
ABSTRACT
Metformin, as the first-line oral drug for type 2 diabetes, has proven benefits against aging, cancer and cardiovascular diseases. But the influence of metformin to the immune response and its molecular mechanisms remain obscure. Metformin increases resistance to not only the Gram-negative pathogens Pseudomonas aeruginosa and Salmonella enterica but also the Gram-positive pathogens Enterococcus faecalis and Staphylococcus aureus. Meanwhile, metformin protects the animals from the infection by enhancing the tolerance to the pathogen infection rather than by reducing the bacterial burden. Through the screening of classical immune pathways in C. elegans, we find metformin enhances innate immunity through p38 MAPK pathway. Furthermore, activated p38/PMK-1 by metformin acts on the intestine for innate immune response. In addition, metformin-treated mice have increased resistance to P. aeruginosa PA14 infection and significantly increased the levels of active PMK-1. Therefore, promoted p38/PMK-1-mediated innate immunity by metformin is conserved from worms to mammals. Our work provides a conserved mechanism by which metformin enhances immune response and boosts its therapeutic application in the treatment of pathogen infection.
Introduction
With the development of antibiotic-resistant bacteria pathogens, identification of a chemical or pharmacological drug that could influence human immune response and decrease the risks of pathogen infection has become a key goal of innate immunity research. Metformin, an antiglycemic biguanide drug and the first-line drug for the treatment of type 2 diabetes, has been proven to extend lifespan in Caenorhabditis elegans [Citation1–Citation5] and mice [Citation6,Citation7]. Furthermore, metformin has been reported to the benefit of the cardiovascular diseases in patients [Citation8]. Metformin also has been used in the treatment of metabolic syndrome and cancer [Citation9,Citation10].
Recent studies have shown that metformin has antimicrobial properties for the treatment of pathogen infection, including Trichinella spiralis [Citation11], Staphylococcus aureus [Citation12], Pseudomonasaeruginosa [Citation13,Citation14], hepatitis B virus [Citation15], hepatitis C virus [Citation16,Citation17], and human immunodeficiency virus [Citation18]. For example, Calu-3 airway epithelial cocultured cells with metformin treatment inhibited the basolateral glucose-induced apical P. aeruginosa growth. Treatment with metformin increased the expression of claudin-1 protein and occludin, which were reduced by P. aeruginosa infection [Citation13]. In addition, hyperglycemic mice with metformin treatment led to a significant reduction of airway P. aeruginosa bacterial load and a decrease in airway glucose concentrations [Citation14]. Although the mechanism of underlying metformin’s metabolic effects not fully understood, it had been widely attributed to AMPK activation [Citation13,Citation19–Citation21]. Furthermore, metformin inhibited the mitochondrial complex I activity against drug-resistant strains of tuberculosis [Citation22–Citation24]. In addition, metformin decreased the mitochondrial H2O2 emission in the skeletal muscle of obese rats [Citation25]. Overall, despite the beneficial functions for metformin in multiple cellular processes, its contribution to innate immunity in animals is unknown.
The innate immune system represents the first line of defense against invading pathogens and it is evolutionarily conserved from worms to mammals [Citation26]. During infection, the innate immune system is activated, resulting in antimicrobial response to invading pathogens [Citation26–Citation30]. Caenorhabditis elegans has been developed as a valuable genetic model for research on the animal immune response. Through using this tractable model, researchers uncover several signaling pathways that have important roles in controlling the innate immunity, such as the PMK-1/p38 MAPK pathway [Citation31,Citation32], the DAF-2/DAF-16 pathway [Citation33], the MPK-1/ERK MAPK pathway [Citation34], the protein kinase D DKF-2 [Citation35], the G protein-coupled receptor FSHR-1 Citation36, and the G protein GqαEGL-30 [Citation37].
In this study, we investigated the ability of metformin to modulate C. elegans host defense. Through the screening of classical immune pathways in C. elegans, we found that metformin protects the host against pathogens through governing p38/PMK-1 activation. In addition, p38/PMK-1 functioned in the intestine for innate immunity activation by metformin. This ancient metformin response pathway was conserved from worms to mammals. These findings revealed that the metformin-promoted innate immunity would significantly boost its application to improve the patients of pathogen infection.
