
ABSTRACT
The complement system is pivotal in the defense against invasive disease caused by Neisseria meningitidis (Nme, meningococcus), particularly via the membrane attack complex. Complement activation liberates the anaphylatoxins C3a and C5a, which activate three distinct G-protein coupled receptors, C3aR, C5aR1 and C5aR2 (anaphylatoxin receptors, ATRs). We recently discovered that C5aR1 exacerbates the course of the disease, revealing a downside of complement in Nme sepsis. Here, we compared the roles of all three ATRs during mouse nasal colonization, intraperitoneal infection and human whole blood infection with Nme. Deficiency of complement or ATRs did not alter nasal colonization, but significantly affected invasive disease: Compared to WT mice, the disease was aggravated in C3ar−/- mice, whereas C5ar1−/- and C5ar2−/- mice showed increased resistance to meningococcal sepsis. Surprisingly, deletion of either of the ATRs resulted in lower cytokine/chemokine responses, irrespective of the different susceptibilities of the mice. This was similar in ex vivo human whole blood infection using ATR inhibitors. Neutrophil responses to Nme were reduced in C5ar1−/- mouse blood. Upon stimulation with C5a plus Nme, mouse macrophages displayed reduced phosphorylation of ERK1/2, when C5aR1 or C5aR2 were ablated or inhibited, suggesting that both C5a-receptors prime an initial macrophage response to Nme. Finally, in vivo blockade of C5aR1 alone (PMX205) or along with C5aR2 (A8Δ71−73) resulted in ameliorated disease, whereas neither antagonizing C3aR (SB290157) nor its activation with a “super-agonist” peptide (WWGKKYRASKLGLAR) demonstrated a benefit. Thus, C5aR1 and C5aR2 augment disease pathology and are interesting targets for treatment, whereas C3aR is protective in experimental meningococcal sepsis.
Introduction
Although usually a harmless colonizer of the upper respiratory tract, the Gram-negative bacterium Neisseria meningitidis (Nme, meningococcus) is feared for its potential to cause rapidly progressing invasive diseases such as meningitis and septicemia [Citation1]. Initial symptoms of invasive meningococcal diseases (IMD) are often not timely recognized before the disease rapidly progresses, thereby limiting the time window for successful treatment; in fact, most deaths caused by meningococcal septicemia occur within 24 h after hospital admission [Citation2]. The incidence of IMD is highest in infants and toddlers (<5 years), with a fatality rate around 5–20%. Approximately 20% of survivors suffer permanent damage, including neuronal and cognitive defects, deafness, chronic pain and limb amputations [Citation2].
In order to survive and replicate in the bloodstream, Nme must avoid killing by the complement system. In fact, Nme evolved intricate mechanisms to counteract complement effector functions [Citation3]. The major virulence factor of Nme in protecting against complement is a polysaccharide capsule, by which 12 different serogroups can be distinguished [Citation4]. Furthermore, Nme sequester the complement regulator fH via the outer membrane proteins fHbp [Citation5] and NspA [Citation6]. Both, capsule and fHbp are used as antigens in meningococcal vaccines [Citation7,Citation8].
The complement system is by far the most important branch of the innate immune system protecting against IMD. Protection critically depends on membrane attack complex (MAC) mediated lysis, as evidenced by the extremely enhanced risk of infection in individuals with defects in terminal complement components required for the MAC assembly [Citation9]. While the role of the MAC in meningococcal disease is well characterized, there is only limited knowledge about the impact of other effector mechanisms triggered by the complement system. Particularly, the roles of the pro-inflammatory mediators C3a and C5a, which are small cleavage fragments liberated during complement activation, have drawn very little attention thus far. These so-called “anaphylatoxins” bind to and thereby activate their corresponding cellular receptors, the C3aR, C5aR1 and C5aR2 (formerly called C5L2) [Citation10]. These “anaphylatoxin receptors” (ATRs) belong to the superfamily of G-protein-coupled receptors, and their activation triggers a multitude of innate immune and inflammatory responses. These include granulocyte and monocyte chemotaxis as well as degranulation, mast cell degranulation [Citation10], vasodilation and endothelial activation [Citation11,Citation12], modulation of cytokine release [Citation13], modulation of T-helper cell polarization [Citation14] and tissue macrophage activation [Citation15].
C5a is the most potent anaphylatoxin, driving a predominantly pro-inflammatory potential response through C5aR1 activation [Citation16]. C5aR1 is a G-protein coupled receptor highly expressed on neutrophil granulocytes and its activation aggravates chronic and acute inflammatory conditions, such as sepsis [Citation17], ischemia-reperfusion injury [Citation18], inflammatory bowel disease [Citation19] and others. In accordance, C5aR1 has been suggested as a key therapeutic target to treat inflammatory disorders [Citation20,Citation21].
In addition to C5aR1, C5a can also bind to C5aR2, which is uncoupled from G-protein signaling, mainly due to a mutation of the crucial DRY motif [Citation22]. In fact, the functional role of C5aR2 is controversially debated as it has been described as a non-signaling scavenger receptor [Citation23], an anti-inflammatory molecule [Citation24,Citation25], a pro-inflammatory receptor-like C5aR1 [Citation26,Citation27], or as a modulator of C5aR1 and C3aR function [Citation28,Citation29]. In addition, there is data suggesting that C5aR2 is involved in metabolic processes including stimulation of triglyceride synthesis [Citation30].
C3aR is activated only by C3a, thereby inducing chemotaxis of eosinophils, mast cells and macrophages [Citation31,Citation32]. Furthermore, C3a, like C5a, increases vascular permeability [Citation33,Citation34] and contributes to airway hyper-responsiveness [Citation35]. C3aR is implicated in the systemic response to LPS challenge, as mice lacking C3aR display enhanced susceptibility and higher levels of IL-1β [Citation36]. Similarly, C3aR deficient mice are more susceptible to infection with the Gram-positive bacterium Listeria monocytogenes [Citation37]. C3aR expression on neutrophils appears to be predominantly anti-inflammatory, by inhibiting neutrophil mobilization into the blood [Citation38]. Thus, although often termed a pro-inflammatory receptor-like C5aR1, C3aR has many anti-inflammatory facets. Therefore, it has recently been suggested to refer to C3aR as an inflammatory modulator [Citation39].
Multiple inflammatory pathways are activated during meningococcal disease [Citation40], which could be modulated by the anaphylatoxins and their receptors. A contribution of the anaphylatoxin receptor family members to the inflammation-driven disease pathology in meningococcal disease appears likely. In support of this, our previous work demonstrated a significant C5aR1-driven aggravation of experimental N. meningitidis sepsis in a mouse model as well as in human whole blood [Citation41].
Here, we aimed to extend our analyses to the relative contributions of all three anaphylatoxin receptors, C3aR, C5aR1 and C5aR2 to meningococcal nasopharyngeal colonization as well as sepsis pathology using in vivo and ex vivo models of disease.
Results
Asymptomatic Nme colonization is not significantly affected by complement or ATRS in CEACAM1-humanized mice
The complement system is the major contributor to the defense against Nme in the blood, both in human disease [Citation3] as well as in the mouse model [Citation41]. Yet, IMD is preceded by Nme colonization of the nasopharyngeal mucosa, and it is currently unknown whether mechanisms of serum resistance in Nme also benefit their mucosal colonization. Thus, we wanted to test whether complement and the anaphylatoxins as important attractors of phagocytes impact on Nme mucosal colonization in vivo. We, therefore, used our CEACAM1-humanized mouse model [Citation42–Citation44] in combination with deficiencies in C3 (C3−/-), C5 (C5−/-), C3aR1 (C3ar1-/-) C5aR1 (C5ar1−/-) or C5aR2 (C5ar2−/-) to monitor Nme nasal colonization following intranasal infection with 105 CFU. Colonization levels of the mice were similar across all genotypes at day 1 and day 3 (). At day 14, there was a trend to lower bacterial burden and colonization frequency across all mouse lines carrying any complement deficiency compared to the complement-sufficient control strain, but this was not statistically significant. Of note, none of the mice showed signs of disease or a positive blood culture taken 4 h after i.n. infection. In all, complement or the ATRs apparently do not significantly affect nasopharyngeal colonization in the CEACAM1-humanized mouse model.
Figure 1. Impact of complement and ATRs on asymptomatic nasal colonization following i.n. infection versus invasive disease following i.p. injection of Nme in mice. Nasal colonization (a,b): Interbreeds of mice expressing human CEACAM1 with mice lacking C3 (C3−/-), C5 (Hc°/0), C3aR (C3ar1−/-), C5aR1 (C5ar1−/-) or C5aR2 (C5ar2−/-) were intranasally infected with 105 CFU of Nme and sacrificed at indicated time points (day 1, 3, 14) to sample nasal tissues for viable Nme (n = 11 animals per cohort). (a) Recovered Nme expressed as CFU per individual animal at indicated time points after infection. (b) Percentage of mice colonized at each time point. Invasive disease (c-h): (c,e) 11 to 15 mice of each genotype were i.p. infected with a total dose of 105 CFU of serogroup B Nme strain MC58 per mouse and followed for 48 h before cessation of the experiment. (f-h) Seven WT and 5 C3ar1−/- mice were subjected to a sub-lethal dose of 5 × 104 CFU per animal. (c,f): Survival rates after infection. (d,g) Clinical scoring over time. Note that only the first 24 h is included in the analysis, since only a single animal in the WT and C3ar1−/- cohort survived past this time point. (e,h) Bacteremia in tail vein blood samples during the course of infection as indicated by the time points below the x-axis. Bacteremia plotted at ’18–27 h’ includes values at 24 h of all mice surviving longer than 27 h and the values of the non-survivors at their time of death (by sacrificing at the “humane endpoint”) within this time window. Closed circles indicate surviving animals, open circles indicate non-survivors. Limit of detection was 500 CFU/ml.
