
ABSTRACT
Using the USA300, methicillin-resistant Staphylococcus aureus strain LAC, we previously examined the impact of regulatory mutations implicated in biofilm formation on protease production and virulence in a murine sepsis model. Here we examined the impact of these mutations in the USA200, methicillin-sensitive strain UAMS-1. Mutation of agr, mgrA, rot, sarA and sigB attenuated the virulence of UAMS-1. A common characteristic of codY, rot, sigB, and sarA mutants was increased protease production, with mutation of rot having the least impact followed by mutation of codY, sigB and sarA, respectively. Protein A was undetectable in conditioned medium from all four mutants, while extracellular nuclease was only present in the proteolytically cleaved NucA form. The abundance of high molecular weight proteins was reduced in all four mutants. Biofilm formation was reduced in codY, sarA and sigB mutants, but not in the rot mutant. Eliminating protease production partially reversed these phenotypes and enhanced biofilm formation. This was also true in LAC codY, rot, sarA and sigB mutants. Eliminating protease production enhanced the virulence of LAC and UAMS-1 sarA, sigB and rot mutants in a murine sepsis model but did not significantly impact the virulence of the codY mutant in either strain. Nevertheless, these results demonstrate that repressing protease production plays an important role in defining critical phenotypes in diverse clinical isolates of S. aureus and that Rot, SigB and SarA play critical roles in this regard.
Introduction
Staphylococcus aureus is among the most prominent of bacterial pathogens and is a serious public health threat. The prominence of S. aureus as a pathogen can be attributed to its ability to produce a diverse array of virulence factors in a carefully orchestrated fashion mediated through a complex and highly interactive regulatory circuit [Citation1,Citation2]. The combination of its arsenal of virulence factors, and the ability to precisely control their production to meet the demands of different microenvironments within the human host, allow S. aureus to cause a diverse array of infections ranging from skin and soft tissue infections to sepsis and disseminated secondary infections of essentially any tissue [Citation3–5]. The treatment of these infections is increasingly compromised by the persistent emergence of S. aureus strains that are resistant to front-line antibiotics, most notably oxacillin [Citation6]. Indeed, the Infectious Disease Society of America (IDSA) has recognized S. aureus as one of the ESKAPE pathogens [Citation7] owing to its prominence as a cause of human infection and the decreasing availability of antibiotics to treat these infections. Additionally, S. aureus has the capacity to form a robust biofilm, which at least in part accounts for its prominence as a cause of osteomyelitis and infections associated with indwelling medical devices [Citation8]. These biofilms confer a therapeutically relevant level of intrinsic resistance to antibiotic therapy and host defenses, thus necessitating an interdisciplinary clinical approach that most often includes surgical debridement augmented by some form of local antibiotic delivery, and even then, recurrent infections and even amputation are an all-too-common occurrence [Citation9,Citation10].
These factors have created an urgent need for alternative strategies to combat S. aureus infections. Although a considerable effort has been put forth to develop an effective vaccine, thus far, these efforts have failed and at present, no such vaccine exists [Citation11]. Therapies based on the use of bacteriophage and/or phage-associated lytic proteins have recently garnered attention but have inherent limitations with respect to strain specificity and have not yet achieved widespread clinical use [Citation12]. Nanotechnology is also being explored as a means of combatting infectious disease. For example, in the specific context of S. aureus, we examined laser irradiation of antibody-conjugated gold nanoparticles, including gold nanocages containing daptomycin, to achieve immediate photothermal killing and controlled antibiotic release directly at the bacterial cell surface [Citation13–17]. Given the arsenal of S. aureus virulence factors, another approach has been to target key elements within the regulatory circuit controlling the production of these virulence factors [Citation18,Citation19]. The two best-studied regulatory loci in this regard are the accessory gene regulator (agr) and the staphylococcal accessory regulatory (sarA), and putative inhibitors of the function of these loci have been described [Citation20–22].
Based on our specific interest in the clinical problem of biofilm-associated orthopedic infections, we initially focused our efforts on identifying regulatory loci that are required for biofilm formation. This led us to focus on sarA, mutation of which limits biofilm formation in diverse clinical isolates of S. aureus [Citation23]. We subsequently evaluated an array of S. aureus regulatory mutants based on their capacity to form a biofilm, and the results confirmed that mutation of sarA limits biofilm formation to a greater extent than mutation of any other regulatory locus we examined [Citation24]. Moreover, in regulatory mutants that exhibited an enhanced capacity to form a biofilm, mutation of sarA reversed this effect [Citation24,Citation25]. Given the diversity of infections caused by S. aureus, we then extended these studies to examine the impact of sarA and other regulatory loci found to impact biofilm formation on pathogenesis in a murine sepsis model [Citation26]. These studies demonstrated that mutation of codY, sarA, and sigB in the USA300 strain LAC limited virulence, and phenotypic characterization of these mutants suggested that this may be due to the increased production of extracellular proteases and a corresponding decrease in the accumulation of high molecular weight extracellular proteins.
