
ABSTRACT
Lipopolysaccharide (LPS) is an essential structural component of the outer membrane (OM) of most Gram-negative bacteria. In the model organism Escherichia coli, LPS transport to the OM requires seven essential proteins (LptABCDEFG) that form a continuous bridge across the cell envelope. In Pseudomonas aeruginosa the recently-demonstrated essentiality of LptD and LptH, the P. aeruginosa LptA homologue, confirmed the crucial role of the Lpt system and, thus, of LPS in OM biogenesis in this species. Surprisingly, independent high-throughput transposon mutagenesis studies identified viable P. aeruginosa insertion mutants in the lptE gene, suggesting that it might be dispensable for bacterial growth. To test this hypothesis, we generated an lptE conditional mutant in P. aeruginosa PAO1. LptE depletion only slightly impairs P. aeruginosa growth in vitro. Conversely, LptE is important for cell envelope stability, antibiotic resistance and virulence in an insect model. Interestingly, the maturation and OM localization of LPS is only marginally affected in LptE-depleted cells, while the levels of the OM component LptD are strongly reduced. This suggests that P. aeruginosa LptE might not be directly involved in LPS transport, although it is clearly essential for the maturation and/or stability of LptD. While poor functionality of LptD caused by LptE depletion is somehow tolerated by P. aeruginosa, this has a high cost in terms of cell integrity, drug resistance and virulence, highlighting LptE function(s) as an interesting target to weaken P. aeruginosa defenses and reduce its infectivity.
Introduction
Gram-negative bacteria are characterized by a cell envelope consisting of two concentric membranes, the inner (IM) and outer membrane (OM), which show distinct composition, and structural and functional properties [Citation1]. The two lipid bilayers delimit an aqueous compartment, the periplasm, containing a thin layer of peptidoglycan [Citation2]. While the IM is a typical phospholipids bilayer, the OM is an asymmetric membrane composed of glycerophospholipids in the inner leaflet, and lipopolysaccharide (LPS) in the outer leaflet [Citation1]. LPS is a negatively charged glycolipid; in the presence of divalent cations LPS molecules form a tightly packed layer at the outer leaflet thus reducing OM fluidity and permeability, which is selectively controlled by dedicated OM proteins [Citation3]. Perturbation of LPS organization at the cell surface by defects in LPS biogenesis and/or impaired assembly of OM components result in loss of membrane integrity due to i) migration of phospholipids from the inner to the outer leaflet of the OM causing locally symmetric bilayer rafts, and ii) generation of transient imperfections or “cracks” which can allow the entry of both lipophilic and hydrophilic compounds [Citation4,Citation5]. The asymmetry of the OM is generated by the Lpt (Lipopolysaccharide transport) system, a molecular machine composed of seven essential proteins in Escherichia coli (LptABCDEFG) that ferries LPS from the periplasmic side of the IM, across the periplasm to the cell surface [Citation6]. The Lpt proteins assemble to form a multiprotein complex that spans the entire cell envelope [Citation7]. This is organized in two sub-assemblies, LptB2CFG and LptDE, located at the IM and at the OM, respectively [Citation8–Citation11], which are connected by the periplasmic protein LptA [Citation12–Citation14]. At the IM the LptB2FG ATP-binding cassette (ABC) transporter, associated to the bitopic protein LptC, powers the LPS export to the cell surface [Citation15,Citation16]. At the OM, the β-barrel protein LptD and the lipoprotein LptE constitute the OM translocon, characterized by a peculiar plug-and-barrel architecture [Citation17–Citation19]. LPS extracted from the IM by the LptB2FG ABC transporter is sequentially transferred to LptC and then to LptA in an energy-dependent process [Citation15,Citation16]. Lastly, LPS is thought to be delivered to the LptDE OM translocon for its final assembly at the outer leaflet [Citation20]. It is well established that LptE plays an essential role in the assembly of functional LptD [Citation20–Citation24]. However, more recently LptE has been shown to play a role also in the LPS export process in E. coli, as witnessed by its ability to bind LPS [Citation20] and disaggregate it in vitro [Citation25]. While the LPS transport machinery has been extensively characterized in E. coli, little is known about its functional conservation in other Gram-negative bacteria. At present, comprehensive investigation of Lpt components has only been carried out in Neisseria meningitidis, an LPS-producing Gram-negative bacterium for which LPS is however not essential for viability and OM biogenesis [Citation26–Citation28]. Systematic analysis of LPS transport genes in N. meningitidis revealed that, despite being dispensable for cell viability in line with the non-essential role of LPS in this species, LPS transport proteins are all essential for LPS transfer to the OM, with the only exception of LptE [Citation24,Citation29,Citation30]. Indeed, deletion of the lptE gene in N. meningitidis does not impair transport of LPS to the cell surface, although it affects total levels of LptD, suggesting a conserved chaperone-like role for LptE in LptD biogenesis [Citation24]. Evidences on the role of the Lpt machinery have also been accumulated in the Gram-negative bacterium Pseudomonas aeruginosa, an opportunistic pathogen responsible for severe infections in immunocompromised and cystic fibrosis patients which are poorly responsive to antibiotic therapies [Citation31]. The first experimental proof of the relevance of the P. aeruginosa Lpt complex came from the serendipitous finding that LptD was the molecular target of a peptidomimetic antibiotic with potent anti-P. aeruginosa activity [Citation32], and from subsequent confirmation of the essentiality of LptD in this species by conditional mutagenesis [Citation33]. More recently, a reverse-genetic screening for uncharacterized essential periplasmic proteins revealed that LptH, the P. aeruginosa homologue of E. coli LptA [Citation34], is crucial for P. aeruginosa growth, cell envelope biogenesis and pathogenicity in different animal models [Citation35]. While these works clearly confirmed the importance of the Lpt machinery, in line with the essentiality of LPS biosynthesis genes and, thus, of LPS in P. aeruginosa [Citation36,Citation37], the role of other Lpt components in LPS transport remains to be determined. Interestingly, although previous projects aimed at generating saturating libraries of sequence-defined transposon insertion mutants proposed lptE as a putative essential gene in this bacterium [Citation38,Citation39], two recent transposon-sequencing (Tn-seq) studies detected viable P. aeruginosa lptE transposon insertion mutants under certain growth conditions [Citation40,Citation41]. This finding suggests that the lptE gene might be dispensable for P. aeruginosa growth. However, considering that Tn-seq has some limitations, including the inability to distinguish mutants whose phenotypes are “complemented” by other bacteria in the transposon-mutant pool [Citation40], confirmatory experiments with individual mutants are crucial to verify Tn-seq findings. In this work, we employ a conditional mutagenesis approach to investigate the effect of LptE depletion on the physiology of P. aeruginosa. LptE-depleted P. aeruginosa PAO1 cells are only slightly impaired in in vitro growth, while they are strongly defective in the ability to cause infection in an animal model. LPS transport in P. aeruginosa is only marginally affected by LptE depletion, although LptE is confirmed to play an important role as LptD chaperone. Notably, detergent and antibiotics sensitivity is drastically increased in LptE-depleted cells, likely because of improperly folded and/or un-plugged LptD channels in the OM.
Materials and methods
Bacterial strains and growth conditions
Bacterial strains and plasmids used in this study are listed in Table S1. Bacteria were cultured in Lysogeny Broth, Lennox formulation (LB; Acumedia) for genetic manipulation, while growth assays were performed in Mueller-Hinton broth (MH; Difco), LB or M9 minimal medium supplemented with 20 mM succinate as carbon source and 50 µM FeCl3 [Citation42]. When specified, growth media were supplemented with L-arabinose, or IPTG at the indicated concentrations. Growth assays in liquid cultures were performed in 96-well microtiter plates (200 µL of medium in each well) or in flasks at 37°C and vigorous shaking (200 rpm). Growth was measured as the optical density at 600 nm (OD600) of bacterial cultures in a Victor plate reader (Wallac) for microtiter plates or of appropriate dilutions in sterile growth medium in a spectrophotometer for flask cultures. Growth assays on solid media were performed by spotting 5 µL of serial ten-fold dilutions from bacterial suspensions in saline normalized to an OD600 = 1, from late-exponential cultures grown in the presence or in the absence of 0.5% (w/v) arabinose on media solidified with 1.5% (w/v) agar. When required, antibiotics were added at the following concentration for E. coli (the concentration used for P. aeruginosa is shown between brackets): ampicillin 100 µg/mL, nalidixic acid 20 µg/mL, chloramphenicol 30 µg/mL (350 µg/mL), tetracycline 12 µg/mL (50–100 µg/mL).