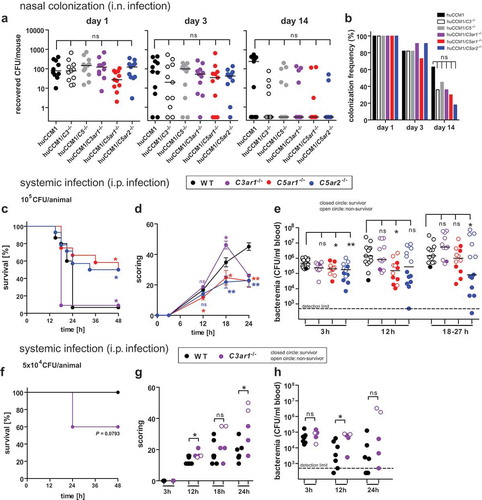
Deficiency of C5ar1 or C5ar2 ameliorates the course of experimental meningococcal sepsis in mice, whereas deficiency of C3ar worsens it
Previously, we uncovered a significantly increased resistance of C5ar1−/- mice over wild-type (WT) mice in a murine sepsis model of N. meningitidis [Citation41]. To determine whether the other anaphylatoxin receptors, C3aR and C5aR2, also impact on the outcome of experimental meningococcal sepsis, we compared the course of disease in cohorts of WT, C3ar1−/-, C5ar1−/- and C5ar2−/- mice. In accordance with our earlier work, C5ar1−/- mice showed significantly enhanced survival in comparison to WT mice (). Interestingly, a similar beneficial effect was observed with C5ar2−/- mice, whereas, in striking contrast, C3ar1−/- mice succumbed even faster to the disease. Accordingly, C5ar1−/- and C5ar2−/- mice displayed reduced clinical scores than WT mice, whereas C3ar1−/- showed aggravated symptoms during the course of the disease (). Past 18 h, there was only one single survivor in the C3ar1−/- cohort, which displayed only moderate symptoms. Aggravation of disease was paralleled by the increased bacterial burden in the diseased mice (). At 3 h, C5ar1−/- and C5ar2−/- showed reduced meningococcemia as compared to WT, and this was also seen at 12 h for C5ar1−/- and around 24 h for C5ar2−/-. In contrast, meningococcemia was not significantly different between WT and C3ar1−/- mice at any time point. In general, higher levels of meningococcemia were found in non-survivors (indicated by open symbols) than in survivors (filled symbols), reflecting that active replication of N. meningitidis correlates with disease outcome. In survivors, the bacterial counts decreased further after 24 h.
In order to elaborate the enhanced susceptibility of C3ar1−/- mice, an infection with a lower inoculum was conducted, demonstrating higher mortality (), aggravated symptoms () and enhanced bacterial burden in the blood () among C3ar1−/- mice compared to WT mice.
Taken together, mice lacking C3aR are more vulnerable to experimental meningococcal sepsis than WT mice, whereas mice lacking either of the two C5a-receptors are more resistant.
C3ar, C5ar1 and C5ar2 impact similarly on cytokine response during experimental meningococcal sepsis in mice
Anaphylatoxin receptors are known to modulate the cytokine response of immune cells [Citation45–Citation47] as well as endothelial cells [Citation13]. Our previous study showed that mice lacking C5aR1 mounted a significantly weaker cytokine response during N. meningitidis sepsis than WT mice, indicating a positive modulation of cytokine release by C5aR1 during infection [Citation41]. Here, we analyzed the cytokine profiles in tail vein blood plasma samples taken 12 h post infection from WT, C3ar1−/-, C5ar1−/- and C5ar2−/- mice (). Consistent with our earlier work [Citation41], C5ar1−/- mice displayed significantly reduced levels of CXCL-1, IL-6, TNF-α, IFN-γ, and MCP-1, in comparison to WT mice. C5ar2−/- mice showed significantly lower levels of CXCL-1 and IL-6 than WT mice, whereas all other tested cytokines and chemokines were similar to WT mice. Given that C3ar1−/- mice showed the highest susceptibility to experimental sepsis, with aggravated symptoms and high levels of bacteremia (), we were surprised that their cytokine/chemokine response was lower than, or similar to, that in WT mice. However, the differences were only significant for CXCL-1 and IL-6, whereas a trend was observed throughout the entire panel of mediators released in response to infection. Interestingly though, the difference in the plasma levels of CXCL-1 between WT and either of the knockout mice was already seen as early as 3 h after infection (supplementary Fig. S1). This indicates that early events in innate immune recognition prime a differential ATR-dependent chemokine/cytokine response in the mice.
Figure 2. Cytokine and chemokine response of C3ar1−/-, C5ar1−/- and C5ar2−/- mice 12 h after intraperitoneal infection with N. meningitidis. Tail vein blood samples from the same mouse cohorts as in were analyzed for inflammatory cytokines and chemokines at 12 h after infection. Controls were samples from the same mice one week prior to the experiment. Plotted are means ± standard deviation.
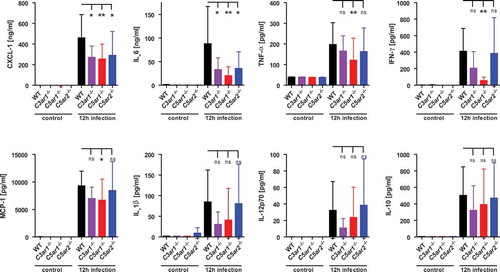
These data point towards a similar net contribution of all three anaphylatoxin receptors to the cytokine/chemokine output in the mice, suggesting that the exacerbated inflammatory response per se is not strictly determining the course of disease in the mice.
N. Meningitidis driven neutrophil activation is attenuated in C5ar1-/- mice, but not in C3ar1-/- or C5ar2-/- mice.
In searching for mechanisms explaining the disparate outcomes of Nme sepsis in the individual ATR-knockout mice, we first focused on the functional responses of neutrophils, i.e. the cell type featuring the highest expression of C5aR1 and C5aR2 [Citation10]. In fact, unbridled neutrophil activation is deemed detrimental during sepsis, and this has been particularly attributed to the C5a/C5aR1 axis [Citation17,Citation48]. In order to determine differences in neutrophil responses in the ATR-deficient mouse strains, we used a hirudin-based whole blood model for ex vivo infection, as this anticoagulant does not interfere with complement activation or Nme growth [Citation49,Citation50]. Upon infection with Nme, the neutrophils showed an oxidative burst response (), degranulation (), and they engulfed GFP-expressing Nme (). Consistent with our earlier observations [Citation41], neutrophils from C5ar1−/- mice showed a significant reduction in oxidative burst and degranulation, whereas there were no significant differences between the mouse genotypes when stimulation was done with PMA as a positive control. However, C3ar1−/- and C5ar2−/- neutrophils mounted comparable oxidative burst and degranulation responses as those from WT mice. Phagocytosis of Nme by neutrophils was similar for all four mouse genotypes, and likewise, bacterial counts rose similarly among all mouse genotypes in blood incubated ex vivo for 4 h. In order to ascertain the role of complement in phagocytic uptake of Nme by neutrophils in the murine model, we compared Nme phagocytosis in WT versus C3−/- whole mouse blood. In agreement with our recent reports [Citation41,Citation51], phagocytosis is severely impaired in the absence of complement (supplementary Fig. S2). The fact that Nme can actually grow in mouse blood despite ongoing neutrophil responses and phagocytosis demonstrates that neutrophil responses alone are insufficient to clear the infection, despite their significant in vivo contribution to Nme clearance reported earlier [Citation41]. In accordance with the attenuated cytokine response in vivo (), IL-6 responses in the ex vivo whole blood infection model showed a (non-significant) down-trend for all ATR-knockout mice compared to WT ().
Figure 3. Neutrophil responses of C3ar1−/-, C5ar1−/- and C5ar2−/- mice in ex vivo whole blood infection. Hirudin-anticoagulated whole blood was infected either with Nme or stimulated with PMA versus PBS as a negative control. Samples were incubated at 37°C rotating over top. (a) The oxidative burst response of mouse neutrophils as assessed by the DHR123-assay. Blood samples were treated for 1 h either with PBS, 107 CFU/ml of Nme or with 100 nM PMA as a positive control. Values of the PBS-condition for each individual mouse sample were subtracted from the others in order to correct for the intrinsic GFP-signal of cells from C5ar2−/- mice. Thus, the y-axis plots ΔMFI values. (b) Neutrophil degranulation as measured by an increase of CD11b on the cell surface. Blood samples were treated for 1 h with PBS (negative control), 107 CFU/ml Nme or with 100 mM PMA (positive control). Values obtained with the isotype-control for the anti-CD11b antibody were subtracted in order to correct for the intrinsic GFP-signal of cells from C5ar2−/- mice. (c) Phagocytosis of GFP-expressing Nme by neutrophils in mouse whole blood. Blood samples were infected with 108 CFU/ml Nme and incubated for 1 h and GFP-signal of the cells measured by flow cytometry. In order to correct for the intrinsic GFP-signal of C5ar2−/- neutrophils, the mean MFI difference between WT and C5ar2−/- was subtracted from all C5ar2−/- neutrophils. (d) Survival of Nme in mouse blood. Whole blood samples were infected with 105 to 106 Nme and samples were taken immediately (0 h) or after 4 h of incubation rotating at 37°C (4 h) and serial dilutions plated on blood agar plates for CFU enumeration. (e) IL-6 plasma levels at 4 h after infection of mouse whole blood with 106 CFU/ml of Nme strain MC58.
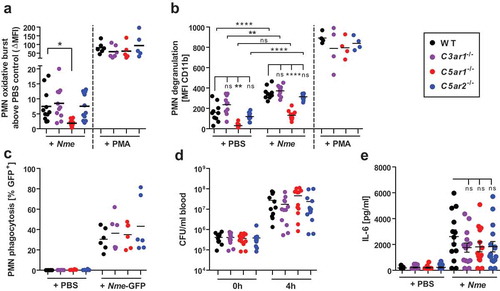
Taken together, we found that only C5ar1−/- mice showed reduced neutrophil responses to Nme whole blood infection, whereas C3ar1−/- and C5ar2−/- neutrophils responded comparably to those from WT mice.