Results
Metformin enhances pathogen resistance
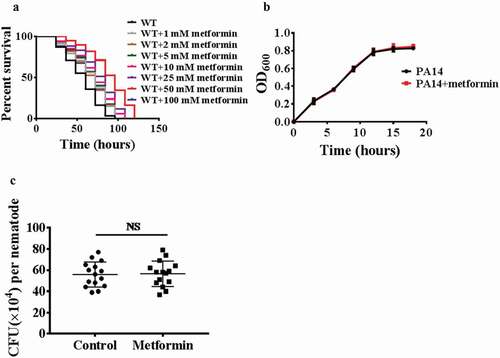
To determine whether metformin promotes the innate immunity, worms were exposed to the human opportunistic pathogen Pseudomonas aeruginosa(PA14), and we found that wild-type animals treated with metformin (1 mM, 2 mM, 5 mM, 10 mM, 25 mM, 50 mM, and 100 mM) exhibited increased resistance to P. aeruginosa in dose-dependent manner (; Table S2). Like lifespan [Citation2], metformin shows a saturating effect on pathogen resistance, maximal at 50 mM drug, and declining at 100 mM drug (; Table S2). After metformin treatment, worms exposed to Staphylococcus aureus, Enterococcus faecalis, or Salmonella enterica have a higher survival rate (Figure S1A, 1B, and 1C; Table S3). These results suggest that metformin enhances the innate immunity in C. elegans. Previous study has shown that metformin inhibits E. coli proliferation through a dose-dependent manner [Citation2]. To test whether metformin promotes host immune response via inhibiting the growth of pathogenic bacteria, we used the bacterial growth assay and demonstrated that metformin did not inhibit the proliferation of P. aeruginosa PA14 (), S. aureus, E. faecalis, S. enterica (Figure S2A, 2B, and 2C). Taken together, these results indicate that metformin action on immune response is not caused by inhibition of bacterial growth. GacA is a virulence-related gene in pathogen Pseudomonas aeruginosa [Citation38]. Worms were exposed to the human opportunistic pathogen Pseudomonas aeruginosa(PA14GacA mutant), and we found that wild-type animals treated with metformin (50 mM) exhibited increased resistance to P. aeruginosa (PA14GacA mutant) (Figure S3; Table S3). The clearance of the bacterial load is part of host defense against pathogen infection [Citation39,Citation40]. Thus, we then examined whether metformin influenced the bacterial accumulation. Metformin did not affect the colony-forming units (CFUs) of bacteria in WT worms after P. aeruginosa PA14 infection (). Overall, these results suggest that metformin enhances host tolerance to pathogen infection rather than reduces the bacterial burden.
Figure 1. Metformin enhances pathogen resistance
Metformin promotes innate immunity through the p38 MAPK pathway
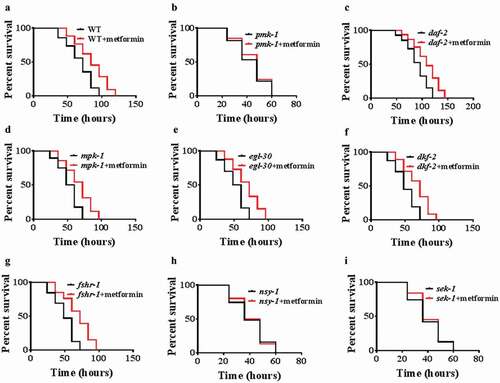
To investigate the molecular mechanisms by which metformin confers protection against pathogens, we screened several signaling pathways, which involved in innate immunity in C. elegans, such as p38 MAPK/PMK-1 [Citation31], DAF-2/DAF-16 [Citation33], ERK MAPK/MPK-1 [Citation34], EGL-30 [Citation37], protein kinase D/DKF-2 [Citation35], and FSHR-1 [Citation36]. We found that metformin failed to enhance resistance to P. aeruginosa PA14 infection in pmk-1(km25) mutants, compared with WT worms (, ; Table S2). However, metformin promoted the survival rates of daf-2(e1370), mpk-1(n2521), egl-30(n686), dkf-2(ok1704), and fshr-1(ok778) mutants after P. aeruginosa PA14 infection (-g; Table S2). Furthermore, we then tested the core components of the p38 MAPK signaling, the MAPK kinase NSY-1 and the MAPK kinase SEK-1. We found that metformin could not confer resistance to PA14 infection in nsy-1(ag3) and sek-1(ag1)mutants (, ; Table S2). VHP-1 MAPK phosphatase (MKP), a homolog of mammalian MKP7, also regulates pathogen resistance through the modulation of PMK-1 activity [Citation41]. Vhp-1RNAi was non-additively beneficial with metformin (Figure S4A; Table S3). Furthermore, knockdown of vhp-1did not further increase the level of K08D8.5p::GFP(p38 target gene) under metformin treatment (Figure S4B). Taken together, these results suggest that metformin acts on the VHP-1/p38 MAPK pathway to enhance innate immunity in C. elegans.