Generation of plasmids and recombinant strains
Recombinant DNA procedures have been described elsewhere [Citation42]. All DNA fragments for cloning were amplified by PCR with Pfu (Promega) or Q5 Hot Start High-Fidelity (New England Biolabs) DNA Polymerases using genomic DNA of P. aeruginosa PAO1 as the template. Primers and restriction enzymes used for cloning are described in Table S2. All constructs were verified by DNA sequencing. The LptE expressing construct pMElptE was generated by cloning the lptE coding sequence into the IPTG-inducible shuttle vector pME6032 [Citation43], and introduced in P. aeruginosa by transformation using chemically competent cells. The integration-proficient construct mini-CTX1-araCPBADlptE was generated by replacing the tolB gene in mini-CTX1-araCPBADtolB [Citation44] with the coding sequence of the lptE gene. In this construct, the lptE coding sequence is cloned downstream of an arabinose-dependent regulatory element araC-PBAD optimized for P. aeruginosa by modification of the ribosome-binding site [Citation36]. The deletion mutagenesis construct pDM4ΔlptE was obtained by directionally cloning two DNA regions upstream and downstream of the lptE coding sequence into pBluescript II KS+ (Stratagene), and then subcloning the DNA fragment encompassing the lptE upstream and downstream regions into the suicide vector pDM4 [Citation45]. The lptE conditional mutant was generated using a recently-described strategy [Citation44]. Briefly, the lptE coding sequence under the control of modified arabinose-dependent regulatory element araC-PBAD (see above) was integrated into the attB neutral site of the P. aeruginosa chromosome, and excision of the mini-CTX1 plasmid backbone from the attB neutral site was achieved by Flp-mediated recombination as described [Citation46]. In-frame deletion of the endogenous copy of lptE was obtained using the sacB-based suicide construct pDM4ΔlptE as previously described [Citation44] under permissive condition (i.e. growth in the presence of 0.5% arabinose). The gene replacement construct pDM4lptD-6his was generated by individually cloning two 500-bp DNA regions encompassing either the 3ʹ region of the lptD coding sequence (upstream region) or the 5ʹ region of the adjacent gene surA (downstream region) into pBluescript II KS+, and then subcloning the upstream and downstream regions into the suicide vector pDM4 by triple ligation. Note that the reverse primer used to amplify the upstream region (lptD-6his_UP_RV) contains an additional sequence encoding the 6His tag (Table S2), and that the resulting gene replacement construct pDM4lptD-6his includes a 64-bp duplication of the lptD 3ʹ end. This was necessary to preserve the ribosome-binding site and the coding sequence of the adjacent gene surA, which partially overlaps with lptD in the P. aeruginosa PAO1 genome (Fig. S1). PAO1 and the lptE conditional mutant expressing the recombinant protein LptD-6His (PAO1 lptD-6his and lptE lptD-6his, respectively; Table S1) were generated by insertion of the sacB-based suicide construct pDM4lptD-6his followed by sucrose-mediated selection of second recombination events [Citation45]. Clones carrying the 6his-tag insertion were screened by PCR and then verified by DNA sequencing.
Detergent sensitivity assay
Sensitivity to the lytic effect of sodium dodecyl sulphate (SDS) was assessed by determining the turbidity (OD600) of bacterial cell suspensions in saline after 5-min incubation at room temperature in the presence of increasing concentrations of SDS (0–5%, w/v) [Citation44].
Antibiotics sensitivity assay
Resistance to the growth inhibitory activity of several antibiotics was assessed by the Kirby-Bauer disc diffusion test. Bacterial cell suspensions in saline were normalized at 0.5 McFarland Standard and swabbed onto MH agar plates supplemented or not with arabinose at the indicated concentration, using disks containing streptomycin (10 µg), ciprofloxacin (5 µg), imipenem (10 µg), colistin (10 µg), novobiocin (30 µg), erythromycin (15 µg), rifampicin (5 µg) and tobramycin (10 µg) (Becton Dickinson). Growth inhibition halo diameters were measured after 20 h of growth at 37°C.
Fluorescence microscopy
Microscopy images were obtained with a Zeiss Axiovert 200M microscope through a 63 × 1.45 oil objective coupled to a AxioCam Mrm device 290 camera (Zeiss). PAO1, lptH and lptE conditional mutant cells grown in MH in the absence and/or presence of arabinose were collected from a total amount corresponding to an OD600 of 4, and a 1:10 ratio of fixation solution [fixation solution: formaldehyde 37% (v/v) – glutaraldehyde 25% (v/v) in PBS] was added. Cells were incubated for 30 min at 37°C with shaking, washed with PBS and resuspended in 500 µL of PBS. 5 µL of the cell suspensions were spotted onto an agarose-coated glass slide [1% (w/v) agarose], and the samples were covered with a glass coverslip. To stain cell membranes, FM5–95 (ThermoFisher) was added to the agarose solution to a final concentration of 2 µg/mL.
Cell fractionation and membrane isolation
For cell fractionation and membrane isolation P. aeruginosa strains were grown in MH, as described above. The IM and OM were separated by sucrose gradient ultracentrifugation according to published procedures [Citation7,Citation12] with the following modifications. A total amount of 200 OD600 of cells were resuspended in 5 mL of 10 mM Tris/HCl buffer (pH 7.8) containing 25% (w/v) sucrose, 50 µg/mL DNAse, 1 mM PMSF, 100 µg/mL lysozyme. The resuspended cells were lysed by a single passage through cell disruptor (Constant system) at 16,000 psi. The cell lysate was clarified by centrifugation at 3,000 × g for 20 min. Cell membranes were collected by centrifuging the supernatant for 1 h at 37,000 rpm in a Sorvall Discovery 90SE with T-890 rotor. Total membranes were resuspended in 1 mL of 10 mM Tris/HCl buffer (pH 7.8) containing 25% (w/v) sucrose and 800 µL were layered on top of the sucrose gradient as described [Citation7]. Fractions (400 µL) were collected from the top of the gradient. The distribution profiles of LPS and of XcpY and OprI proteins across the gradient were estimated by western blot as described below. The analysis of LptD levels in LptE-depleted cells was performed on membranes isolated from a total of 20 OD600 according to a published protocol [Citation47]. Collected cells were resuspended in 50 mM Tris-HCl, 5 mM EDTA (pH 8.0) containing 1 mM PMSF. After disintegration in cell disruptor (Constant system, 1 passage at 16,000 psi), unbroken cells were removed by centrifugation (3,000 × g, 15 min, 4°C). Membrane samples were collected by ultracentrifugation (37,000 in a Sorvall Discovery 90SE with T-890 rotor, 1 h, 4°C) and dissolved in 2 mM Tris-HCl (pH 7.6).
Lipid A extraction and analysis
Lipid A was extracted from bacterial cell pellets using the ammonium hydroxide-isobutyric acid-based procedure as previously described [Citation48], with few modifications. Briefly, 2 mL of early stationary phase cultures were pelleted and resuspended in 400 μL of 70% (v) isobutyric acid and 1 M ammonium hydroxide (5:3). Samples were incubated for 1 hour at 100ºC and centrifuged at 2,000 x g for 15 min. Supernatants were added to 400 μL of endotoxin-free water, frozen at −80°C, and lyophilized for 4–5 h in a centrifugal vacuum concentrator. The resultant pellet was washed with 1 mL methanol, and the lipid A was extracted using 100 μL of chloroform, methanol, and water (3:1:0.25). After centrifugation at 2,000 x g for 15 min, 2 μL of the supernatants were mixed with 2 μL of 10 mg/mL norharmane matrix in chloroform:methanol (2:1), and 0.5 μL of the mixtures were spotted on a matrix-assisted laser desorption-time of flight (MALDI-TOF) plate (5800 MALDI TOF/TOF Analyzer, Sciex, Ontario, Canada). Samples were analyzed in the negative-ion mode with reflectron mode. Calibration was performed using default calibration originated by five standard spots. Spectral data were analyzed with the 4000 Series Explorer software Version 4.1.0 (Sciex, Ontario, Canada), and used to estimate lipid A forms based on their predicted structures and molecular weights.
3-Deoxy-d-manno-oct-2-ulosonic acid (KDO) assay
LPS was extracted from a total of 7 OD600 of cells. Cells were collected by centrifugation, the cell pellets were washed twice with 2 mL of sterile phosphate-buffered saline (PBS), resuspended in 500 µL of Hitchcock and Brown lysis buffer [2% (w/v) SDS, 10% (v/v) glycerol and 1 M Tris-HCl, pH 6.8], and heated at 100°C for 30 min. Then, samples were cooled to 20°C and incubated at 56°C for 3 h with 30 µg/mL proteinase K, and proteinase K was inactivated by heating at 95°C for 30 min [Citation49]. The isolated crude LPS was quantified by determining the amount of KDO as previously described [Citation50], using standard curves generated from purified KDO, and normalized to mg of total proteins.