C5ar1 and C5ar2 modulate phosphorylation of ERK1/2 in early macrophage activation
C5aR1 and C5aR2 differ fundamentally in their ability to signal via G-proteins to trigger cellular responses (e.g. neutrophil chemotaxis) [Citation22], which is reflected by the distinct neutrophil responses in whole blood (). In attempting to find a common denominator for the homogenously ameliorated disease phenotype of C5ar1−/- and C5ar2−/- mice () as compared to WT mice, we measured ERK1/2 phosphorylation, a central hub in cellular signaling events upon extracellular stimuli. Indeed, C5aR1 and C5aR2 both act on ERK phosphorylation in neutrophils, although for C5aR2, there is conflicting data regarding whether it is activating or inhibiting ERK1/2 phosphorylation [Citation22,Citation28,Citation29]. We challenged bone marrow-derived murine macrophages with 10 nM C5a and/or 108 CFU/ml heat-inactivated Nme. Robust ERK1/2 phosphorylation in response to C5a alone occurred in WT and C5ar2−/- macrophages within 5 min, but not in C5ar1−/- macrophages (). ERK1/2 phosphorylation in response to C5a was of short duration and declined to baseline levels within 15 min. In contrast, there was no significant ERK1/2 phosphorylation within 5 min when the cells were exposed to Nme alone. Yet, WT macrophages showed a significant increase of ERK1/2 phosphorylation when C5a along with Nme was added, whereas this was not observed with C5ar1−/- or C5ar2−/- macrophages. Also, blocking both C5a-receptors with A8Δ71−73 did not significantly heighten ERK1/2-phosphorylation above the C5a alone condition. Thus, at this early timepoint, both C5a-receptors appear to function in concert to prime a fast and robust ERK1/2 phosphorylation in macrophages. At later timepoints, there was no difference among the genotypes upon stimulation with Nme alone or Nme plus C5a. Interestingly, at 15 min and 30 min, there was a slight (not significant) reduction in ERK1/2 phosphorylation when the cells were stimulated with Nme plus C5a as compared to Nme alone. Also, treatment with A8Δ71−73, limited the duration of ERK1/2 phosphorylation, as it was significantly reduced in comparison to the WT control at 30 min.
Figure 4. ERK1/2 phosphorylation in bone marrow-derived murine macrophages in response to C5a and/or Nme. Macrophages were derived from WT, C5ar1−/- or C5ar2−/- mice and differentiated for 8 days in L929-conditioned medium and their phenotype (CD68+CD11b+F4/80+) verified by flow cytometry. Cells were stimulated with murine C5a (10 nM final concentration), Nme (heat-inactivated; corresponding to 108/ml), or both, for the indicated duration (5 min, 15 min, 30 min) before harvesting samples. (a) Western blots probed with anti-phospho-ERK1/2 (left panels) or with anti-ERK1/2 (right panels) as a loading control. (b) Densitometric analysis of the Western blot bands from four independent experiments.
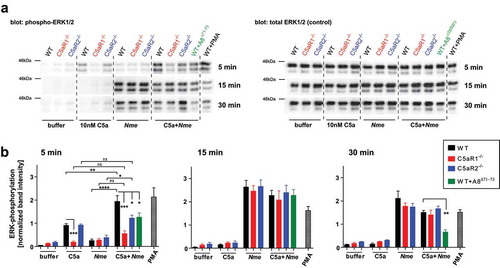
From these data, we conclude that ERK1/2 phosphorylation in macrophages is positively influenced by C5aR1 and C5aR2 during the very early phase of their activation by C5a and Nme. This provides a possible approach to explain the similar outcome of in vivo disease despite different abilities of the two C5a-receptors in coupling to G-proteins and activation of neutrophil responses.
Pharmacologic targeting of C5a-receptors, but not C3aR ameliorates experimental meningococcal sepsis
Given the differential susceptibility of C3ar1−/-, C5ar1−/- and C5ar2−/- mice to experimental meningococcal sepsis (), and the successful treatment of mouse meningococcal sepsis with a C5aR1-specific antagonist, PMX-205, in our earlier work [Citation41], we speculated that pharmacologic targeting of C3aR or C5aR2 might as well influence the course of disease in WT mice. To test this hypothesis, we injected mice with the antagonist SB290157 to block C3aR, or with the superagonistic peptide WWGKKYRASKLGLAR to stimulate C3aR [Citation52], or with the antagonist A8Δ71−73 to simultaneously block C5aR1 and C5aR2, or with PMX205 to block C5aR1 alone before intraperitoneal challenge with a lethal dose of 105 CFU of Nme. The control cohort received vehicle only. Consistent with earlier results, PMX205 treatment resulted in a significantly higher survival rate (), reduced levels of bacteremia () and lower levels of inflammatory cytokines (). A similar effect was observed upon blockade of C5aR1 together with C5aR2 using A8Δ71−73. Notably, the effect on bacteremia and cytokine response was even more pronounced with A8Δ71−73 as with PMX205, and this difference was statistically significant for bacteremia at 3 h. Thus, there may be a slight-added benefit in blocking both C5a-receptors simultaneously. In contrast, neither antagonizing (SB290157) nor activation (superagonist) of C3aR seemed to significantly alter the course of disease in the treated mice in comparison to the vehicle control.
Figure 5. Experimental Nme sepsis in WT mice treated with agonists or antagonists of the anaphylatoxin receptors. Mice were subjected to intraperitoneal treatment with antagonists to C5aR1 (PMX205), C5aR1 plus C5aR2 (A8Δ71−73), C3aR (SB290157), or with a super-agonist (WWGKKYRASKLGLAR) of C3aR, or with vehicle alone (sterile 5% glucose solution) starting prior to infection and continued throughout the experiment (see Methods). Each mouse was intraperitoneally infected with a total of 105 CFU of Nme. Mice were monitored for 48 h before the experiment cessation. (a) Survival curves of mice. (b) Bacteremia at indicated time points of the mice. Bacteremia plotted at ’18–27 h’ includes values at 24 h of all mice surviving longer than 27 h and the values of the non-survivors at their time of death (by sacrificing at the “humane endpoint”) within this time window. Closed circles indicate surviving animals, open circles indicate non-survivors. Limit of detection was 500 CFU/ml. (c) Inflammatory mediators IL-6 and CXCL-1 as measured by ELISA in tail vein blood samples obtained from the same mice as in A at 12 h after infection. Plotted are means ± SEM.
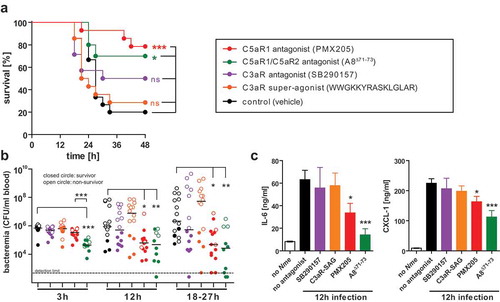
Considering the small differences between sepsis outcome after blockade of C5aR1 alone (PMX205) versus C5aR1 plus C5aR2 (A8Δ71−73), the additional effect of blocking C5aR2 seems to be minor. Unfortunately, no specific C5aR2 inhibitor is available to individually assess the role of C5aR2 during Nme sepsis.
Taken together, we conclude from these data that interference with the C5a/C5a-receptor axes ameliorates disease, whereas our results do not support the notion that C3aR is a promising candidate for pharmacologic targeting during IMD.
Blockade of C3ar, C5ar1, and C5ar2 reduces IL-8 secretion and neutrophil oxidative burst in human whole blood
In order to consider the human-specific tropism of Nme, we conducted human whole blood infection experiments to determine the influence of the individual anaphylatoxin receptors on cytokine secretion and neutrophil responses. Therefore, we blocked the individual anaphylatoxin receptors in hirudin-anticoagulated blood and measured secretion of IL-8 and neutrophil oxidative burst response upon ex vivo Nme infection. The IL-8 secretion in infected human blood () was reduced upon blockade of either C3aR (using SB290157), or C5aR1 (using PMX53), or C5aR2 (using IgG2a mAb clone 1D9-M12), an observation well matching the reduced cytokine/chemokine response of in vivo infected knockout mice (). In addition, a significant reduction of IL-8 secretion was observed when C5aR1 together with C5aR2 was blocked using A8Δ71−73 [Citation53], but the effect did not exceed that of the individual blockade of the individual C5a-receptors. Also, when complement was blocked at the stage of C3 convertase by compstatin Cp20, a significant reduction (36%) was seen, which as well was comparable with the other treatments, which inhibited IL-8 secretion by 29–42%. Compstatin efficiently blocked complement activation, as indicated by the near-complete ablation of C5a liberation (reduction by 87%) in Nme infection whole blood from the same donors (supplementary figure S3). Triggering C3aR using the superagonistic peptide WWGKKYRASKLGLAR [Citation52] enhanced IL-8 release in whole blood.
Figure 6. Innate immune responses in human whole blood during infection with Nme in presence of specific C3 or ATR antagonists. Hirudin-anticoagulated human blood samples (n = 6 donors) were incubated for 10 min with the indicated antagonists before the addition of Nme. The target structures are indicated above the graphs, with the agent indicated in brackets. In the graphs, values obtained from the same individual are represented as dots connected with lines; the bar graphs plot mean ± SEM of the same data. The average reduction of the analyzed response relative to no antagonist addition is given as percentage ± SD in each graph. (a) IL-8 response at 90 min of infection with 106 CFU/ml Nme at 37°C rotating over top. (b) Neutrophil oxidative burst as measured by DHR123 assay in human whole blood after infection with 107 CFU/ml Nme at 1 h of incubation at 37°C rotating over top. Values are expressed as mean fluorescence intensity (MFI). Representative histograms from the flow cytometric analysis are shown directly below the graphs. (c) Neutrophil degranulation assessed as surface CD11b by flow cytometry. Data are plotted as MFI, representative histograms are shown directly below the graphs. (d) Neutrophil phagocytosis of GFP-expressing Nme (108 CFU/ml) as assessed by flow cytometry after incubation in whole blood for 1 h. Date are plotted as % of GFP-positive neutrophils; representative histograms and gating are shown directly below the graphs.
