppresses survival of human melanoma cells associated with inhibition of STAT3 and akt signaling. Cancer Biol Ther. 2012;13:1255–1261.
- Xingi E, Smirlis D, Myrianthopoulos V, et al. 6-br-5methylindirubin-3ʹoxime (5-Me-6-BIO) targeting the leishmanial glycogen synthase kinase-3 (GSK-3) short form affects cell-cycle progression and induces apoptosis-like death: exploitation of GSK-3 for treating leishmaniasis. Int J Parasitol. 2009;39:1289–1303.
- Durieu E, Prina E, Leclercq O, et al. From drug screening to target deconvolution: a target-based drug discovery pipeline using leishmania casein kinase 1 isoform 2 to identify compounds with antileishmanial activity. Antimicrob Agents Chemother. 2016;60:2822–2833.
- Grant KM, Dunion MH, Yardley V, et al. Inhibitors of leishmania mexicana CRK3 cyclin-dependent kinase: chemical library screen and antileishmanial activity. Antimicrob Agents Chemother. 2004;48:3033–3042.
- Brancaglion GA, Toyota AE, Cardoso Machado JV, et al. In vitro and in vivo trypanocidal activities of 8-methoxy-3-(4-nitrobenzoyl)-6-propyl-2H-cromen-2-one, a new synthetic coumarin of low cytotoxicity against mammalian cells. Chem Biol Drug Des. 2018
- MacLean LM, Thomas J, Lewis MD, et al. Development of trypanosoma cruzi in vitro assays to identify compounds suitable for progression in chagas’ disease drug discovery. PLoS Negl Trop Dis. 2018;12:e0006612.
- Bastos TM, Barbosa MI, Da Silva MM, et al. Nitro/nitrosyl-ruthenium complexes are potent and selective anti-trypanosoma cruzi agents causing autophagy and necrotic parasite death. Antimicrob Agents Chemother. 2014;58:6044–6055.
- Zong WX, Thompson CB. Necrotic death as a cell fate. Genes & Dev. 2006;20:1–15.
- Juliany Cola Fernandes Rodrigues SHSaWdS. APOPTOSIS-LIKE DEATH IN PARASITIC PROTOZOA. Braz J morphol Sci. 2006;23:87–89.
- Tessarolo LD, de Menezes R, Mello CP, et al. Nanoencapsulation of benznidazole in calcium carbonate increases its selectivity to trypanosoma cruzi. Parasitology. 2018;145:1191–1198.
- Maya JD, Cassels BK, Iturriaga-Vasquez P, et al. Mode of action of natural and synthetic drugs against trypanosoma cruzi and their interaction with the mammalian host. Comp Biochem and Physiol Part A, Mol & Integr Physiol. 2007;146:601–620.
- Manallack DT, Prankerd RJ, Yuriev E, et al. The significance of acid/base properties in drug discovery. Chem Soc Rev. 2013;42:485–496.
- Sousa MC, Varandas R, Santos RC, et al. Antileishmanial activity of semisynthetic lupane triterpenoids betulin and betulinic acid derivatives: synergistic effects with miltefosine. PloS one. 2014;9:e89939.
- Alirol E, Schrumpf D, Amici Heradi J, et al. Nifurtimox-eflornithine combination therapy for second-stage gambiense human African trypanosomiasis: medecins sans frontieres experience in the democratic republic of the congo. Clin Infect Dis: Off Publ Infect Dis Soc Am. 2013;56:195–203.
- Ferandin Y, Bettayeb K, Kritsanida M, et al. 3ʹ-substituted 7-halogenoindirubins, a new class of cell death inducing agents. J Med Chem. 2006;49:4638–4649.
- Meijer L, Skaltsounis AL, Magiatis P, et al. GSK-3-selective inhibitors derived from tyrian purple indirubins. Chem Biol. 2003;10:1255–1266.
- Silva LHNV. Sobre uma cepa de Trypanosoma cruzi virulenta para o camundongo branco. Folia Clin Biol. 1953;20:191–207.
- Brener Z. Therapeutic activity and criterion of cure on mice experimentally infected with trypanosoma cruzi. Rev Inst Med Trop Sao Paulo. 1962;4:389–396.