Western blot analyses
For western blot analysis of whole cell extracts, appropriate volumes of exponentially-growing bacterial cultures in MH in flasks were centrifuged, and pellets were suspended in SDS-PAGE loading buffer [0.25 M Tris-HCl pH 6.8, 2% (w/v) SDS, 10% (v/v) β-mercaptoethanol, 20% (v/v) glycerol]. Pellets from identical culture volumes were also collected to determine the cellular protein concentration for each sample by using the DC protein assay kit (Bio-Rad) and bovine serum albumin as the standard. Volumes of SDS-PAGE samples corresponding to 20 μg of proteins were loaded onto the gels. Proteins resolved by SDS-PAGE were electrotransferred onto a nitrocellulose filter (Hybond-C extra, Amersham) and probed for LptE or LptC using custom polyclonal rabbit antibodies and a goat anti-rabbit IgG HRP-conjugated secondary antibody (Sigma-Aldrich). Anti-LptE and anti-LptC antibodies were generated at GenScript (http://www.genscript.com/custom-polyclonal-antibody-production-services.html) using Keyhole Limpet Hemocyanin (KLH)-conjugated peptides as antigens (LptE epitope: RAAEPQQSPIEFPT; LptC epitope: NAHSLQYQEDGSLD). The epitopes were selected using the Optimum Antigen™ Design Tool (GenScript). Filters were developed with ECL chemiluminescent reagents (Amersham) and visualized in a ChemiDoc XRS+ system (Bio-Rad). When specified ()), filters were developed with the Cyanagen Westar ηC Ultra 2.0 reagent and detected using an Odissey system (Li-Cor) system. To analyze the protein and LPS profiles across the sucrose gradient, aliquots from each fraction were subjected to immunoblot analysis. For the protein profile of XcpY, 20 µL of each fraction were loaded on 12.5% SDS-PAGE gels and analyzed by immunoblotting using anti-XcpY antibodies [Citation51]. To estimate the OprI and LPS distribution, 5 µL and 20 µL of each fraction, respectively, were separated by 18% Tricine-SDS-PAGE and immunoblotted using anti OprI antibodies [Citation52] and anti-P. aeruginosa outer core specific antibodies (MEDIMABS). For western blot analysis of LptD-6His levels in LptE-depleted cells, 20 µL of isolated membranes were diluted in 5× SDS-PAGE loading buffer and proteins were separated by 10% SDS-PAGE followed by immunoblot analysis using anti-6His antibodies (Sigma-Aldrich), according to the manufacturer instruction. To analyze the oxidation state of LptD, equal amounts of membrane samples were treated with 5× SDS loading buffer with or without 5% (v/v) β-mercaptoethanol. Samples were boiled for 10 min before SDS-PAGE. For immunoblots with antibodies against XcpY, OprI, LPS and 6His, filters were visualized by immunofluorescence using IRDye secondary antibodies (Li-Cor) on an Odissey system (Li-Cor). Densitometric analysis of band intensities was performed using the Image Lab 3.0 software (Bio-Rad).
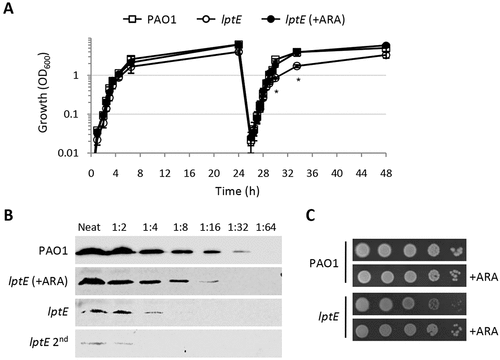
Figure 1. Growth curves, plating efficiency and LptE levels of the wild type strain P. aeruginosa PAO1 and the lptE conditional mutant. (a) Stationary phase PAO1 and lptE conditional mutant (lptE) cells grown in MH with 0.5% arabinose (ARA) were diluted 1:1000 in flasks containing MH with or without 0.5% ARA (first refresh). After 24 h of growth, PAO1 and lptE cells grown in the absence of ARA were sub-cultured (dilution 1:200) in flasks in fresh medium containing or not 0.5% ARA (second refresh). As the growth curves of PAO1 with or without ARA were basically superimposable, for clarity only the curve in the absence of ARA is shown. Values are the mean (±SD) of three independent experiments. Asterisks indicate the time-points at which the growth of lptE without ARA was significantly different from that of other samples (P < 0.05, Kruskal-Wallis) (b) Western blot analysis to quantify LptE levels in whole cell lysates (20 µg of proteins; neat), or serial 1:2 dilutions as indicated, from PAO1 or lptE conditional mutant cells grown for 14 h in MH with or without ARA for one or two passages (lptE 2nd). Filters were developed with Cyanagen Westar ηC Ultra 2.0 reagent and visualized in a Odissey system (Li-Cor) system. (c) Colony growth of PAO1 and lptE on MH agar plates supplemented or not with ARA. Exponential phase cultures in MH with 0.5% ARA were normalized to OD600 = 1 in saline, and 5 μL of the 10−2–10−6 dilutions were spotted onto the plates and incubated for 20 h at 37°C. Images in panels B-C are representative of at least three experiments giving similar results.
Galleria mellonella infection assay
P. aeruginosa strains were grown in MH with 0.5% arabinose, and serial dilutions of bacterial cell suspensions in saline were injected into G. mellonella larvae as described [Citation53]. Ten larvae were infected with each infecting dose in at least three independent experiments, and infected larvae were incubated at 30°C for up to 4 days to monitor mortality. Survival curves, LD90 and R2 values were determined using GraphPad Prism as previously described [Citation54].
Statistical analysis
Statistical analysis was performed with the software GraphPad Instat, using Kruskal-Wallis test followed by Dunn test for selected pairs of columns.
Results
LptE depletion only marginally affects P. aeruginosa growth
In order to investigate the role of LptE in LPS transport and cell viability in P. aeruginosa, we first attempted to generate a clean in-frame deletion in the lptE gene. However, several independent attempts failed, as the recombinant clones obtained after the insertion of the deletion mutagenesis plasmid into the chromosome always reverted to the wild type genotype during the second recombination event selected by the mutagenesis procedures (data not shown). We therefore decided to use a recently-developed strategy [Citation44] to construct a conditional mutant carrying a copy of the lptE coding sequence in a neutral site of the genome (attB) under the control of an arabinose-dependent araC-PBAD regulatory element and an in-frame deletion in the endogenous lptE gene. Surprisingly, we found that this conditional mutant was able to grow in MH in the absence of arabinose, although growth yields were slightly reduced as compared to those of the wild type PAO1 strain or the lptE conditional mutant grown in the presence of arabinose ()). Growth of LptE-depleted cells was further reduced if these cells were subsequently refreshed in the absence of arabinose (second refresh in )), while no additional growth defects were observed after further refreshes (data not shown). As lptE conditional mutant cells grown for two passages in the absence of the inducer showed a more severe growth deficiency as compared to cells directly refreshed from arabinose-containing medium ()), also these cells (hereafter named lptE 2nd) were analyzed in subsequent assays. The effect of arabinose on the growth kinetics of the lptE conditional mutant showed a clear dose-dependent response (Fig. S2), indicating that the growth of this mutant is proportional to LptE expression levels. Western blot analysis with a polyclonal anti-LptE antibody revealed that LptE was strongly reduced in whole cell extracts of the lptE conditional mutant cultured under non-inducing conditions as compared to the wild-type strain (Fig. S3). Using a very sensitive detection system and appropriate sample dilutions, the LptE intracellular levels in the conditional mutant grown in the absence of arabinose were tentatively quantified as 8-fold lower (ca. 12.5%) than those of the wild type, while the amount of LptE further fell down to less than 6% of wild type levels in lptE mutant cells cultured for two subsequent passages in the absence of arabinose (lptE 2nd) ()). It must be noted that LptE levels in the lptE conditional mutant cultured in the presence of 0.5% arabinose were only about 25–40% of the wild type levels () and S3), although growth of the mutant was restored to wild type rates () and S2). No further increase in LptE levels was observed with higher arabinose concentrations (up to 2%; data not shown). The partial restoration of LptE levels by arabinose can be explained by the fact that our conditional mutant is based on an araC-PBAD regulatory element with a very weak ribosome-binding site (see Materials and Methods and ref. 44 for details). While this regulatory element is effective in lowering the basal, non-induced expression from araC-PBAD in P. aeruginosa [Citation36], it can also decrease its maximal induced activity, as recently demonstrated for similar constructs [Citation55]. Interestingly, the amount of a cytoplasmic membrane protein involved in LPS transport (LptC) was comparable between LptE-replete and -depleted cells (Fig. S3), suggesting that LptE depletion does not negatively impact on the stability of the whole Lpt machinery. To investigate whether the residual growth of LptE-depleted cells is a general feature of the whole population or an adaptation occurring in a sub-population of cells, we determined the plating efficiency of the lptE conditional mutant in the presence or absence of arabinose. As shown in ), the number of colonies obtained for the lptE conditional mutant was comparable in MH agar plates with or without arabinose, although colonies were visually smaller in non-inducing conditions, confirming that also colony growth rates were slightly reduced for LptE-depleted cells. Similar results were obtained in a different complex medium (LB) and in the minimal medium M9 (Fig. S4). In addition, to rule out that the growth of the lptE conditional mutant under non-inducing conditions could be at least partly influenced by the appearance of sub-populations of fast-growing cells (carrying suppressor mutations), the strain was sequentially streaked on MH agar plates for several passages. Notably, no decrease in colony yields was observed over passages, nor did we detect colonies showing faster growth (Fig. S5).