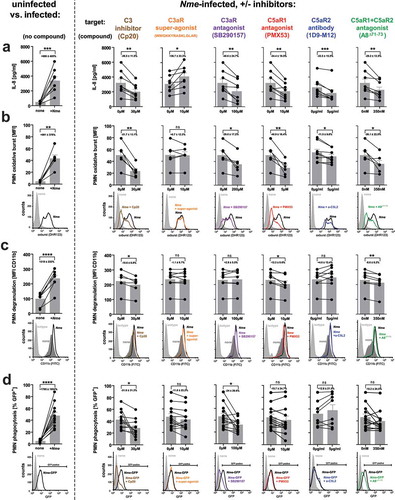
In addition to IL-8 secretion, we found a significant reduction of the neutrophil oxidative burst response () upon inhibition of C3aR, C5aR1, or C5aR2. Simultaneous inhibition of C5aR1 and C5aR2 with A8Δ71−73, yielded a significantly decreased burst response. In contrast, the C3aR super-agonist had no effect on the oxidative burst response. When complement activation was blocked using compstatin Cp20, a significant decrease of the neutrophil oxidative burst was observed.
Similar to the impact on oxidative burst, blockade of C3, C5aR1 or simultaneous blockade of C5aR1 and C5aR2 significantly reduced the neutrophil degranulation in whole blood upon Nme infection; however, no effect was seen when C3aR was either triggered or inhibited, and C5aR2 inhibition as well did not impact the degranulation response ().
Finally, we assessed the uptake of GFP-expressing meningococci by neutrophils in human whole blood. Here, a significant reduction of phagocytosis was only evident upon inhibition of C3 using compstatin Cp20 or blockade of the C3aR, whereas there was no significant effect with any of the other treatments ().
Of note, we did not observe a bactericidal effect of any of the used agents to block or activate the ATRs (supplementary Fig. S4).
Taken together, the results obtained in human blood are in good accordance with those of in vivo mouse infections and mouse ex vivo whole blood infections when considering the effect of all three ATRs in their contribution to cytokine/chemokine release, and, additionally, for C5aR1 with neutrophil activation (oxidative burst, degranulation). However, while we did not observe an effect of C3aR or C5aR2 on mouse neutrophil activation during mouse blood infection, this is apparently the case in human whole blood.
Discussion
We employed in vivo models to analyze the contribution of anaphylatoxin receptors to nasal colonization and invasive disease with Nme. While we found no significant contribution of complement in general (C3, C5), or of the ATRs during asymptomatic Nme nasal colonization, we identified a significant contribution of the ATRs to the course of Nme sepsis in mice. Thus, we expanded upon our recent observation that C5aR1 aggravates Nme sepsis pathophysiology [Citation41] in showing that C3aR is protective, whereas C5aR2, like C5aR1, is detrimental in the mouse Nme sepsis model. We found that pharmacological blockade of C5aR1 alone, and particularly in combination with C5aR2, significantly ameliorated disease symptoms in mice, whereas tampering with the C3a/C3aR-axis did not benefit the infected mice. Interestingly, invalidation of any of the three ATRs in vivo and/or ex vivo similarly reduced the cytokine/chemokine levels in mouse and human blood, suggesting the involvement of pathomechanisms beyond inflammatory mediator release disease progression and outcome. Neutrophil responses in mouse and human whole blood showed a complex and partially species-specific pattern of ATR influences on neutrophil effector functions. When C5aR1 and/or C5aR2 were invalidated in mouse macrophages, early ERK1/2 phosphorylation in response to Nme along with C5a was significantly reduced. Hence, we suggest that C5aR1 and C5aR2 triggered ERK1/2 phosphorylation initiates a macrophage response early on during experimental disease, which may underlie the similar disease phenotype found in C5ar1−/- and C5ar2−/- mice.
Considering the nasal infection model, earlier work with the CEACAM1-humanized mouse model implied a connection between Nme nasal colonization exogenous complement activation using Cobra venom factor (CVF) when administered shortly (4 h) but not long (24 h) before intranasal instillation of Nme [Citation42]. Thus, we were surprised to see no effect on Nme colonization in mice devoid of functional complement (C3 or C5 deficient) here. Thus, we conclude that in the previous work, the injection of CVF may have caused changes to the epithelial environment which transiently (4 h only) affected Nme colonization.
Considering the invasive disease model, this study is in agreement with our own previous work [Citation41] showing a deleterious effect of C5aR1 during meningococcal sepsis, since C5ar1−/- mice showed significantly reduced mortality, morbidity, cytokine/chemokine levels and reduced neutrophil activation patterns in mouse and human blood. Overall, this is consistent with the pro-inflammatory capacity of C5aR1 as the canonical receptor for C5a which is well known to exacerbate sepsis-induced pathomechanisms [Citation48,Citation54].
While the pro-inflammatory potential of C5aR1 is very clear-cut, the biologic effects of its sibling receptors, C3aR and C5aR2, are usually more subtle and their actual contribution to inflammation and immunity is less pronounced [Citation39,Citation55]. In fact, the functional role of C5aR2 has been heavily debated over the past 20 years [Citation55–Citation57]. The detrimental role of C5aR2 during experimental Nme sepsis supports the notion of a functionally relevant, pro-inflammatory role for this receptor, as described by Rittirsch et al. using the cecum ligation and puncture (CLP) model [Citation26]. The detrimental effects of C5aR1 during sepsis have been linked to neutrophil dysfunction [Citation58]. Yet neutrophil activation can be beneficial (pathogen clearance) as well as deleterious (organ damage) during sepsis [Citation59]. Our data show reduced neutrophil responses in C5ar1−/- mice, suggesting that C5aR1 might trigger adverse responses in Nme sepsis. In contrast, C5ar2−/- neutrophils responded similar to WT neutrophils in our ex vivo analyses, therefore, neutrophils are likely not the cell type that mediates the C5aR2-dependent phenotype in our mouse model. From infections of clodronate liposome-treated mice, we know that macrophages are crucial to control Nme sepsis [Citation41]. Given that C5ar1−/- and C5ar2−/- macrophages (and WT macrophages treated with A8Δ71−73) show decreased ERK1/2 phosphorylation at early timepoints of C5a/Nme stimulation, we speculate that the beneficial outcome of experimental Nme sepsis in C5ar1−/- and C5ar2−/- mice (or A8Δ71−73 treated WT) is partially due to this early macrophage activation event. Thus, future work will need to dissect the impact of the C5a-receptors on early macrophage activation during Nme sepsis.
In contrast to C5ar1−/- and C5ar2−/- mice, C3aR−/- mice were more susceptible to Nme sepsis. Similar observations were made in other infection models, such as LPS challenge [Citation36], Listeria monocytogenes infection [Citation60] and Chlamydia psittaci infection [Citation61]. Yet, this disease aggravation is not a general feature, since C3ar1−/- mice are more resistant to Pseudomonas aeruginosa pneumonia [Citation60], indicating distinct mechanisms involved in the C3aR-dependent response towards different pathogens. Pharmacological targeting the C3a/C3aR-axis, either by activating (“superagonist”) or by antagonizing (SB293157) C3aR, did not significantly alter the course of the disease as expected based on the results with the C3ar1−/- mice. Yet, there are some caveats to be considered. If C3aR activation was already at maximum during Nme sepsis, the “superagonist” could not increase it any further to alter the disease outcome. Further, the “superagonist” is a linear peptide [Citation52], thereby subject to proteolytic inactivation by serum carboxypeptidase N; hence, its biologic activity could be limited below a therapeutic threshold in vivo. On the other hand, the C3aR-antagonist SB290157 has reported off-target activities in vivo, causing neutropenia independent to C3aR, as well as having C3aR agonist activity in certain cell settings [Citation62,Citation63]. Thus, we consider our data from C3ar1-/- mice most reliable.
Unexpectedly, C3ar1−/- mice mounted lower chemokine/cytokine responses as WT mice (CXCL-1 and IL-6) despite aggravated disease symptoms. This finding contrasts previous reports for C3ar1−/- mice, in which their susceptibility was correlated with inflammatory mediators [Citation36,Citation37,Citation60]. Thus, the functional interrelationship between C3aR and the chemokine/cytokine system differs significantly between infection with Nme and other bacterial pathogens. In addition, this suggests that the poor outcome of C3ar1−/- mice is not a direct consequence of the aggravated systemic inflammatory response syndrome.
Although mouse neutrophils have been suggested to express C3aR [Citation28,Citation38], recent evidence shows that mouse neutrophils – in contrast to human neutrophils [Citation64] – do not express C3aR [Citation65]. This would explain the lack of a C3aR-dependent phenotype in the mouse whole blood model. Hence, we can only speculate which cell types mediate the beneficial C3aR-dependent effects during Nme sepsis. Macrophages of mice and humans express C3aR [Citation47,Citation65] and could be the driving force of C3aR-dependent disease outcome, potentially in a fashion opposing that triggered by the C5a-receptors. Endothelial cells are activated during septic settings and respond to C3a [Citation66] and are crucial interfaces for the interaction between Nme and the host [Citation67]. Thus, by modulation of vascular permeability [Citation60] or intravascular coagulation in combination with C3aR-bearing platelets [Citation68], C3aR might impact on the course of Nme sepsis in a cytokine/chemokine-independent fashion.
In order to consider the species-restriction of Nme, we performed human whole blood infection experiments. While many aspects were well correlated between human and mouse whole blood, few others were in disalignment: The differential effects of C3aR-inhibition on the oxidative burst response and phagocytosis by mouse versus human neutrophils are explained by the lack of C3aR expression in mouse neutrophils [Citation65]. The difference in the oxidative burst response in dependence of C5aR2 may reflect a functional difference in C5aR2 on neutrophils among mice and humans.