Figure 2. Metformin promotes innate immunity through the p38 MAPK pathway
Metformin activates p38 MAPK signaling in C. elegans
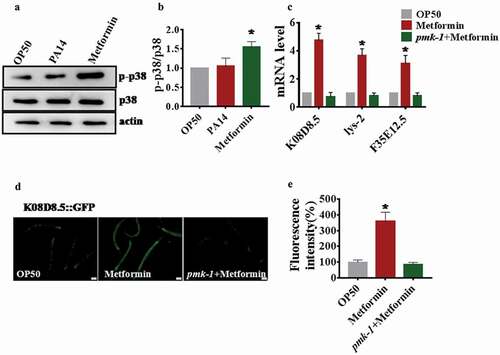
To determine whether metformin activates p38 MAPK signaling or not, we tested the level of phosphorylation PMK-1, which is a signature of its activation [Citation35,Citation39]. We found that P. aeruginosa PA14 infection did not heighten the phosphorylation levels of p38 MAPK in C.elegans (), which is consistent with a recent study [Citation42]. In contrast, metformin significantly increased the protein levels of active PMK-1 in C.elegans(). Next, we tested the expression of PMK-1-targeted genes, K08D8.5, lys-2, and F35E12.5 [Citation43]. Quantitative real-time PCR analysis demonstrated that PMK-1-dependent genes were up-regulated in metformin-treated animals compared with OP50 (). However, metformin failed to enhance their expression in the pmk-1(km25)worms (). Furthermore, we detected the expression of K08D8.5 through using the transgenic worms expressing K08D8.5p::GFP. We observed higher levels of GFP in metformin-treated animals, but not in pmk-1(km25) worms (). In addition, to find out whether metformin activated the PMK-1 through a canonical metformin target aak-2, skn-1, mitochondrial complex I(gas-1), mTOR(let-363). Using the K08D8.5::GFP reporter, we found that knockdown of aak-2, skn-1, gas-1, let-363 did not influence the level of K08D8.5::GFP in metformin-treated animals (Figure S4B and 4C) indicating that the activation of PMK-1 by metformin did not depend on canonical targets. In conclusion, these findings indicate that metformin activates the p38 MAPK pathway in C. elegans.
Figure 3. Metformin activates p38 MAPK signaling in C. elegans
Intestinal PMK-1 enhances resistance to pathogen infection after metformin treatment
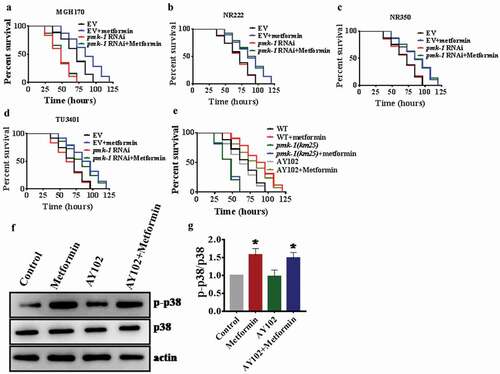
To determine tissue-specific actions of PMK-1 in response to P. aeruginosa infection after metformin treatment, we performed tissue of knockdown experiments using MGH170 strain in the intestine [Citation44], NR222 strain in hypodermis [Citation45], NR350 strain in muscle [Citation45], and TU3401 strain in neuron [Citation46]. We found that knockdown of pmk-1 in the intestine of MGH170 animals completely abolished the protection conferred by metformin (; Table S2). However, after metformin treatment, other knockdowns did not prevent metformin action on promoting pseudomonas resistance (; Table S2). These results demonstrate that metformin- induced immune response is necessary to the intestinal activity of PMK-1. To further determine whether metformin depends on the intestinal PMK-1 to regulate immunity, we utilized the AY102 transgenic strain, which carried pmk-1 under the control of the intestinal vha-6 in pmk-1(km25) worms. We found that expression of pmk-1 in the intestine after metformin treatment fully rescued the survival rate of pmk-1(km25) animals exposed to P. aeruginosa PA14 (; Table S2). Furthermore, pre-treatment of AY102 worms with metformin restored the level of active PMK-1 (). Taken together, these results suggest that metformin triggered immunity requires the intestinal activity of PMK-1.