LptE depletion drastically impairs cell envelope integrity
LptE appears to be dispensable for P. aeruginosa growth and viability, nevertheless its depletion could impact on cell envelope integrity. In order to test this hypothesis, we determined the effect of LptE depletion on detergent and antibiotic resistance. Cell envelope integrity was first investigated through an SDS sensitivity assay [Citation44]. Under non-permissive conditions, SDS resistance of the lptE conditional mutant was decreased as compared to the wild type, and was strongly defective in lptE cells cultured for two subsequent passages in the absence of arabinose (). The SDS sensitivity profile of lptE cells at the second refresh under non-permissive conditions was almost comparable to that of LptH-depleted cells (), which were collected using the culturing strategy described in Fig. S6, and used as control for a mutant with severe defects in envelope biogenesis due to the lack of an essential LPS transport protein [Citation35]. Notably, arabinose completely restored SDS resistance in lptE conditional mutant cells to wild type levels (). Microscopic analysis revealed that the morphology of LptE-depleted cells at the first refresh was overall comparable to that of wild type cells, while after two subsequent passages in the absence of arabinose LptE-depleted cells were slightly shorter and a small proportion of them appeared to grow as short cell chains (). The chaining phenotype was much more severe in cells impaired in growth because of LptH depletion, which formed very long chains (). This phenotype parallels that previously observed in E. coli mutants defective in LPS transport [Citation12]. Both lptH and lptE conditional mutants showed a normal phenotype when cultured in the presence of arabinose (Fig. S7). To further investigate the contribution of LptE to the integrity of the OM barrier, the susceptibility of LptE-replete and -depleted cells to different antibiotics was investigated through the Kirby-Bauer disc diffusion assay (). As control we also tested antibiotic resistance of LptH-depleted cells, by culturing the lptH conditional mutant in the presence of the lowest concentration of arabinose, which supports residual growth (0.03%) [Citation35]. Interestingly, the lptE conditional mutant grown without arabinose was much more sensitive to all antibiotics tested than the wild type strain, with the only exception of colistin (). In line with the SDS resistance profile (), LptE-depleted cells cultured for two subsequent passages in the absence of arabinose showed further defects in resistance to many antibiotics, including colistin (). In spite of antibiotic-specific differences, the drug resistance profiles of LptE- and LptH-depleted cells were overall comparable (), suggesting that the OM barrier is strongly defective upon LptE depletion. Notably, addition of 0.5% arabinose to the growth medium only partially ameliorated the antibiotic susceptibility profile of the lptE conditional mutant, while it almost completely restored antibiotic resistance to wild type levels in the lptH conditional mutant (). Considering that our immunoblot assays showed that arabinose supplementation only partially re-establishes LptE levels in the lptE conditional mutant (), it is worth hypothesizing that such reduced expression of LptE could account for the failure of arabinose to fully restore antibiotic resistance in the conditional mutant. To confirm this hypothesis, a plasmid for IPTG-inducible ectopic expression of LptE (pMElptE; Table S1) was introduced in the lptE conditional mutant. Immunoblot analysis confirmed that LptE levels in the lptE mutant harboring pMElptE were restored to wild type levels in the presence of 1 mM IPTG and 0.5% arabinose (Fig. S8). Notably, under these culturing conditions, susceptibility to most antibiotics was basically comparable between the lptE mutant harboring pMElptE and the wild type carrying the empty plasmid, although a slight decrease in resistance to streptomycin and rifampicin was still observed in the conditional mutant (). Overall, these assays clearly show that LptE-depleted cells are strongly defective in detergent and antibiotic resistance, which is indicative of major defects in cell envelope stability.
Figure 2. Effect of LptE depletion on cell envelope stability. Lytic effect of different SDS concentrations (0–5%), measured as decrease in cell suspension turbidity (OD600), on PAO1 and lptE conditional mutant cells grown in the presence and/or absence of 0.5% arabinose (ARA), corresponding to the first (lptE) or second refresh (lptE 2nd) described in the legend of . As control, LptH-deficient cells (lptH), obtained through the culturing strategy described in Fig. S6, were included in the analysis. Values are the mean (±SD) of three independent experiments performed in duplicate.
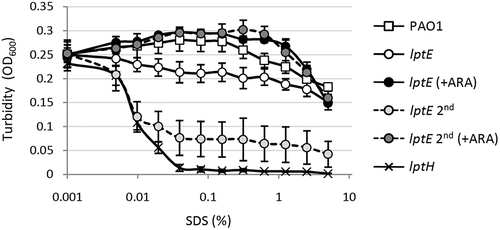
Figure 3. Phase contrast and fluorescence microscopy images of P. aeruginosa wild type PAO1, lptH and lptE conditional mutant cells grown in MH without arabinose, and stained with the membrane-binding dye FMTM 5–95. lptE 2nd corresponds to cells cultured for two subsequent passages in the absence of arabinose (see the legend of for details). Images are representative of several fields (≥10) showing comparable results. Scale bar: 3 µm.
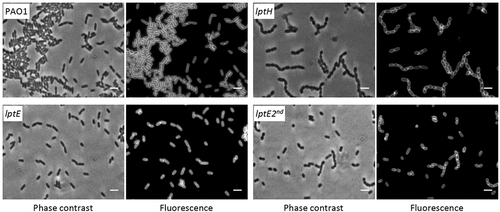
Table 1. Antibiotic susceptibility of lpt mutant strains by the Kirby-Bauer disk diffusion testa.