Nme phagocytosis by neutrophils was similarly unaffected by both C5a-receptors in mice and men, whereas only in human cells, C3aR-blockade had a negative impact. In contrast to other reports on the phagocytosis of Nme by human neutrophils [Citation69,Citation70], we did not observe an impact of C5aR1 in this or in our previous study [Citation41]. This may be due to the assay system used (e.g. heat-inactivated Nme [Citation70] vs. viable Nme) or the immune status of the donors (vaccinated [Citation69] vs. unknown immune status). However, we recently reported slightly reduced Nme phagocytosis upon C5aR1-inhibition using purified human neutrophils in the presence of immune serum [Citation51], indicating that the whole blood assay may be, although biologically the most relevant, not the most sensitive assay to measure Nme phagocytosis. Overall, the importance of neutrophil phagocytosis during Nme sepsis is not clear, and future work will have to address this.
In summary, our work demonstrates that the course of experimental Nme sepsis is significantly influenced by the anaphylatoxins and their receptors, expanding our knowledge about complement in Nme disease. While C3aR activation is protective during experimental Nme sepsis, triggering C5aR1 or C5aR2 is harmful. Thus, our data suggest that targeting the C5a/C5aR1-C5aR2 axis could prove useful as an adjunctive treatment in addition to antibiotics to treat human IMD. Also, our data support emerging studies [Citation38,Citation71,Citation72] indicating that the C3a/C3aR-axis acts protectively in many disease contexts.
Materials and methods
Animals
Holding of animals was done in accordance with all legal requirements under the German animal welfare act (TierSchVersV). Animal handling and experimentation were approved by the appropriate authorities (government of Lower Franconia) under files 55.2.2531.01–90/13 and 55.2–2532-2–165. All efforts were made to keep animal distress and suffering to a minimum. Animals were housed with a 12 h bright/dark cycle at 22–24°C and 55 ± 10% relative humidity. Testing for specified pathogens as recommended by the Federation for European Laboratory Animal Science Associations (FELASA) was conducted quarterly to ascertain SPF-status of the mice. The mice had ad libitum access to water and standard rodent diet and enrichment was placed in all cages. All mice used were on C57Bl/6J genetic background. WT mice were either bred in-house or purchased from Charles-River or Envigo (formerly Harlan). The mouse strains lacking the individual ATRs, namely B6.129S4-C3ar1tm1RWe (termed C3ar1−/-) [Citation36], B6.129S4-C5ar1tm1Cge (termed C5ar1−/-) [Citation73] and B6.129S4-C5ar2tm1Cge (termed C5ar2−/-) [Citation24], and the mouse strain lacking C3 (B6.129S4-C3tm1Crr/J, termed C3−/-) [Citation74], were bred in-house. The mouse strain lacking C5 (B6.FVB-Hc°; here termed C5−/-) was obtained by crossing the defective gene for C5 (Hc°) from the FVB mouse line for 10 generations onto the C57Bl/6J background, as previously described [Citation41]. The transgenic mouse line expressing human CEACAM1 (termed huCCM1) [Citation75] was backcrossed onto C57Bl/6J background for 10 generations and was bred in-house.
Bacteria
Nme were cultivated in all assays described below, by plating on Columbia agar containing 5% sheep blood (further referred to as “blood agar”; bioMérieux) and incubated in water-saturated conditions at 37°C with 5% CO2. For our experiments, the Nme strain MC58, serogroup B (typed B:15:P1.7,16–2:F1-5: ST-74, ST-32 clonal complex) or its capsule-deficient mutant expressing a green fluorescent protein, MC58Δcsb-GFP strain [Citation76] was used.
Nme inocula preparation
Meningococci were grown on blood agar plates overnight and transferred onto fresh plates for 4 h to obtain log-phase grown bacteria. The bacteria were then harvested with a cotton swab, resuspended in brain heart infusion broth (BHI). Using a spectrophotometer, OD600 of the bacterial suspension was adjusted to 1.0, corresponding to 1.5 × 109 CFU/ml. Further dilutions were made to adjust the concentration to the desired density, as indicated in the specific protocols. For accuracy confirmation, serial dilutions of the inocula were prepared, plated and grown colonies enumerated.
Nme intranasal infection of mice
The CEACAM1-humanized mouse model for Nme colonization was described previously [Citation42,Citation44]. In brief, the Nme inoculum (strain MC58) was prepared as described above, but in PBS with 1 mM MgCl2 instead of BHI, and adjusted to 107 CFU/ml. 10 μl hereof were instillated by dropwise application to both nares of the alert mouse. Mice were monitored for bacteremia at 4 h after infection by spreading 5 µl of tail vein blood onto blood agar plates. Mice were scored using the following scheme to assess clinical scores, which included body weight loss (score “1”: >3% to 5%; score “5”: ≥5% to 10%; score “10”: ≥10% to 20%; score “20”: ≥20%), general physical appearance (score “1”: small aberrations such as slight lack of grooming; score “5”: dull fur, slightly hunched posture; score “10”: dirty fur, hunched posture, smudgy perianal region, dull eyes; score “20”: tremors or shivering, immobility, difficulty to rise from lateral position), behavior (score “1”: minor aberrations; score “5”: impaired motoric mobility; score “10”: self-isolation, lethargy, strongly impaired coordinative abilities; score “20”: unconsciousness, no/minimal defensive reaction, when picked up) and clinical condition (score “1”: small aberrations; score “10”: ±30% changes in breathing frequency; score “20”: ±50% changes in breathing frequency, blood flow impairments). At indicated time points, mice were sacrificed by CO2 asphyxiation. Through a tracheal incision, 200 µl PBS with 1 mM MgCl2 were applied to collect a nasal wash sample for plating and CFU enumeration. Then, the nose tip was cut off and the nasal mucosa exposed by lateral cuts through the premaxilla. Nasal mucosa was probed by inserting an ultra-fine polyester-tipped aluminum applicator swab (Puritan) wetted with PBS with 1 mM MgCl2 and thorough rubbing. The adherent nasal tissues were resuspended in 500 µl of PBS with 1 mM MgCl2 to yield a “nasal swab” sample. Nasal wash and nasal swab samples were spread onto Neisseria-selective modified Thayer-Martin agar plates and incubated overnight at 37°C, 5% CO2 and water-saturated atmosphere before the enumeration of resultant Nme colonies.
Nme induced sepsis in mice
The 6–8-week old male mice were injected intraperitoneally (i.p.) with 200 µl BHI containing 105 CFU/ml (or 5 × 104 CFU/ml, as indicated) of Nme and 200 µl iron dextran (Santa Cruz) diluted 1:3.33 in sterile isotonic saline. A second i.p. injection of iron dextran was given 12 h post infection. Mice were weighed and monitored every 3 h for 48 h following the scoring scheme described above. In accordance with the German Animal Welfare Law, mice were humanely sacrificed by CO2 inhalation upon reaching the human endpoint criteria (tremors/shivering or inability to rise from lateral position) and were not left to die. To assess plasma cytokine levels, 10 µl tail vein blood was drawn 12 h post infection, mixed with 90 µl PBS containing 5 mM EDTA, centrifuged and the plasma was then frozen at −80°C until ELISA. For bacterial burden assessment, 5 µl tail vein blood was drawn 3 h and 12 h post infection and then every 12 h. The blood was diluted in 45 µl PBS containing 5 U/ml heparin to avoid clotting. Serial 1:10 dilutions were made and plated on Columbia sheep blood agar plates to enumerate colonies after overnight incubation. Additional samples were taken after mouse euthanasia upon reaching the human endpoint.
Nme sepsis experiments using antagonists and agonists
Mice were infected and monitored as described above. For the blockade of the C5ar1, WT mice received i.p. 3 mg PMX205 per kg of body weight in a total volume of 100 µl per injection at the following time points relative to the infection start: −12 h, −6 h, 0 h, and then every 12 h post infection. In order to block C3aR, WT mice received i.p. 1 mg/kg body weight SB290157 in a total volume of 100 µl per injection every 12 h starting at −12 h. In order to simultaneously block C5aR1 and C5aR2, WT mice received 200 µl of A8Δ71−73 at 88,7 µM at −6 h, 0 h, 6 h, 12 h and then every 12 h. In order to stimulate C3aR, WT mice received 1 mg per kg body weight of the C3aR-superagonist peptide (WWGKKYRASKLGLAR) in a total volume of 100 µl per injection at −3 h, 0 h, 6 h, 12 h and then every 12 h. Control WT mice were treated with vehicle alone at −12 h, −6, 0 h, 6 h and then every 12 h. All agonist/antagonist injections were freshly prepared in sterile 5% glucose solution (vehicle) and sterile filtered through 0.2 µm syringe-mounted devices. Sterility of injected compounds was monitored by plating onto Columbia sheep blood agar plates and incubation for 48 h with none of the preparations ever yielding bacterial contamination.
Mouse whole blood assays to assess oxidative burst in neutrophils:
Mice were euthanized by CO2 inhalation and whole blood was drawn via cardiac puncture using hirudin monovettes (525 antithrombin units/ml; Sarstedt). Hirudin was chosen as anticoagulant since it does not interfere with complement or with Nme viability [Citation50]. Mouse whole blood samples were infected with 107 CFU/ml of Nme; 100 nM PMA (Sigma Aldrich/Merck) served as a positive control, whereas PBS was used as a negative control. Samples were incubated, rotating for 15 min at 37°C. Then, 20 µg/ml dihydrorhodamine 123 (DHR123) (Sigma Aldrich/Merck) were added and the samples were incubated rotating for 1 h at 37°C. Then, samples were placed on ice and stained for 30 min with anti-mouse Ly6G-APC (Clone: 1A8; BioLegend). After hypotonic lysis of the erythrocytes, samples were washed with cold PBS and fixated with 4% formaldehyde for 30 min at RT in the dark. Flow cytometric analysis was done on a BD FACSCalibur (Becton Dickinson) and data analyzed and plotted with FlowJo v10 (FlowJo, LLC). Cell clusters were gated out with an FSC/SSC gate. Neutrophils were gated as Ly6G-APChigh and fluorescence intensity of oxidized DHR123 assessed in the FL1-H channel. For further analysis, the PBS background signal in FL1-H was subtracted from the corresponding signals in the PMA and the Nme samples to correct for the slight GFP-dependent shift of cells isolated from C5ar2−/- mice, which express low levels of GFP.