Figure 4. Intestinal PMK-1 enhances resistance to pathogen infection after metformin treatment
Metformin protected mice against P. aeruginosa infection
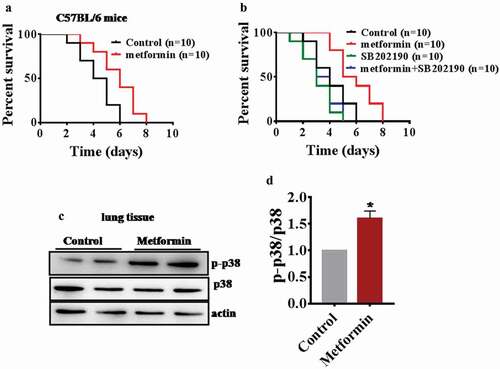
Next, we examined whether metformin enhances the innate immunity in mice. The mice were treated with 200 mg/kg body weight metformin and control mice were subjected to infection (1.0 × 106 CFUs/mouse) with P. aeruginosa PA14. The result showed that metformin-treated mice had increased resistance to P. aeruginosa PA14 infection compared with control mice (; Table S4). The p38 MAPK pathway is conserved from worms to mammals. To test whether metformin acts on the p38 MAPK pathway to enhance innate immunity in mice, the treating mice with 200 mg/kg body weight metformin or 20 μg/kg body weight p38 inhibitor SB202190 [Citation47] and control mice were subjected to infection (1.0 × 106 CFUs/mouse) with P. aeruginosa PA14. The results showed that SB202190 increased the susceptibility to P. aeruginosa PA14 infection compared with control mice (). In contrast, SB202190 suppressed the enhancement resistance to P. aeruginosa PA14 upon metformin treatment (; Table S4). Meanwhile, we also found that metformin significantly increased the levels of active PMK-1 in mice (). Taken together, metformin may also activate immunity in mice through p38 MAPK pathway.
Figure 5. Metformin protected mice against P. aeruginosa infection and increased p-p38 in the lung
Discussion
In this study, we identify that preventive application of metformin protects C. elegans against Gram-negative pathogens P. aeruginosa, S. enterica and the Gram-positive pathogens E. faecalis, S. aureus. This effect is associated with a conserved mechanism that regulates the innate immunity in C. elegans, including the p38/PMK-1 MAPK pathway. In addition, our results indicate that the enhancement of immune responses by metformin requires intestinal PMK-1 and that it does not protect the worms from the infection by reducing the bacterial burden but rather by making the worms more tolerant to the infection. Furthermore, metformin also activates immunity in mice through p38 MAPK pathway. Taken together, these results uncover an alternative mechanism by which antibiotic action-like metformin may protect the host from bacterial infections.
Previous work has shown that metformin reduced the levels of pro-inflammatory cytokines and improved survival in several endotoxemia models of mice [Citation19]. Furthermore, Metformin was active against T. spiralis, S. aureus, P. aeruginosa, and hepatitis B virusin vitro [Citation19]. These researches imply that metformin may have a potential role in the therapy for pathogen infection. However, whether the metformin influences the immune response and the underlying molecular mechanisms remain obscure. Here, using model organism C. elegans, we investigate the ability of metformin to modulate host defense. Through the screening of classical immune pathways in C.elegans, we find that metformin protects the host against pathogens through governing p38/PMK-1 activation. Animal studies also show that metformin protects mice against P. aeruginosa infection and it activates the p38/PMK-1. These results presented clearly show that metformin acts as an activator of conserved immune pathways. Intriguingly, metformin may function on AMPK to prevent the P. aeruginosa-induced increase in glucose permeability and hyperglycemia-induced growth [Citation13]. In this study, our work indicates that metformin regulates the innate immunity independent of AMPK(AAK-2) in C. elegans.