P. aeruginosa LptE only moderately influences LPS transport
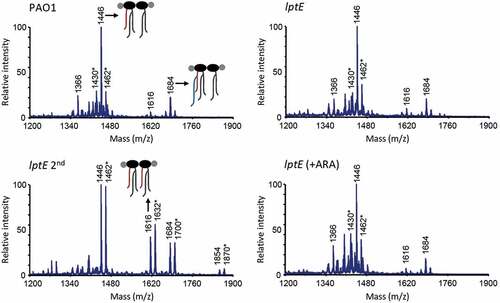
In order to verify whether the observed defects in cell envelope stability upon LptE depletion may correlate with impaired LPS transport, we performed membrane fractionation experiments to compare the amount and localization of LPS in LptE-replete and -depleted cells. We also used LptH-depleted cells as a positive control for the effect of an essential LPS transport protein on LPS localization [Citation34,Citation35]. Membranes were fractionated by sucrose density gradient centrifugation, as described in Materials and Methods, and the fractions were immunoassayed for LPS, as well as for XcpY and OprI as IM and OM markers, respectively [Citation51,Citation52]. As shown in , in wild type cells the IM equilibrated around fractions 5–9 as judged by the distribution profile of XcpY, whereas the OM equilibrated around fractions 17–25 where the majority of both LPS and OprI fractionated. The LPS fractionation profile markedly changed in LptH-depleted cells, and was overall shifted towards lighter fractions as witnessed by the presence of LPS even in fractions where the IM XcpY protein equilibrates. Notably, the OprI fractionation profile paralleled that of LPS, indicating that the IM and OM could not be properly separated by buoyant-density centrifugation. A very small amount of LPS was shifted towards lighter fractions also in lptH mutant cells cultured under permissive conditions (0.5% arabinose), while no changes in the localization of the IM and OM markers XcpY and OprI were observed ()). It thus appears that LPS transport is not fully restored to wild type levels in the lptH mutant grown in the presence of arabinose, albeit its antibiotic susceptibility profile was overall comparable to that of the PAO1 strain (). Collectively, these results indicate that LPS transport is severely impaired upon LptH depletion and are indicative of major defects in cell envelope organization, in line with the hypersensitivity of LptH-depleted cells to antibiotics and detergents ( and ) [Citation35]. The fractionation profile of LptE-depleted cells was more similar to that of lptH mutant cells grown with arabinose. Indeed, some defects in LPS transport appeared in the lptE conditional mutant (lptE and lptE 2nd) as witnessed by appearance of small amount of LPS in lighter fractions. Nevertheless, LptE-replete and-depleted cells did not undergo the huge OM reorganization observed in LptH-depleted cells, since the fractionation profile of the IM and OM markers paralleled that observed in PAO1 ()). Notably, the fractionation profile of the lptE conditional mutant was basically independent of the presence of arabinose in the culture medium, suggesting that the level of LptE does not impact on LPS localization. Although not quantitative, the immunoblots shown in ) suggest that LPS levels were somewhat reduced in LptE-depleted cells at the first refresh as compared to all other samples. We therefore quantified LPS levels by analyzing the amount of KDO, a specific component of LPS core oligosaccharide. The amount of KDO was overall comparable among wild type cells, LptH-depleted cells, and LptE-depleted cells at the second refresh, as well as all conditional mutants cultured in the presence of arabinose ()). In contrast, a significant reduction (almost 40%) in KDO levels was observed in the lptE condition mutant at the first refresh without arabinose, in line with western blot results ()). Finally, to confirm the correct localization of LPS in the outer leaflet of the OM, we analyzed the lipid A from wild type and lptE mutant cells by MALDI-TOF mass spectrometry (MS), to verify the presence of OM-specific lipid A modifications, e.g. PagL-mediated deacylation and PagP-mediated palmitoylation [Citation56]. The MS spectra are shown in , while the chemical structures of the identified lipid A forms are reported in Fig. S9. The lipid A profiles of wild type, LptE-replete mutant cells and LptE-depleted mutant cells (at the first refresh) were almost identical, with a major peak corresponding to mono-hydroxylated penta-acylated lipid A (m/z = 1446). This represents the main lipid A form in laboratory P. aeruginosa strains (e.g. PAO1 and PA14), and derives from PagL-mediated removal of the O-linked 3-OH-C10 acyl chain at position 3 of the hexa-acylated precursor (Fig. S9) [Citation57,Citation58]. Less abundant peaks have been identified as (i) different hydroxylation states of the penta-acylated lipid A (m/z = 1430 and 1462), (ii) penta-acylated lipid A palmitoylated by PagP (m/z = 1684), or (iii) the hexa-acylated lipid A precursor (m/z = 1616) ( and S9). Some differences were instead observed in the lipid A profile of lptE mutant cells grown for two subsequent passages without arabinose (lptE 2nd). While the penta-acylated molecule remained the main lipid A form also in these cells, there was an increase in the hexa-acylated precursor and, to a minor extent, in the palmitoylated penta-acylated form (). In addition, we also observed a relevant increase in the hydroxylation state of all these molecules (m/z = 1462, 1632 and 1700, respectively). Overall, this analysis strongly suggests that the majority of lipid A in LptE-depleted cells is localized in the outer leaflet of the OM, as ≥ 75% of peaks intensity corresponds to molecules modified by PagL or PagP (penta-acylated and palmitoylated lipid A), whose active sites face the external surface of the OM [Citation56]. In conclusion, these data indicate that LptE is not directly involved in the transport and assembly of LPS into the OM and strongly suggest that lack of envelope integrity/stability in lptE mutant cells is not simply due to impaired LPS transport.
Figure 4. Localization and quantification of LPS. (a) Membrane fractionation of wild type PAO1, lptH and lptE conditional mutants. PAO1 and the lptE conditional mutant (lptE) were cultured in MH with or without 0.5% arabinose (ARA) as described in the legend to . The sample lptE 2nd corresponds to the lptE mutant pre-cultured in the absence of ARA (second refresh in ). PAO1 and lptE cells were collected for membrane fractionation when the cultures reached an OD600 = 0.8. As control, LptH-deficient cells (lptH), obtained through the culturing strategy described in Fig. S6, were included in the analysis. Membranes were fractionated by sucrose density gradient, and the different fractions were immunoblotted using antibodies against the IM protein XcpY, the OM protein OprI or LPS, as indicated on the right of each blot. Fractions corresponding to the IM or OM on the basis of the results obtained with the wild type PAO1 are indicated on the top of the figure. Images are representative of two experiments giving similar results. (b) Quantification of KDO in the above-mentioned strains cultured as described in panel A. Values are the mean (± SD) of three independent experiments. The asterisk indicates a statistically-significant decrease with respect to all other samples (P < 0.05, Kruskal-Wallis).
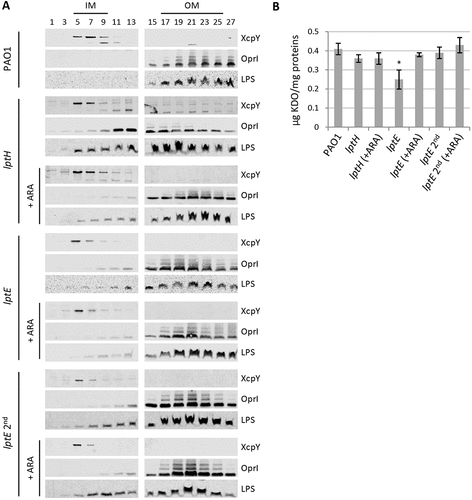
Figure 5. MALDI-TOF analysis of lipid A extracted from wild type (PAO1) and lptE mutant cells cultured in MH with or without 0.5% arabinose (ARA). lptE 2nd corresponds to the lptE conditional mutant cultured for two subsequent passages in the absence of ARA (see the legend of for details). Spectra were obtained in the ion negative mode, thus m/z values correspond to (molecular mass – 1)/1. Spectra are representative of three biological replicates. Relevant lipid A forms are shown with cartoons: m/z = 1646, penta-acylated lipid A; m/z = 1616, hexa-acylated lipid A; m/z = 1684, penta-acylated lipid A with the addition of a palmitoyl group (in blue). Asterisks indicate lipid A forms that vary due to the removal (m/z −16) or addition (m/z + 16) of a hydroxyl group to the secondary C12 acyl chains. The chemical structures of the identified lipid A forms are shown in Fig. S9.
LptE affects LptD levels
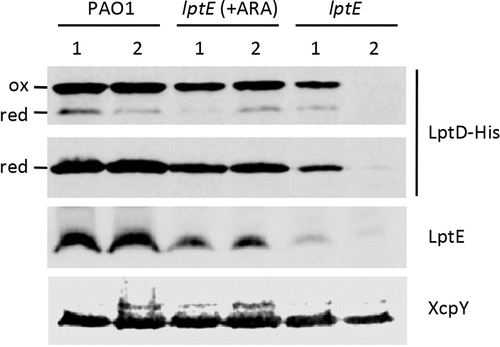
LptE directly participates to and is essential for LPS transport in E. coli [Citation8], while it is dispensable for LPS transfer to the OM in N. meningitidis [Citation24]. However, in both bacteria LptE was found to play an important role in the maturation of the integral OM component of the LPS transport machinery LptD, acting as a chaperone to stabilize LptD and/or assist LptD in folding and OM insertion [Citation20–Citation22,Citation24]. We therefore sought to verify whether this chaperone function of LptE was also conserved in P. aeruginosa. To this aim, we first engineered the genome of the PAO1 and lptE conditional mutant strains to express a 6-His tagged LptD instead of the wild type protein (Table S1 and Fig. S1), and then compared the amounts of LptD-6His in membrane fractions from LptE-replete and -depleted cells by western blot analysis with an anti-6His antibody. LptD-6His levels were reduced in LptE-depleted cells as compared to wild type PAO1, and they fell to barely detectable levels in the lptE conditional mutant grown for two passages in the absence of arabinose (). Growth of the lptE conditional mutant in the presence of arabinose partially restored LptD-6His levels, accounting for about 40% of the wild type levels (). The majority of LptD was found in the oxidized (mature) form in all strains, with the only exception of lptE 2nd samples, in which the amount of LptD was too low to draw conclusions about its oxidation state (). Together, these results indicate that LptE is crucial for LptD maturation and/or stability also in P. aeruginosa, and suggest that, as previously observed in N. meningitidis [Citation24], very low levels of LptD may be sufficient to allow insertion of LPS into the OM and, thus, to sustain growth of P. aeruginosa.