Mouse whole blood assays to assess degranulation in neutrophils
To measure neutrophil degranulation, mouse whole blood was obtained and infected/stimulated as described for the oxidative burst assay. The samples were incubated for 60 min at 37°C, then placed on ice and then stained either with anti-mouse Ly6G-APC (clone 1A8; BioLegend) plus anti-mouse/human CD11b-FITC (clone M1/70; BioLegend) or with anti-mouse Ly6G-APC (Clone: 1A8) plus FITC labeled rat IgG2b, κ Isotype Ctrl Antibody (clone RTK4530; BioLegend) Neutrophils were gated as Ly6G-APChigh and assessed for their expression of CD11b in the FL1-H channel. The values were corrected by subtracting the isotype-FITC background signal of the PBS-controls in FL1-H from the CD11b-signal in all corresponding samples; this subtraction was necessary to correct for GFP-expression in C5ar2−/- neutrophils.
Mouse whole blood assay to assess phagocytosis capacity in neutrophils
For the phagocytosis assay, cardiac blood was obtained as described in the oxidative burst assay. The samples were infected with 108 CFU/ml of GFP-expressing Nme; to the negative controls, RPMI was added. After incubation for 60 min rotating at 37°C, samples were stained for neutrophils with anti-Ly6G-APC antibody (clone 1A8, BioLegend) for 30 min on ice followed by hypotonic lysis of erythrocytes and fixation of the sample with 4% formaldehyde. Phagocytosis was assessed by flow cytometry on a FACSCalibur (Becton Dickinson) as an increase in FL1-H relative to PBS control. The background subtraction to correct for GFP-expression in C5ar2−/- neutrophils was done by subtracting the difference between the FL1-H background signal between WT and C5ar2−/- neutrophils, from the signals in our PBS and Nme samples of C5ar2−/- neutrophils.
Mouse whole blood assays to assess Nme survival:
Whole blood samples were collected as described for the oxidative burst above and infected with 106 CFU/ml of Nme, or treated with PBS as a negative control. Amount of bacteria present in the samples was assessed by dilution plating and colony enumeration at 0 h and 4 h of infection.
Cytokine analysis using ELISA or bead-based LEGENDPlextm assay
Plasma samples from infected mice or untreated animals (controls) were either measured for IL-6 and CXCL-1 using DuoSet® ELISA kits (R&D Systems) or using the LEGENDPlexTM mouse inflammation panel immunoassay (BioLegend,) as per the manufacturer’s instructions. Human plasma samples were measured for IL-8 or C5a using DuoSet® ELISA kits (R&D Systems) as per the manufacturer’s instructions.
ERK1/2-phosphorylation in mouse bone marrow-derived macrophages
Mice were euthanized by CO2 asphyxiation and femurs were obtained, placed for 1 min in 70% ethanol and then the opened bones flushed with 15 ml PBS. Cell aggregates were passed through a 25 G needle to obtain single cells and the suspension was passed through a 40 µm cell strainer to remove residual cell clumps and bone fragments. Cells were pelleted at 1000 g for 5 min and then subjected to hypotonic lysis of erythrocytes before washing with 30 ml PBS and centrifugation. Cells were taken up in macrophage differentiation serum (DMEM with 10% fetal calf serum (all Gibco) plus 10% of L929 conditioned medium) [Citation77] and seeded at 106 cells per well in 48 well plates (Corning). Medium was exchanged at days 3 and 6 and macrophage differentiation ascertained as CD68+CD11b+F4/80+ at day 8 by flow cytometry. For analysis of ERK1/2 phosphorylation, cells were used on day 8, when they were confluent. Cells were PBS washed and reaction buffer added (KBR-buffer: 10 mM HEPES, 125 mM NaCl, 1 mM MgCl2, 1 mM CaCl2, pH 7.3 (Virion\Serion) plus 0.1% BSA). After incubation for 30 min for the cells to adjust, the final concentration of 10 nM mouse C5a (R&D Systems), 108/ml heat-inactivated Nme, or both, were added. After incubation for 5 min, 15 min or 30 min, the supernatant was removed and cells lysed directly with 1x Laemmli buffer (BioRad) and samples boiled for 10 min. Samples were analyzed on 10% SDS polyacrylamide gels, transferred onto nitrocellulose membranes and probed with rabbit-anti-ERK1/2 (Cell Signaling #9102) or with rabbit-anti-phospho-ERK1/2 (Cell Signaling #9101), followed by goat-anti-rabbit-HRP conjugate (Jackson ImmunoResearch) and enhanced chemoluminescence imaged on a ChemiDoc MP imaging system (BioRad). Analysis of band intensities was done using ImageLab software (BioRad). For data analysis, band intensities were normalized by the division of the phospho-ERK band intensity by the corresponding relative band intensity of the total-ERK blot.
Human whole blood models
Human whole blood was drawn from healthy donors by venipuncture using hirudin Monovettes (Sarstedt) and used immediately. For measurement of oxidative burst, samples were pre-incubated for 10 min with 20 µg/ml DHR123 (Sigma Aldrich/Merck) and treated by addition of substances as follows: 1 µM PMA (Sigma Aldrich/Merck) as positive control, or 1 µg/ml of C3aR-superagonistic peptide (WWGKKYRASKLGLAR); termed C3aR-SAG here to stimulate C3aR [Citation52], 200 µM SB29015 to inhibit C3aR, 5 µM PMX53 (Tocris Bioscience) to block C5aR1, 50 µg/ml C5aR2-antibody (clone 1D9-M12; mouse IgG2a; sodium azide was removed by repeated dilution of the antibody in PBS followed by concentration using Amicon Ultra centrifugal devices (3 kDa cutoff) (Merck Millipore) to block C5aR2, 350 nM A8Δ71−73 to simultaneously block C5aR1 and C5aR2, 30 µM compstatin Cp20 to block complement at the level of C3 convertase [Citation78], or plain RPMI medium (Life Technologies) as negative control. Blood samples were infected with 107 CFU/ml of Nme in a final volume of 100 µl and incubated rotating at 37°C for 1 h. After hypotonic lysis of erythrocytes, samples were washed with cold PBS and fixated with 4% formaldehyde for 30 min at RT in the dark and then analyzed by flow cytometry on a FACSCalibur. Cell clusters were gated out with an FSC/SSC gate. Polymorphonuclear cells (PMNs) were gated based on their distinct FSC/SSC profile and oxidative burst assessed as changes in the MFI in the FL1-H channel.
For measurement of IL-8, human whole blood samples treated with antagonists as above were infected with 106 CFU/ml Nme and incubated for 90 min before addition of 1 volume ice-cold PBS with 40 mM EDTA to 1 volume of blood, centrifugation at 10,000 g for 5 min at 4°C before harvesting the supernatant. Samples were frozen at −80°C until ELISA analysis.
For measurement of any bactericidal effect of any of the antagonists above, blood samples equipped with concentrations of the antagonists as stated in were infected with Nme and incubated for 30 min before serial dilutions were made in PBS and plated onto Columbia blood agar plates for incubation and subsequent colony enumeration.
Ethics statement
For human whole blood experiments, healthy adult donors without any history of meningococcal disease, acute or chronic inflammatory disorders, or antibiotic treatment in the previous 2 weeks were used. The study was conducted in compliance with the Helsinki Declaration. The study protocol was approved by the Ethics Committee of the Medical Faculty of the University of Würzburg (file 181/16-ge) and was carried in accordance with all relevant local guidelines and was in compliance with the Helsinki Declaration. All donors expressed their informed consent in written form.
Statistics
Statistical analyses of the data were performed using Prism version 6 (GraphPad). For survival analyses () and ), the Mantel–Cox test was used. Non-parametric data (clinical scores) were analyzed using the Kruskal–Wallis test for multiple groups applying Dunnett’s post hoc test. Parametric data from more than two cohorts were analyzed by one-way ANOVA applying either Dunnett’s post hoc test to compare genotypes or treatments to the corresponding controls (e.g. WT in ), , ) or applying Bonferroni’s post hoc test to compare all cohorts (). For the analysis of bacteremia data (), , data were log-transformed to achieve normal distribution of the values. For comparison of two cohorts (), the paired, two-tailed Student’s T-test was used. For the analysis of band intensities in , one-way ANOVA applying Bonferroni’s post hoc test was chosen. Differences were considered to be significant at P < 0.05. In all figures, ns denotes not significant, * denotes P < 0.05, ** denotes P < 0.01, *** denotes P < 0.001, **** denotes P < 0.0001.
Author contributions:
M.M. and K.O.J. designed the experiments, conceived the project, analyzed the data and wrote the manuscript. M.M., L.S., and K.O.J conducted the experiments. A.K., C.G., R.A.W., J.D.L., T.M.W., and J.K. provided mouse strains and reagents and intellectual input to the study. All authors reviewed and approved the manuscript.
Competing interests
The authors declare that they have no competing interests.
Data availability
All data and materials associated with this publication are available upon request.
Supplemental Material
Download PDF (1.1 MB)Acknowledgments
This work was financed by the Deutsche Forschungsgemeinschaft grant JO 1204/2-1 issued to K.O. Johswich and by the University of Würzburg through the funding program Open Access Publishing. We would also like to thank Professor John D. Lambris, University of Pennsylvania School of Medicine, Philadelphia, USA, for providing compstatin Cp20 and Professor Craig Gerard, Children’s Hospital, Harvard Medical School, Boston, USA, for providing mouse strains.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Rosenstein NE, Perkins BA, Stephens DS, et al. Meningococcal disease. N Engl J Med. 2001 May 3;344(18):1378–1388. PubMed PMID: 11333996 .