It is noteworthy that based on the immunomodulatory properties, certain antibiotics improve the long-term outcome of patients with some chronic diseases [Citation48,Citation49]. For example, azithromycin, a macrolide, has beneficial effects of the patients with chronic obstructive pulmonary disease and cystic fibrosis (CF) [Citation50,Citation51]. Therefore, using immunomodulatory properties to develop new anti-infective drugs is an effective way. However, a role for antibiotic action-like metformin in the control of immunity has not been reported. Here we show that a short, nontoxic treatment with metformin can activate the innate immunity in C. elegans and mice through the p38/PMK-1-dependent manner. Our work put forward an ancient and conserved mechanism that metformin protects worms and mice from infection by pathogens.
Materials and methods
Worm strains and cultivation
Worms were maintained and propagated under standard conditions as previously described [Citation52,Citation53]. The following nematode strains were obtained from the Caenorhabditis Genetics Center (CGC), which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440): N2 Bristol wild-type, KU25 pmk-1(km25), AU3 nsy-1(ag3), AU1 sek-1(ag1), CB1370 daf-2(e1370), SD184 mpk-1(n2521), MT1434 egl-30(n686), RB911 fshr-1(ok778), RB1468 dkf-2(ok1704), SAL144(K08D8.5p::gfp), AY102 pmk-1(km25);acEx102, NR222 rde-1(ne219);kzIs9, NR350 rde-1(ne219);kzIs20, TU3401 (sid-1(pk3321)V;uIs69V) for neuronal RNAi. The nematode strain for intestinal RNAi (sid-1(qt9); Is [vha-6pr::sid-1]; Is [sur-5pr:: GFPNLS]) were kindly provided by Dr. Gary Ruvkun (Massachusetts General Hospital, Harvard Medical School, Boston, MA). C. elegans mutants were backcrossed three times into the WT strain (N2) and used in the laboratory.
RNA interference
The strains of E.coliused for RNAi were obtained from the Ahringer library [Citation54]. RNAi feeding experiments were performed on synchronized L1 to L2 larvae at 20°C. Briefly, E. coli strain HT115(DE3) expressing dsRNA was grown overnight in LB broth containing 100 μg/ml ampicillin at 37°C, and then spread to NGM plates containing 100 μg/ml ampicillin and 5 mM isopropyl 1-thio-β-D-galactopyranoside (IPTG). The RNAi-expressing bacteria were grown overnight at 25°C. Synchronized L1 to L2 larvae were placed on RNAi plates until they reach maturity at 20°C. Unc-22 RNAi was included as a positive control in all experiments to account for RNAi efficiency.
Infection assay
E.coli OP50, S. enterica SL1344, E. faecalis ATCC 29,212, and P. aeruginosa PA14 were grown overnight in LB broth at 37°C, and S. aureus NCTC8325 was grown overnight in tryptic soy broth (TSB, BD, Sparks, MD) at 37°C, were then spread to NGM plates. All infection assays were performed on NGM agar plates or NGM plates supplemented with or without 50 mM of metformin. Synchronized populations of worms were cultivated on E.coli OP50 at 20°C until the young adult stage (i.e., within 12 h beyond the L4 stage). About 50–60 worms were transferred to NGM agar plates containing S. enterica SL1344, S. aureus NCTC8325, and P. aeruginosa PA14 at 25°C, respectively. The number of living worms was counted at 12 h intervals. Immobile adult worms unresponsive to touch were scored as dead. Three plates were tested per assay and all experiments were performed three times independently.
Fluorescence microscopy
Synchronized L1 worms of the K08D8.5::GFP strain were transferred to agar plates supplemented with or without 50 mM of metformin. The images were obtained by using a Zeiss Axioskop 2 plus fluorescence microscope (Carl Zeiss, Jena, Germany) with a digital camera. Fluorescence intensity was quantified by using the ImageJ software (NIH). Three plates of about 40 animals per plate were tested per assay and all experiments were performed three times independently.