Figure 6. LptD levels in LptE-depleted cells. Total membrane samples were obtained from PAO1 lptD-6his and lptE lptD-6his cells grown as described in the legend of . Cells were collected at OD600 = 0.8, both for the first (lanes 1) and the second refresh (lanes 2). Total membrane proteins corresponding to an OD600 = 1.6 were loaded in each lane. Filters were immunoblotted using antibodies raised against the 6His tag to detect LptD-6His, against LptE, or against XcpY, as indicated on the right of each blot. To analyze the oxidation state of LptD (red, reduced; ox, oxidized), membrane samples were treated with SDS loading buffer containing (lower panel) or not 5% β-mercaptoethanol (upper panel). Images are representative of three independent experiments giving similar results.
LptE is important for P. aeruginosa virulence
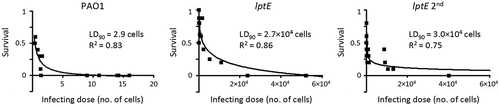
The above results demonstrate that LptE-depleted cells of P. aeruginosa are able to grow under in vitro conditions ( and S4) even if they appear to be strongly defective in cell envelope stability ( and ). To evaluate the impact of LptE depletion on P. aeruginosa virulence, we tested the lptE conditional mutant in a simple model of acute infection based on the larvae of the insect G. mellonella [Citation53]. This infection model was successfully used by our group to verify and compare the pathogenicity of P. aeruginosa arabinose-dependent conditional mutants in different essential genes [Citation35,Citation44,Citation59]. G. mellonella larvae were infected with different doses of either the wild type or the lptE conditional mutant, pre-cultured either in MH with 0.5% arabinose or in the absence of arabinose (lptE 2nd in ), and lethality was monitored for 4 days. The LD90 (i.e. the number of bacterial cells that is theoretically required to kill 90% of the larvae) inferred from survival curves was about 3 cells for the wild type PAO1, in line with previous reports [Citation35,Citation44,Citation59], while it was about 9,000-and 10,000-fold higher for the lptE conditional mutant pre-cultured with or without arabinose, respectively (). This infection assay demonstrates that LptE depletion severely impairs the pathogenicity of P. aeruginosa in G. mellonella larvae, suggesting that this protein can play a relevant role during the P. aeruginosa infectious process. Notably, the defect in infectivity of LptE-depleted cells is however not comparable to that of a mutant completely impaired in LPS transport, i.e. the lptH conditional mutant, which showed an LD90 of 1.6 × 107 cells in the same animal model [Citation35], more than 500-fold higher than that of the lptE conditional mutant ().
Figure 7. Effect of LptE depletion on P. aeruginosa virulence in G. mellonella. Survival curves of G. mellonella larvae infected with different doses of P. aeruginosa PAO1 or the lptE conditional mutant cultured in MH with 0.5% arabinose (PAO1 and lptE) or in MH without arabinose (lptE 2nd). Ten larvae were infected with each infecting dose in at least three independent experiments. LD90 and R2 values for each curve are shown.
Discussion
In the last decade, several research groups have made significant efforts to understand the molecular and biochemical bases of LPS transport across the cell envelope and insertion into the OM. However, the majority of the information gained on this crucial OM biogenesis process derives from studies involving only two model Gram-negative bacteria, E. coli and the non-LPS-dependent N. meningitidis (reviewed in [Citation6,Citation60–Citation64]). Notably, detailed investigation of LPS transport in only two organisms was sufficient to identify a major distinctive feature of the Lpt machinery. Indeed, while in E. coli the Lpt machinery consists of seven essential components, the N. meningitidis Lpt machinery can work as a six-component complex, since lptE gene deletion does not impair LPS transport to the cell surface [Citation24]. In this work, we sought to verify the role of LptE in LPS transport in P. aeruginosa through conditional mutagenesis. In our P. aeruginosa lptE conditional mutant, we found that LptE depletion only marginally affects LPS localization and maturation ( and ). Moreover, arabinose-mediated increase in LptE expression does not change the LPS profile of the lptE conditional mutant (), indicating that LPS localization is not strictly dependent on LptE levels. These results suggest that LptE could not be directly involved in LPS transport in P. aeruginosa, similarly to what observed in N. meningitidis [Citation24] but in contrast with its proposed LPS-binding role in E. coli [Citation8,Citation20,Citation25]. This hypothesis would be indirectly supported by a recent report showing that P. aeruginosa LptE has very weak affinity for LPS in vitro [Citation65]. On the other hand, as in E. coli and N. meningitidis [Citation20–Citation22,Citation24], LptE appears to be crucial for LptD biogenesis and/or stability also in P. aeruginosa (), indicating that the chaperone function of LptE may be conserved in Gram-negative bacteria. It is interesting to note that both bacterial growth and LPS transport were only slightly impaired in P. aeruginosa LptE-depleted cells in spite of the low levels of LptD ( and ), as previously reported also for the N. meningitidis lptE mutant [Citation24]. Considering that the essentiality of LptD in P. aeruginosa has been experimentally demonstrated [Citation33], this evidence suggests that strongly reduced amounts of LptD (and LptE) may be sufficient to sustain LPS transport and growth in P. aeruginosa, as previously observed in N. meningitidis [Citation24]. Notably, LptE-depleted cells were proficient in growth under in vitro conditions, but strongly defective in detergent and antibiotics resistance ( and ). This is suggestive of major defects in cell envelope biogenesis and/or integrity, which could reasonably account for the significantly reduced virulence of LptE-depleted cells in the G. mellonella model of infection (). Notably, no relevant differences were observed in the infectivity of LptE-depleted cells pre-cultured or not with arabinose (), even if these cells displayed different susceptibility levels to SDS and antibiotics ( and ). Although we are aware that is not easy to directly correlate results from in vitro assays with the outcome of infection models, this finding suggests that either maximum LptE depletion is reached earlier during in vivo growth as compared to in vitro conditions or, alternatively, that the strongly reduced infectivity of LptE-depleted cells might (also) be due to some additional role(s) of LptE during infection other than its relevant effect on cell envelope stability. It is also interesting to note that P. aeruginosa sensitivity to detergents and antibiotics showed differential dependence on LptE and LptD levels. Indeed, partial complementation of LptE and LptD expression in the lptE conditional mutant grown in the presence of arabinose (, S3 and ) was sufficient to fully re-establish wild type levels of SDS resistance (), but only moderately restored antibiotics resistance (). Antibiotics resistance in the lptE conditional mutant was however re-established at almost wild type levels upon complete restoration of LptE levels through plasmid-mediated expression (). Moreover, the LPS profile of the lptE conditional mutant was very similar to that of the lptH conditional mutant grown in the presence of 0.5% arabinose (), whereas only the former was strongly defective in drug resistance (), implying that the high antibiotic sensitivity of the lptE mutant reasonably does not depend on its slightly impaired LPS transport. Overall, our data suggest that the detrimental effect on cell envelope integrity caused by LptE depletion could involve two different mechanism(s). First, reduced LptE expression strongly impairs LptD maturation, and although P. aeruginosa seems to tolerate low LptD levels, LptD depletion likely causes slight defects in LPS transport () and, consequently, in cell division (), that could represent the basis for the high SDS sensitivity of LptE-depleted cells, as previously observed for other P. aeruginosa mutants defective in cell division [Citation44]. Second, LptE depletion may result in a fraction of LptD channels in the OM that are not properly stabilized and plugged by LptE. Improperly folded and/or un-plugged LptD channels in the OM could compromise the OM barrier, ultimately leading to impaired antibiotic resistance, as previously reported in E. coli [Citation66]. This would also be in agreement with a recent work showing that the replacement of the P. aeruginosa lptD gene with defective alleles, encoding LptD variants with a deletion in a loop predicted to interact with the LptE plug, drastically increases the susceptibility of P. aeruginosa to several classes of antibiotics [Citation67]. Recently, the structure of the P. aeruginosa LptD/E has been solved and compared to that of other Gram-negative bacteria [Citation68]. LptD/E structures exhibit an identical fold, although the P. aeruginosa LptD/E complex slightly differs due to (i) a larger luminal volume, composed of two cavities in contrast to only one in other structures, and (ii) the presence of additional insertion loops in the extracellular region of the barrel, conferring a larger molecular