- van Deuren M, Brandtzaeg P, van der Meer JW. Update on meningococcal disease with emphasis on pathogenesis and clinical management. Clin Microbiol Rev. 2000 Jan;13(1):144–66, table of contents. PubMed PMID: 10627495; PubMed Central PMCID: PMC88937.
- Lewis LA, Ram S. Meningococcal disease and the complement system. Virulence. 2014 Jan 1;5(1):98–126. PubMed PMID: 24104403; PubMed Central PMCID: PMC3916388.
- Harrison OB, Claus H, Jiang Y, et al. Description and nomenclature of Neisseria meningitidis capsule locus. Emerg Infect Dis. 2013 Apr;19(4):566–573. PubMed PMID: 23628376; PubMed Central PMCID: PMC3647402.
- Madico G, Welsch JA, Lewis LA, et al. The meningococcal vaccine candidate GNA1870 binds the complement regulatory protein factor H and enhances serum resistance. J Iimmunol. 2006 Jul 01;177(1):501–510. PubMed PMID: 16785547; PubMed Central PMCID: PMC2248442.
- Lewis LA, Ngampasutadol J, Wallace R, et al. The meningococcal vaccine candidate neisserial surface protein A (NspA) binds to factor H and enhances meningococcal resistance to complement. PLoS Pathog. 2010 Jul 29;6(7):e1001027. PubMed PMID: 20686663; PubMed Central PMCID: PMC2912398.
- Gorringe AR, Pajon R. Bexsero: a multicomponent vaccine for prevention of meningococcal disease. Hum Vaccin Immunother. 2012 Feb;8(2):174–183. PubMed PMID: 22426368. .
- McCarthy PC, Sharyan A, Sheikhi Moghaddam L. Meningococcal vaccines: current status and emerging strategies. Vaccines (Basel). 2018 Feb 25;6(1):12. PubMed PMID: 29495347; PubMed Central PMCID: PMC5874653.
- Figueroa JE, Densen P. Infectious diseases associated with complement deficiencies. Clin Microbiol Rev. 1991 Jul;4(3):359–395. PubMed PMID: 1889047; PubMed Central PMCID: PMC358203.
- Klos A, Tenner AJ, Johswich KO, et al. The role of the anaphylatoxins in health and disease. Mol Immunol. 2009 Sep;46(14):2753–2766. PubMed PMID: 19477527; PubMed Central PMCID: PMC2725201.
- Jagels MA, Daffern PJ, Hugli TE. C3a and C5a enhance granulocyte adhesion to endothelial and epithelial cell monolayers: epithelial and endothelial priming is required for C3a-induced eosinophil adhesion. Immunopharmacology. 2000 Mar;46(3):209–222. PubMed PMID: 10741901.
- Albrecht EA, Chinnaiyan AM, Varambally S, et al. C5a-induced gene expression in human umbilical vein endothelial cells. Am J Pathol. 2004 Mar;164(3):849–859. PubMed PMID: 14982839; PubMed Central PMCID: PMC1613300.
- Monsinjon T, Gasque P, Chan P, et al. Regulation by complement C3a and C5a anaphylatoxins of cytokine production in human umbilical vein endothelial cells. Faseb J. 2003 Jun;17(9):1003–1014. PubMed PMID: 12773483.
- Hawlisch H, Wills-Karp M, Karp CL, et al. The anaphylatoxins bridge innate and adaptive immune responses in allergic asthma. Mol Immunol. 2004 Jun;41(2–3):123–131. PubMed PMID: 15159057.
- Strey CW, Markiewski M, Mastellos D, et al. The proinflammatory mediators C3a and C5a are essential for liver regeneration. J Exp Med. 2003 Sep 15;198(6):913–923. PubMed PMID: 12975457; PubMed Central PMCID: PMC2194207.
- Gerard NP, Gerard C. The chemotactic receptor for human C5a anaphylatoxin. Nature. 1991 Feb 14;349(6310):614–617. PubMed PMID: 1847994.
- Ward PA. The harmful role of C5a on innate immunity in sepsis. J Innate Immun. 2010;2(5):439–445. PubMed PMID: 20588003; PubMed Central PMCID: PMC2968761.
- Arumugam TV, Magnus T, Woodruff TM, et al. Complement mediators in ischemia-reperfusion injury. Clin Chim Acta. 2006 Dec;374(1–2):33–45. PubMed PMID: 16872589.
- Johswich K, Martin M, Bleich A, et al. Role of the C5a receptor (C5aR) in acute and chronic dextran sulfate-induced models of inflammatory bowel disease. Inflamm Bowel Dis. 2009 Dec;15(12):1812–1823. PubMed PMID: 19714742.
- Monk PN, Scola AM, Madala P, et al. Function, structure and therapeutic potential of complement C5a receptors. Br J Pharmacol. 2007;152(4):429–448.
- Köhl J. Drug evaluation: the C5a receptor antagonist PMX-53. Curr Opin Mol Ther. 2006 Dec;8(6):529–538. PubMed PMID: 17243489.
- Okinaga S, Slattery D, Humbles A, et al. C5L2, a nonsignaling C5A binding protein. Biochemistry. 2003 Aug 12;42(31):9406–9415. PubMed PMID: 12899627.
- Scola AM, Johswich KO, Morgan BP, et al. The human complement fragment receptor, C5L2, is a recycling decoy receptor. Mol Immunol. 2009 Mar;46(6):1149–1162. PubMed PMID: 19100624; PubMed Central PMCID: PMC2697321.
- Gerard NP, Lu B, Liu P, et al. An anti-inflammatory function for the complement anaphylatoxin C5a-binding protein, C5L2. J Biol Chem. 2005 Dec 02;280(48):39677–39680. PubMed PMID: 16204243.
- Wang R, Lu B, Gerard C, et al. C5L2, the second C5a anaphylatoxin receptor, suppresses LPS-induced acute lung injury. Am J Respir Cell Mol Biol. 2016 Nov;55(5):657–666. PubMed PMID: 27285858; PubMed Central PMCID: PMC5105183.
- Rittirsch D, Flierl MA, Nadeau BA, et al. Functional roles for C5a receptors in sepsis. Nat Med. 2008 May;14(5):551–557. PubMed PMID: 18454156; PubMed Central PMCID: PMC2753858.
- Hao J, Wang C, Yuan J, et al. A pro-inflammatory role of C5L2 in C5a-primed neutrophils for ANCA-induced activation. PloS One. 2013;8(6):e66305. PubMed PMID: 23785491; PubMed Central PMCID: PMC3681967.
- Chen NJ, Mirtsos C, Suh D, et al. C5L2 is critical for the biological activities of the anaphylatoxins C5a and C3a. Nature. 2007 Mar 08;446(7132):203–207. PubMed PMID: 17322907.
- Bamberg CE, Mackay CR, Lee H, et al. The C5a receptor (C5aR) C5L2 is a modulator of C5aR-mediated signal transduction. J Biol Chem. 2010 Mar 5;285(10):7633–7644. PubMed PMID: 20044484; PubMed Central PMCID: PMC2844210.
- Kalant D, MacLaren R, Cui W, et al. C5L2 is a functional receptor for acylation-stimulating protein. J Biol Chem. 2005 Jun 24;280(25):23936–23944. PubMed PMID: 15833747.
- Hartmann K, Henz BM, Kruger-Krasagakes S, et al. C3a and C5a stimulate chemotaxis of human mast cells. Blood. 1997 Apr 15;89(8):2863–2870. PubMed PMID: 9108406.
- Daffern PJ, Pfeifer PH, Ember JA, et al. C3a is a chemotaxin for human eosinophils but not for neutrophils. I. C3a stimulation of neutrophils is secondary to eosinophil activation. J Exp Med. 1995 Jun 1;181(6):2119–2127. PubMed PMID: 7760001; PubMed Central PMCID: PMC2192052.
- Cochrane CG, Müller-Eberhard HJ. The derivation of two distinct anaphylatoxin activities from the third and fifth components of human complement. J Exp Med. 1968 Feb 1;127(2):371–386. PubMed PMID: 4383923; PubMed Central PMCID: PMC2138443.
- Williams TJ Vascular permeability changes induced by complement-derived peptides. Agents Actions. 1983 Aug; 13 (5–6): 451–455. PubMed PMID: 6195896.
- Humbles AA, Lu B, Nilsson CA, et al. A role for the C3a anaphylatoxin receptor in the effector phase of asthma. Nature. 2000 Aug 31;406(6799):998–1001. PubMed PMID: 10984054.
- Kildsgaard J, Hollmann TJ, Matthews KW, et al. Cutting edge: targeted disruption of the C3a receptor gene demonstrates a novel protective anti-inflammatory role for C3a in endotoxin-shock. J Iimmunol. 2000 Nov 15;165(10):5406–5409. PubMed PMID: 11067891.
- Mueller-Ortiz SL, Morales JE, Wetsel RA. The receptor for the complement C3a anaphylatoxin (C3aR) provides host protection against Listeria monocytogenes-induced apoptosis. J Iimmunol. 2014 Aug 1;193(3):1278–1289. PubMed PMID: 24981453; PubMed Central PMCID: PMC4122265. .
- Wu MC, Brennan FH, Lynch JP, et al. The receptor for complement component C3a mediates protection from intestinal ischemia-reperfusion injuries by inhibiting neutrophil mobilization. Proc Natl Acad Sci U S A. 2013 Jun 4;110(23):9439–9444. PubMed PMID: 23696668; PubMed Central PMCID: PMC3677481.
- Coulthard LG, Woodruff TM. Is the complement activation product C3a a proinflammatory molecule? Re-evaluating the evidence and the myth. J Iimmunol. 2015 Apr 15;194(8):3542–3548. PubMed PMID: 25848071. .
- Johswich K. Innate immune recognition and inflammation in Neisseria meningitidis infection. Pathog Dis. 2017 Mar 01;75(2). PubMed PMID: 28334203. DOI: 10.1093/femspd/ftx022.