Quantitative real-time PCR
Total RNA was extracted from worms with TRIzolReagent (Invitrogen) as previously described [Citation55]. Random-primed cDNAs were generated by reverse transcription of the total RNA samples with SuperScript II (Invitrogen) and qPCR analysis was conducted by using SYBR Premix-Ex TagTM (Takara, Dalian, China) on an Applied Biosystems Prism 7000 Sequence Detection System (Applied Biosystems, Foster City, CA). Using pmp-3 for an internal control as previously described [Citation55]. The primers used for PCR were listed in Table S1.
Quantification of intestinal bacterial loads
Synchronized populations of worms were cultivated on E.coli OP50 at 20°C until the young adult stage. P. aeruginosa/GFP were grown in LB liquid medium containing ampicillin (100 μg/ml) at 37°C overnight and plated onto NGM plates. Worms then were transferred to NGM agar plates (supplemented with or without 50 mM of metformin) containing P. aeruginosa/GFP for 48 h at 25°C [Citation39]. To eliminate the P. aeruginosa/GFP around the surface of worms, worms were transferred to NGM agar plate seeded with E. coliOP50 for 15 min for three times [Citation39]. Ten worms were transferred into 50-µl PBS plus 0.1% Triton and ground [Citation39]. The lysates were serially diluted by 10-folds in sterilized water and spread onto LB agar plates/ampicillin at 37°C. After 1 d of incubation at 37°C, colonies of P. aeruginosa/GFP were counted. Five plates were tested per assay and all experiments were performed three times independently.
Western blotting
After worms and lung tissues were homogenized in liquid nitrogen, the homogenate was lysed on ice for 60 min in lysis buffer (BioTeKe). The lysates of total protein were loaded (40 μg per well) and separated on a 10% SDS polyacrylamide gel. Proteins were then transferred to immobilon-PSQ transfer PVDF membrane (Millipore, Bedford, MA). Phosphorylated PMK-1 protein was detected using an anti-active p38 polyclonal antibody from rabbit (1:1000 dilution; Abcam, ab4822), and anti-beta-actin antibodies (1:1000 dilution; Abcam, ab227387). The secondary antibody was a peroxidase-coupled anti-rabbit IgG (1:20,000 dilution; Abmart). Blots were developed using the Super Signal chemiluminescence substrate (Pierce). Band intensities were measured using ImageJ software.
Animal studies
C57BL/6 mice were inoculated with P. aeruginosa-laden agarose beads, as previously described [Citation56]. The average 50 µl agar-beads suspension contained 1.0 × 106 CFUs/mouse. The agar-beads suspension was ready for inoculation in the lungs of mice by an intratracheal injection. Meanwhile, some of animals received daily doses of 200 mg/kg body weight metformin (#D150959‑5G) and SB202190 (20 μg/kg/d) through intraperitoneal injection for 5 d.
Statistics
Differences in survival rates were analyzed using the log-rank test. Differences in gene expression and fluorescence intensity were determined by performing a one-way ANOVA followed by a Student-Newman-Keulstest. Data were analyzed using the SPSS17.0 software (IBM, Armonk, New York).
Supplemental Material
Download MS Word (545.7 KB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Onken B, Driscoll M. Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans Healthspan via AMPK, LKB1, and SKN-1. PloS One. 2010;5:e8758.
- Cabreiro F, Au C, Leung KY, et al. Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell. 2013;153:228–239.
- Wu L, Zhou B, Oshiro-Rapley N, et al. An ancient, unified mechanism for metformin growth inhibition in C. elegans and cancer. Cell. 2016;167:1705–18 e13.
- De Haes W, Frooninckx L, Van Assche R, et al. Metformin promotes lifespan through mitohormesis via the peroxiredoxin PRDX-2. Proc Natl Acad Sci U S A. 2014; 111:E2501–9.
- Chen J, Ou Y, Li Y, et al. Metformin extends C. elegans lifespan through lysosomal pathway. eLife. 2017;6:e31268.
- Martin-Montalvo A, Mercken EM, Mitchell SJ, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013;4:2192.
- Anisimov VN, Berstein LM, Egormin PA, et al. Metformin slows down aging and extends life span of female SHR mice. Cell Cycle. 2008;7:2769–2773.
- Scarpello JH. Improving survival with metformin: the evidence base today. Diabetes Metab. 2003;29:6S36–43.