surface [Citation68]. Moreover, P. aeruginosa LptD carries a long (~ 90-aa) insertion at the N-terminus, which is absent in other LptD homologues [Citation34,Citation68]. The biological implication of these unique properties of P. aeruginosa LptD/E is not clear. While it has been proposed that they could be due to the specific structure of the P. aeruginosa LPS [Citation64], it cannot be ruled out that such structural features could (also) account for a specific maturation process and/or additional functions of the P. aeruginosa LptD/E complex. An aspect that deserves further investigation is the effect of LptE depletion on LPS biogenesis and maturation. Our data suggest that reduced LptE expression can cause a reduction in LPS biogenesis, while LPS transport and maturation are not or only slightly affected ( and ). Notably, after prolonged LptE depletion (lptE 2nd samples), P. aeruginosa cells appear to restore normal LPS biosynthesis levels, although the hydroxylation state of lipid A is considerably increased ( and ). P. aeruginosa has two homologues (LpxO1 and LpxO2) of the lipid A-specific hydroxylase LpxO first characterized in Salmonella, which are likely responsible for the hydroxylation of the two secondary acyl chains of lipid A in P. aeruginosa (Fig. S9) [Citation58]. Although the specific role of lipid A hydroxylation in P. aeruginosa has not been directly investigated yet, it has been hypothesized that changes in the hydroxylation state of lipid A could influence the fluidity and/or permeability of the OM [Citation69,Citation70]. At present, however, we cannot state whether the observed increase in lipid A hydroxylation represents a specific strategy of P. aeruginosa to adapt to an LptE/LptD-depleted LPS transport machinery or, alternatively, a general stress response of cells growing with slightly impaired LPS insertion into the OM. Studies involving the generation of lpxO1/2 single and double mutants are clearly required to elucidate the function and/or effect of lipid A hydroxylation in P. aeruginosa. Finally, it should be noted that, while LptE depletion strongly impaired LptD folding and stability (), in our study we did not observe the appearance of any aminorabinosylated lipid A forms ( and S9), which were instead found to accumulate in a P. aeruginosa lptD conditional mutant cultured with a very low inducer concentration [Citation33]. Whether these differences in lipid A modifications are due to experimental/technical issues (e.g. different culture media, different growth rates) or to different adaptive responses to LptE or LptD depletion remains to be determined. In conclusion, this work provides evidence that LptE plays a crucial role in P. aeruginosa due to its importance as chaperone and plug for LptD. It should be noted that, while our lptE conditional mutant is viable and grows in vitro with strongly reduced LptE levels, many attempts to obtain a clean lptE deletion mutant were unsuccessful (see Results). Therefore, it is plausible that the growth phenotype of the lptE conditional mutant is influenced by such residual, low-level expression of LptE, making it impossible to definitely conclude whether LptE represents a bona fide essential protein in P. aeruginosa. To date, the role of LptE in LPS transport and cell physiology has only been investigated in E. coli, N. meningitidis and P. aeruginosa, although bioinformatics analyses have identified LptE homologues in almost all Proteobacteria and in some non-proteobacterial LPS-producing Gram-negatives [Citation24,Citation25,Citation60]. It would therefore be interesting to investigate to what extent the roles of LptE in LPS transport, LptD maturation and/or its LPS transport-independent effect on cell envelope homeostasis are conserved among Gram-negative bacteria. This information could also have important clinical implications, also considering that the LptD/E complex has been pharmacologically validated as a promising antibiotic target in vitro [Citation32,Citation33]. Understanding how the LptD/E translocon works in different bacteria and the effect of its depletion on bacterial pathogenicity, in vivo fitness and resistance could indeed drive the development of further compounds targeting this crucial cell envelope biogenesis step with narrow- or broad-spectrum activity against Gram-negative pathogens.
Supplemental Material
Download PDF (1.2 MB)Acknowledgments
We are grateful to Prof. Alexandre Leitão and Prof. Romé Voulhoux for providing the anti-OprI and anti-XcpY antibodies, respectively. We also thank Prof. Robert Ernst for sharing the protocol and suggestions for the extraction and analysis of lipid A. This work was supported by grants from the Italian Cystic Fibrosis Research Foundation (FFC#10/2013), the Sapienza University of Rome (Ateneo 2015) and the Pasteur Institute-Cenci Bolognetti Foundation to F.I, and from the MIUR_Regione Lombardia (project n. 30190679) to A.P.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary Material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Silhavy TJ, Kahne D, Walker S. The bacterial cell envelope. Cold Spring Harb Perspect Biol. 2010;2:a000414.
- Vollmer W, Blanot D, de Pedro MA. Peptidoglycan structure and architecture. FEMS Microbiol Rev. 2008;32:149–167.
- Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev. 2003;67:593–656.
- Nikaido H. Restoring permeability barrier function to outer membrane. Chem Biol. 2005;12:507–509.
- Ruiz N, Wu T, Khane D, et al. Probing the barrier function of the outer membrane with chemical conditionality. ACS Chem Biol. 2006;1:385–395.
- Sperandeo P, Martorana AM, Polissi A. The lipopolysaccharide transport (Lpt) machinery: a non-conventional transporter for lipopolysaccharide assembly at the outer membrane of Gram-negative bacteria. J Biol Chem. 2017;292:17981–17990.
- Chng SS, Gronenberg LS, Kahne D. Proteins required for lipopolysaccharide assembly in Escherichia coli form a transenvelope complex. Biochemistry. 2010;49:4565–4567.
- Wu T, McCandlish AC, Gronenberg LS, et al. Identification of a protein complex that assembles lipopolysaccharide in the outer membrane of Escherichia coli. Proc Natl Acad Sci U S A. 2006;103:11754–11759.
- Ruiz N, Gronenberg LS, Kahne D, et al. Identification of two inner-membrane proteins required for the transport of lipopolysaccharide to the outer membrane of Escherichia coli. Proc Natl Acad Sci U S A. 2008;105:5537–5542.
- Sperandeo P, Lau FK, Carpentieri A, et al. Functional analysis of the protein machinery required for transport of lipopolysaccharide to the outer membrane of Escherichia coli. J Bacteriol. 2008;190:4460–4469.
- Narita S, Tokuda H. Biochemical characterization of an ABC transporter LptBFGC complex required for the outer membrane sorting of lipopolysaccharides. FEBS Lett. 2009;583:2160–2164.
- Sperandeo P, Cescutti R, Villa R, et al. Characterization of lptA and lptB, two essential genes implicated in lipopolysaccharide transport to the outer membrane of Escherichia coli. J Bacteriol. 2007;189:244–253.
- Freinkman E, Okuda S, Ruiz N, et al. Regulated assembly of the transenvelope protein complex required for lipopolysaccharide export. Biochemistry. 2012;51:4800–4806.
- Sperandeo P, Villa R, Martorana AM, et al. New insights into the Lpt machinery for lipopolysaccharide transport to the cell surface: LptA-LptC interaction and LptA stability as sensors of a properly assembled transenvelope complex. J Bacteriol. 2011;193:1042–1053.
- Okuda S, Freinkman E, Kahne D. Cytoplasmic ATP hydrolysis powers transport of lipopolysaccharide across the periplasm in E.. Coli. Science.. 2012;338:1214–1217.
- Luo Q, Yang X, Yu S, et al. Structural basis for lipopolysaccharide extraction by ABC transporter LptB(2)FG. Nat Struct Mol Biol. 2017;24:469–474.
- Freinkman E, Chng SS, Kahne D. The complex that inserts lipopolysaccharide into the bacterial outer membrane forms a two-protein plug-and-barrel. Proc Natl Acad Sci U S A. 2011;108:2486–2491.
- Qiao S, Luo Q, Zhao Y, et al. Structural basis for lipopolysaccharide insertion in the bacterial outer membrane. Nature. 2014;511:108–111.
- Dong H, Xiang Q, Gu Y, et al. Structural basis for outer membrane lipopolysaccharide insertion. Nature. 2014;511:52–56.
- Chng SS, Ruiz N, Chimalakonda G, et al. Characterization of the two-protein complex in Escherichia coli responsible for lipopolysaccharide assembly at the outer membrane. Proc Natl Acad Sci U S A. 2014;107:5363–5368.
- Ruiz N, Chng SS, Hiniker A, et al. Nonconsecutive disulfide bond formation in an essential integral outer membrane protein. Proc Natl Acad Sci U S A. 2010;107:12245–12250.
- Chimalakonda G, Ruiz N, Chng SS, et al. Lipoprotein LptE is required for the assembly of LptD by the beta-barrel assembly machine in the outer membrane of Escherichia coli. Proc Natl Acad Sci U S A. 2011;108:2492–2497.
- Chng SS, Xue M, Garner RA, et al. Disulfide rearrangement triggered by translocon assembly controls lipopolysaccharide export. Science. 2012;337:1665–1668.
- Bos MP, Tommassen J. The LptD chaperone LptE is not directly involved in lipopolysaccharide transport in Neisseria meningitidis. J Biol Chem. 2011;286:28688–28696.
- Malojčić G, Andres D, Grabowicz M, et al. LptE binds to and alters the physical state of LPS to catalyze its assembly at the cell surface. Proc Natl Acad Sci U S A. 2014;111:9467–9472.