- Herrmann JB, Muenstermann M, Strobel L, et al. Complement C5a Receptor 1 exacerbates the pathophysiology of N. meningitidis sepsis and is a potential target for disease treatment. MBio. 2018 Jan 23;9(1). PubMed PMID: 29362231.
- Johswich KO, McCaw SE, Islam E, et al. In vivo adaptation and persistence of Neisseria meningitidis within the nasopharyngeal mucosa. PLoS Pathog. 2013;9(7):e1003509. PubMed PMID: 23935487; PubMed Central PMCID: PMC3723569.
- Johswich KO, McCaw SE, Strobel L, et al. Sterilizing immunity elicited by Neisseria meningitidis carriage shows broader protection than predicted by serum antibody cross-reactivity in CEACAM1-humanized mice. Infect Immun. 2015 Jan;83(1):354–363. PubMed PMID: 25368118; PubMed Central PMCID: PMC4288889.
- Johswich KO, Gray-Owen SD. Modeling Neisseria meningitidis infection in mice: methods and logistical considerations for Nasal colonization and invasive disease. Methods Mol Biol. 2019;1969:149–168. PubMed PMID: 30877676.
- Seow V, Lim J, Iyer A, et al. Inflammatory responses induced by lipopolysaccharide are amplified in primary human monocytes but suppressed in macrophages by complement protein C5a. J Iimmunol. 2013 Oct 15;191(8):4308–4316. PubMed PMID: 24043889.
- Ember JA, Sanderson SD, Hugli TE, et al. Induction of interleukin-8 synthesis from monocytes by human C5a anaphylatoxin. Am J Pathol. 1994 Feb;144(2):393–403. PubMed PMID: 7508686; PubMed Central PMCID: PMC1887151.
- Asgari E, Le Friec G, Yamamoto H, et al. C3a modulates IL-1beta secretion in human monocytes by regulating ATP efflux and subsequent NLRP3 inflammasome activation. Blood. 2013 Nov 14;122(20):3473–3481. PubMed PMID: 23878142.
- Ward PA. The dark side of C5a in sepsis. Nat Rev Immunol. 2004 Feb;4(2):133–142. PubMed PMID: 15040586.
- Mollnes TE, Brekke OL, Fung M, et al. Essential role of the C5a receptor in E coli-induced oxidative burst and phagocytosis revealed by a novel lepirudin-based human whole blood model of inflammation. Blood. 2002 Sep 01;100(5):1869–1877. PubMed PMID: 12176911; eng.
- Strobel L, Johswich KO. Anticoagulants impact on innate immune responses and bacterial survival in whole blood models of Neisseria meningitidis infection. Sci Rep. 2018;8(1):10225.
- Krüger S, Eichler E, Strobel L, et al. Differential influences of complement on neutrophil responses to Neisseria meningitidis infection. Pathog Dis. 2018 Nov 1;76(8). PubMed PMID: 30476070.
- Ember JA, Johansen NL, Hugli TE. Designing synthetic superagonists of C3a anaphylatoxin. Biochemistry. 1991 Apr 16;30(15):3603–3612. PubMed PMID: 2015217.
- Otto M, Hawlisch H, Monk PN, et al. C5a mutants are potent antagonists of the C5a receptor (CD88) and of C5L2: position 69 is the locus that determines agonism or antagonism. J Biol Chem. 2004 Jan 2;279(1):142–151. PubMed PMID: 14570896.
- Riedemann NC, Guo RF, Neff TA, et al. Increased C5a receptor expression in sepsis. J Clin Invest. 2002 Jul;110(1):101–108. PubMed PMID: 12093893; PubMed Central PMCID: PMC151030.
- Li R, Coulthard LG, Wu MC, et al. C5L2: a controversial receptor of complement anaphylatoxin, C5a. Faseb J. 2013 Mar;27(3):855–864. PubMed PMID: 23239822.
- Johswich K, Klos A. C5L2–an anti-inflammatory molecule or a receptor for acylation stimulating protein (C3a-desArg)? Adv Exp Med Biol. 2007;598:159–180. PubMed PMID: 17892211.
- Zhang T, Garstka MA, Li K. The controversial C5a receptor C5aR2: its role in health and disease. J Immunol Res. 2017;2017:8193932. PubMed PMID: 28706957; PubMed Central PMCID: PMC5494583.
- Riedemann NC, Guo RF, Bernacki KD, et al. Regulation by C5a of neutrophil activation during sepsis. Immunity. 2003 Aug;19(2):193–202. PubMed PMID: 12932353.
- Sonego F, Castanheira FV, Ferreira RG, et al. Paradoxical roles of the neutrophil in sepsis: protective and deleterious. Front Immunol. 2016;7:155. PubMed PMID: 27199981; PubMed Central PMCID: PMC4844928.
- Mueller-Ortiz SL, Hollmann TJ, Haviland DL, et al. Ablation of the complement C3a anaphylatoxin receptor causes enhanced killing of Pseudomonas aeruginosa in a mouse model of pneumonia. Am J Physiol Lung Cell Mol Physiol. 2006 Aug;291(2):L157–65. PubMed PMID: 16461429.
- Dutow P, Fehlhaber B, Bode J, et al. The complement C3a receptor is critical in defense against Chlamydia psittaci in mouse lung infection and required for antibody and optimal T cell response. J Infect Dis. 2014 Apr 15;209(8):1269–1278. PubMed PMID: 24273177; PubMed Central PMCID: PMC3969542.
- Busche MN, Stahl GL Role of the complement components C5 and C3a in a mouse model of myocardial ischemia and reperfusion injury. German Med Sci. 2010 Sep 8;8. PubMed PMID: 20930931; PubMed Central PMCID: PMC2940219. DOI:10.3205/000109.
- Mathieu MC, Sawyer N, Greig GM, et al. The C3a receptor antagonist SB 290157 has agonist activity. Immunol Lett. 2005 Sep 15;100(2):139–145. PubMed PMID: 16154494.
- Martin U, Bock D, Arseniev L, et al. The human C3a receptor is expressed on neutrophils and monocytes, but not on B or T lymphocytes. J Exp Med. 1997 Jul 21;186(2):199–207. PubMed PMID: 9221749; PubMed Central PMCID: PMC2198980.
- Quell KM, Karsten CM, Kordowski A, et al. Monitoring C3aR expression using a floxed tdTomato-C3aR reporter knock-in mouse. J Iimmunol. 2017 Jul 15;199(2):688–706. PubMed PMID: 28626064.
- Wu F, Zou Q, Ding X, et al. Complement component C3a plays a critical role in endothelial activation and leukocyte recruitment into the brain. J Neuroinflammation. 2016 Jan 28;13:23. 10.1186/s12974-016-0485-y. PubMed PMID: 26822321; PubMed Central PMCID: PMC4731990.
- Coureuil M, Bourdoulous S, Marullo S, et al. Invasive meningococcal disease: a disease of the endothelial cells. Trends Mol Med. 2014 Oct;20(10):571–578. PubMed PMID: 25178566.
- Sauter RJ, Sauter M, Reis ES, et al. Functional relevance of the anaphylatoxin receptor C3aR for platelet function and arterial thrombus formation marks an intersection point between innate immunity and thrombosis. Circulation. 2018 Oct 16;138(16):1720–1735. PubMed PMID: 29802205; PubMed Central PMCID: PMC6202244.
- Konar M, Granoff DM. Eculizumab treatment and impaired opsonophagocytic killing of meningococci by whole blood from immunized adults. Blood. 2017 Aug 17;130(7):891–899. PubMed PMID: 28630122; PubMed Central PMCID: PMC5561903. .
- Sprong T, Brandtzaeg P, Fung M, et al. Inhibition of C5a-induced inflammation with preserved C5b-9-mediated bactericidal activity in a human whole blood model of meningococcal sepsis. Blood. 2003 Nov 15;102(10):3702–3710. 10.1182/blood-2003-03-0703. PubMed PMID: 12881318.
- Nabizadeh JA, Manthey HD, Steyn FJ, et al. The complement C3a receptor contributes to melanoma tumorigenesis by inhibiting neutrophil and CD4+ T cell responses. J Iimmunol. 2016 Jun 1;196(11):4783–4792. PubMed PMID: 27183625.
- Wenderfer SE, Wang H, Ke B, et al. C3a receptor deficiency accelerates the onset of renal injury in the MRL/lpr mouse. Mol Immunol. 2009 Apr;46(7):1397–1404. PubMed PMID: 19167760; PubMed Central PMCID: PMC2697606.
- Höpken UE, Lu B, Gerard NP, et al. The C5a chemoattractant receptor mediates mucosal defence to infection. Nature. 1996 Sep 5;383(6595):86–89. PubMed PMID: WOS:A1996VF29500053; English.
- Wessels MR, Butko P, Ma M, et al. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci U S A. 1995 Dec 5;92(25):11490–11494. PubMed PMID: 8524789; PubMed Central PMCID: PMC40427.
- Gu A, Zhang Z, Zhang N, et al. Generation of human CEACAM1 transgenic mice and binding of Neisseria Opa protein to their neutrophils. PubMed PMID: 20404914; PubMed Central PMCID: PMC2852402. eng PloS One. 2010;54:e10067.
- Lappann M, Haagensen JA, Claus H, et al. Meningococcal biofilm formation: structure, development and phenotypes in a standardized continuous flow system. Mol Microbiol. 2006 Dec;62(5):1292–1309. PubMed PMID: 17121595. .
- Weischenfeldt J, Porse B. Bone marrow-derived macrophages (BMM): isolation and applications. CSH Protoc. 2008 Dec 1 PubMed PMID: 21356739:2008. pdbprot5080. DOI:10.1101/pdb.prot5080
- Qu H, Magotti P, Ricklin D, et al. Novel analogues of the therapeutic complement inhibitor compstatin with significantly improved affinity and potency. Mol Immunol. 2011 Jan;48(4):481–489. PubMed PMID: 21067811; PubMed Central PMCID: PMC3014449.