- Dowling RJ, Goodwin PJ, Stambolic V. Understanding the benefit of metformin use in cancer treatment. BMC Med. 2011;9:33.
- Foretz M, Guigas B, Bertrand L, et al. Metformin: from mechanisms of action to therapies. Cell Metab. 2014;20:953–966.
- Othman AA, Abou Rayia DM, Ashour DS, et al. Atorvastatin and metformin administration modulates experimental Trichinella spiralis infection. Parasitol Int. 2016;65:105–112.
- Garnett JP, Baker EH, Naik S, et al. Metformin reduces airway glucose permeability and hyperglycaemia-induced Staphylococcus aureus load independently of effects on blood glucose. Thorax. 2013;68:835–845.
- Patkee WR, Carr G, Baker EH, et al. Metformin prevents the effects of Pseudomonas aeruginosa on airway epithelial tight junctions and restricts hyperglycaemia-induced bacterial growth. J Cell Mol Med. 2016;20:758–764.
- Tzanavari T, Varela A, Theocharis S, et al. Metformin protects against infection-induced myocardial dysfunction. Metabolism. 2016;65:1447–1458.
- Xun YH, Zhang YJ, Pan QC, et al. Metformin inhibits hepatitis B virus protein production and replication in human hepatoma cells. J Viral Hepat. 2014;21:597–603.
- Yu JW, Sun LJ, Liu W, et al. Hepatitis C virus core protein induces hepatic metabolism disorders through down-regulation of the SIRT1-AMPK signaling pathway. Int J Infect Dis. 2013;17:e539–45.
- Hsu CS, Hsu SJ, Lin HH, et al. A pilot study of add-on oral hypoglycemic agents in treatment-naive genotype-1 chronic hepatitis C patients receiving peginterferon alfa-2b plus ribavirin. J Formos Med Assoc. 2014;113:716–721.
- Fitch K, Abbara S, Lee H, et al. Effects of lifestyle modification and metformin on atherosclerotic indices among HIV-infected patients with the metabolic syndrome. Aids. 2012;26:587–597.
- Malik F, Mehdi SF, Ali H, et al. Is metformin poised for a second career as an antimicrobial? Diabetes Metab Res Rev. 2018;34:e2975.
- Shaw RJ, Lamia KA, Vasquez D, et al. The kinase LKB1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science. 2005;310:1642–1646.
- Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174.
- Singhal A, Jie L, Kumar P, et al. Metformin as adjunct antituberculosis therapy. Sci Transl Med. 2014;6:263ra159.
- Madiraju AK, Erion DM, Rahimi Y, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510:542–546.
- Vashisht R, Brahmachari SK. Metformin as a potential combination therapy with existing front-line antibiotics for Tuberculosis. J Transl Med. 2015;13:83.
- Kane DA, Anderson EJ, Price JW 3rd, et al. Metformin selectively attenuates mitochondrial H2O2 emission without affecting respiratory capacity in skeletal muscle of obese rats. Free Radic Biol Med. 2010;49:1082–1087.
- Aballay A, Ausubel FM. Caenorhabditis elegans as a host for the study of host-pathogen interactions. Curr Opin Microbiol. 2002;5:97–101.
- Xiao Y, Liu F, Zhao PJ, et al. PKA/KIN-1 mediates innate immune responses to bacterial pathogens in Caenorhabditis elegans. Innate Immun. 2017;23:656–666.
- Irazoqui JE, Urbach JM, Ausubel FM. Evolution of host innate defence: insights from Caenorhabditis elegans and primitive invertebrates. Nat Rev Immunol. 2010;10:47–58.
- Kim D. Studying host-pathogen interactions and innate immunity in Caenorhabditis elegans. Dis Model Mech. 2008;1:205–208.
- Kurz CL, Ewbank JJ. Caenorhabditis elegans: an emerging genetic model for the study of innate immunity. Nat Rev Genet. 2003;4:380–390.
- Kim DH, Feinbaum R, Alloing G, et al. A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science. 2002;297:623–626.
- Aballay A, Drenkard E, Hilbun LR, et al. Caenorhabditis elegans innate immune response triggered by Salmonella enterica requires intact LPS and is mediated by a MAPK signaling pathway. Curr Biol. 2003;13:47–52.