- Steeghs L, Den Hartog R, Den Boer A, et al. Meningitis bacterium is viable without endotoxin. Nature. 1998;392:449–450.
- Steeghs L, de Cock H, Evers E, et al. Outer membrane composition of a lipopolysaccharide-deficient Neisseria meningitidis mutant. Embo J. 2001;20:6937–6945.
- Bos MP, Tommassen J. Viability of a capsule- and lipopolysaccharide-deficient mutant of Neisseria meningitidis. Infect Immun. 2005;73:6194–6197.
- Bos MP, Tefsen B, Geurtsen J, et al. Identification of an outer membrane protein required for the transport of lipopolysaccharide to the bacterial cell surface. Proc Natl Acad Sci U S A. 2004;101:9417–9422.
- Tefsen B, Bos MP, Beckers F, et al. MsbA is not required for phospholipid transport in Neisseria meningitidis. J Biol Chem. 2005;280:35961–35966.
- Poole K. Pseudomonas aeruginosa: resistance to the max. Front Microbiol. 2011;2:65.
- Srinivas N, Jetter P, Ueberbacher BJ, et al. Peptidomimetic antibiotics target outer-membrane biogenesis in Pseudomonas aeruginosa. Science. 2010;327:1010–1013.
- Werneburg M, Zerbe K, Juhas M, et al. Inhibition of lipopolysaccharide transport to the outer membrane in Pseudomonas aeruginosa by peptidomimetic antibiotics. Chembiochem. 2012;13:1767–1775.
- Bollati M, Villa R, Gourlay LJ, et al. Crystal structure of LptH, the periplasmic component of the lipopolysaccharide transport machinery from Pseudomonas aeruginosa. FEBS J. 2015;282:1980–1997.
- Fernández-Piñar R, Lo Sciuto A, Rossi A, et al. In vitro and in vivo screening for novel essential cell-envelope proteins in Pseudomonas aeruginosa. Sci Rep. 2015;5:17593.
- Mdluli KE, Witte PR, Kline T, et al. Molecular validation of LpxC as an antibacterial drug target in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2006;50:2178–2184.
- Delucia AM, Six DA, Caughlan RE, et al. Lipopolysaccharide (LPS) inner-core phosphates are required for complete LPS synthesis and transport to the outer membrane in Pseudomonas aeruginosa PAO1. MBio. 2011;2:pii: e00142–11.
- Jacobs MA, Alwood A, Thaipisuttikul I, et al. Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2003;100:14339–14344.
- Liberati NT, Urbach JM, Miyata S, et al. An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc Natl Acad Sci U S A. 2006;103:2833–2838.
- Lee SA, Gallagher LA, Thongdee M, et al. General and condition-specific essential functions of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2015;112:5189–5194.
- Turner KH, Wessel AK, Palmer GC, et al. Essential genome of Pseudomonas aeruginosa in cystic fibrosis sputum. Proc Natl Acad Sci U S A. 2015;112:4110–4115.
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: a Laboratory Manual. 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1989.
- Heeb S, Blumer C, Haas D. Regulatory RNA as mediator in GacA/RsmA-dependent global control of exoproduct formation in Pseudomonas fluorescens CHA0. J Bacteriol. 2002;84:1046–1056.
- Lo Sciuto A, Fernández-Piñar R, Bertuccini L, et al. The periplasmic protein TolB as a potential drug target in Pseudomonas aeruginosa. PLoS One. 2014;9:e103784.
- Milton DL, O’Toole R, Horstedt P, et al. Flagellin A is essential for the virulence of Vibrio anguillarum. J Bacteriol. 1996;178:1310–1319.
- Hoang TT, Kutchma AJ, Becher A, et al. Integration-proficient plasmids for Pseudomonas aeruginosa: site-specific integration and use for engineering of reporter and expression strains. Plasmid. 2000;43:59–72.
- Volokhina EB, Beckers F, Tommassen J, et al. The beta-barrel outer membrane protein assembly complex of Neisseria meningitidis. J Bacteriol. 2009;191:7074–7085.
- Liu YY, Chandler CE, Leung LM, et al. Structural modification of lipopolysaccharide conferred by mcr-1 in gram-negative ESKAPE pathogens. Antimicrob Agents Chemother. 2017;61:pii: e00580-17.
- Hitchcock PJ, Brown TM. Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J Bacteriol. 1983;154:269–277.
- Lam JS, Anderson EM, Hao Y. LPS quantitation procedures. Methods Mol Biol. 2014;1149:375–402.
- Michel G, Bleves S, Ball G, et al. Mutual stabilization of the XcpZ and XcpY components of the secretory apparatus in Pseudomonas aeruginosa. Microbiology. 1998;144:3379–3386.
- Basto AP, Piedade J, Ramalho R, et al. A new cloning system based on the OprI lipoprotein for the production of recombinant bacterial cell wall-derived immunogenic formulations. J Biotechnol. 2012;157:50–63.
- Jander G, Rahme LG, Ausubel FM. Positive correlation between virulence of Pseudomonas aeruginosa mutants in mice and insects. J Bacteriol. 2000;182:3843–3845.
- Antunes LC, Imperi F, Carattoli A, et al. Deciphering the multifactorial nature of Acinetobacter baumannii pathogenicity. PLoS One. 2011;6:e22674.
- Meisner J, Goldberg JB. The Escherichia coli rhaSR-PrhaBAD Inducible promoter system allows tightly controlled gene expression over a wide range in Pseudomonas aeruginosa. Appl Environ Microbiol. 2016;82:6715–6727.
- Bishop RE. Structural biology of membrane-intrinsic beta-barrel enzymes: sentinels of the bacterial outer membrane. Biochim Biophys Acta. 2008;1778:1881–1896.
- Knirel YA, Bystrova OV, Kocharova NA, et al. Conserved and variable structural features in the lipopolysaccharide of Pseudomonas aeruginosa. J Endotoxin Res. 2006;12:324–336.
- King JD, Kocíncová D, Westman EL, et al. Review: lipopolysaccharide biosynthesis in Pseudomonas aeruginosa. Innate Immun. 2009;15:261–312.
- Pasqua M, Visaggio D, Lo Sciuto A, et al. The ferric uptake regulator Fur is conditionally essential in Pseudomonas aeruginosa. J Bacteriol. 2017;199:pii: e00472–17.
- Putker F, Bos MP, Tommassen J. Transport of lipopolysaccharide to the Gram-negative bacterial cell surface. FEMS Microbiol Rev. 2015;39:985–1002.
- May JM, Sherman DJ, Simpson BW, et al. Lipopolysaccharide transport to the cell surface: periplasmic transport and assembly into the outer membrane. Philos Trans R Soc Lond B Biol Sci. 2015;370:pii: 20150027.
- Simpson BW, May JM, Sherman DJ, et al. Lipopolysaccharide transport to the cell surface: biosynthesis and extraction from the inner membrane. Philos Trans R Soc Lond B Biol Sci. 2015;370:pii: 20150029.
- Okuda S, Sherman DJ, Silhavy TJ, et al. Lipopolysaccharide transport and assembly at the outer membrane: the PEZ model. Nat Rev Microbiol. 2016;14:337–345.
- Botos I, Noinaj N, Buchanan SK. Insertion of proteins and lipopolysaccharide into the bacterial outer membrane. Philos Trans R Soc Lond B Biol Sci. 2017;372:pii: 20160224.
- Moehle K, Kocherla H, Bacsa B, et al. Solution Structure and Dynamics of LptE from Pseudomonas aeruginosa. Biochemistry. 2016;55:2936–2943.
- Grabowicz M, Yeh J, Silhavy TJ. Dominant negative lptE mutation that supports a role for LptE as a plug in the LptD barrel. J Bacteriol. 2013;195:1327–1334.
- Balibar CJ, Grabowicz M. Mutant Alleles of lptD increase the permeability of Pseudomonas aeruginosa and define determinants of intrinsic resistance to antibiotics. Antimicrob Agents Chemother. 2015;60:845–854.
- Botos I, Majdalani N, Mayclin SJ, et al. Structural and functional characterization of the LPS transporter LptDE from Gram-negative pathogens. Structure. 2016;24:965–976.
- Gibbons HS, Reynolds CM, Guan Z, et al. An inner membrane dioxygenase that generates the 2-hydroxymyristate moiety of Salmonella lipid A. Biochemistry. 2008;47:2814–2825.
- Hittle LE, Powell DA, Jones JW, et al. Site-specific activity of the acyltransferases HtrB1 and HtrB2 in Pseudomonas aeruginosa lipid A biosynthesis. Pathog Dis. 2015;73:ftv053.