- Garsin DA, Villanueva JM, Begun J, et al. Long-lived C. elegans daf-2 mutants are resistant to bacterial pathogens. Science. 2003;300:1921.
- Nicholas HR, Hodgkin J. The ERK MAP kinase cascade mediates tail swelling and a protective response to rectal infection in C. elegans. Curr Biol. 2004;14:1256–1261.
- Ren M, Feng H, Fu Y, et al. Protein kinase D is an essential regulator of C. elegans innate immunity. Immunity. 2009;30:521–532.
- Powell JR, Kim DH, Ausubel FM The G protein-coupled receptor FSHR-1 is required for the Caenorhabditis elegans innate immune response. Proc Natl Acad Sci U S A. 2009; 106:2782–2787.
- Kawli T, Wu C, Tan MW Systemic and cell intrinsic roles of Gqalpha signaling in the regulation of innate immunity, oxidative stress, and longevity in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2010; 107:13788–13793.
- Tan MW, Rahme LG, Sternberg JA, et al. Pseudomonas aeruginosa killing of Caenorhabditis elegans used to identify P. aeruginosa virulence factors. Proc Natl Acad Sci U S A. 1999; 96:2408–2413.
- Sun J, Singh V, Kajino-Sakamoto R, et al. Neuronal GPCR controls innate immunity by regulating noncanonical unfolded protein response genes. Science. 2011;332:729–732.
- Fuhrman LE, Goel AK, Smith J, et al. Nucleolar proteins suppress Caenorhabditis elegans innate immunity by inhibiting p53/CEP-1. PLoS Genet. 2009;5:e1000657.
- Kim DH, Liberati NT, Mizuno T, et al. Integration of Caenorhabditis elegans MAPK pathways mediating immunity and stress resistance by MEK-1 MAPK kinase and VHP-1 MAPK phosphatase. Proc Natl Acad Sci U S A. 2004; 101:10990–10994.
- Ren Z, Ambros VR Caenorhabditis elegans microRNAs of the let-7 family act in innate immune response circuits and confer robust developmental timing against pathogen stress. Proc Natl Acad Sci U S A. 2015; 112:E2366–75.
- Cao X, Aballay A. Neural inhibition of dopaminergic signaling enhances immunity in a cell-non-autonomous manner. Curr Biol. 2016;26:2329–2334.
- Melo JA, Ruvkun G. Inactivation of conserved C. elegans genes engages pathogen- and xenobiotic-associated defenses. Cell. 2012;149:452–466.
- Qadota H, Inoue M, Hikita T, et al. Establishment of a tissue-specific RNAi system in C. elegans. Gene. 2007;400:166–173.
- Calixto A, Chelur D, Topalidou I, et al. Enhanced neuronal RNAi in C. elegans using SID-1. Nat Methods. 2010;7:554–559.
- Grossi V, Liuzzi M, Murzilli S, et al. Sorafenib inhibits p38alpha activity in colorectal cancer cells and synergizes with the DFG-in inhibitor SB202190 to increase apoptotic response. Cancer Biol Ther. 2012;13:1471–1481.
- Tauber SC, Nau R. Immunomodulatory properties of antibiotics. Curr Mol Pharmacol. 2008;1:68–79.
- Cai Y, Cao X, Aballay A. Whole-animal chemical screen identifies colistin as a new immunomodulator that targets conserved pathways. mBio. 2014;5:e01235-14.
- Rubin BK, Henke MO. Immunomodulatory activity and effectiveness of macrolides in chronic airway disease. Chest. 2004;125:70S–8S.
- Saiman L, Marshall BC, Mayer-Hamblett N, et al. Azithromycin in patients with cystic fibrosis chronically infected with Pseudomonas aeruginosa: a randomized controlled trial. Jama. 2003;290:1749–1756.
- Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974;77:71–94.
- Stiernagle T. Maintenance of C. elegans. In: Fay D, editor. WormBook. 2006. p. 1–11.
- Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313–321.
- Lapierre LR, De Magalhaes Filho CD, McQuary PR, et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat Commun. 2013;4:2267.
- Facchini M, De Fino I, Riva C, et al. Long term chronic Pseudomonas aeruginosa airway infection in mice. J Vis Exp. 2014:e51019.