Our previous sepsis studies were done with LAC because it is a prominent cause of serious community-acquired infections [Citation27]. However, yet another factor that complicates the development of alternative therapies to combat S. aureus infections is the genetic and phenotypic diversity among clinical isolates. Given our interest in orthopedic infections, we focused much of our work on an isolate obtained directly from the bone of an osteomyelitis patient during surgical debridement [Citation28]. This isolate, which was designated UAMS-1, is a USA200, methicillin-sensitive strain that is very distinct by comparison to USA300 isolates like LAC [Citation29,Citation30]. At a genetic level, examples of potentially relevant differences include cna, which encodes the primary collagen-binding adhesin produced by S. aureus [Citation31] and is present in in UAMS-1 but not in LAC and, conversely, the absence of the genes encoding the Panton-Valentine leucocidin (PVL) and the regulatory loci sarT and sarU, all of which are present in LAC [Citation29]. Phenotypically, UAMS-1 is characterized by its ability to form a robust biofilm and limited production of extracellular toxins, while LAC is highly toxigenic owing to the increased expression of agr [Citation27]. LAC also produces alpha toxin, while UAMS-1 does not owing to a point mutation in the corresponding gene (hla) [Citation32]. Any alternative therapeutic strategy targeting S. aureus specifically would be most effective if it were not limited by such differences. For this reason, we extended our previous experiments that were limited to LAC to assess the impact of these same regulatory mutations in UAMS-1.
Materials and methods
Bacterial strains and growth conditions
The bacterial strains used in these experiments are listed in . Relevant citations are included for UAMS-1 and LAC regulatory mutants described in previous reports [Citation24,Citation26,Citation28,Citation33,Citation34]. The only exception was the UAMS-1 fur mutant, which was generated by phage-mediated transduction from the existing mutant in the Nebraska Transposon Mutant Library (NTML) into LAC and then into UAMS-1 [Citation35]. A LAC sarA mutant unable to produce aureolysin, ScpA, SspA, SspB, and any of the six spl-encoded proteases (SplA-F) was generated as previously described [Citation36]. The protease-deficient LAC codY, rot, and sigB mutants were generated by phage-mediated transduction of each regulatory mutation into this protease-deficient strain.
Table 1. LAC S. aureus strains used in this study
Table 2. UAMS-1 S. aureus strains used in this study
A derivative of UAMS-1 unable to produce aureolysin or SspA/SspB was generated as previously described [Citation37]. To generate a derivative of this strain unable to produce ScpA, the resistance marker present in the scpA mutant present in the NTML was switched from erythromycin to tetracycline using previously described methods [Citation38]. This mutation was then transduced into the UAMS-1 mutant unable to produce aureolysin and SspA/SspB. Unlike LAC, which encodes the entire six-gene splA-F operon, UAMS-1 encodes a truncated version limited to splC-F (data not shown). To generate a UAMS-1 splC-F mutant, allelic exchange was performed using the protocol previously described [Citation38]. Specifically, primer sets () were used to amplify a 722 bp fragment and a 544 bp fragment upstream and downstream of the spl operon, respectively. These fragments were digested with SacII, ligated together, and PCR amplified using the upstream primer used to amplify the 722 bp fragment together with the downstream primer used to amplify the 544 bp fragment primer sets were used to amplify flanking regions of the UAMS-1 spl gene cluster to generate 722 bp and 544 bp fragments, respectively. These fragments were digested with SacII, ligated together, and PCR amplified to generate a construct containing the regions upstream and downstream of the spl operon but lacking the operon itself. The PCR product was then digested with EcoRI, cloned into the temperature-sensitive vector pJB38, and transformed into TOP10 E. coli (Invitrogen, Cat. # C404010) [Citation38]. Once confirmed, the plasmid was isolated from E. coli and transformed into S. aureus strain RN4220 before being transduced into the derivative of UAMS-1 unable to produce aureolysin, SspA, SspB and ScpA. To induce recombination, the recipient strain was incubated at the non-permissive temperature (42°C) overnight with 10 μg ml−1 chloramphenicol to induce a single crossover event. Bacteria were then sub-cultured and grown at 30°C without antibiotic selection to induce plasmid loss. Bacteria were then recovered on tryptic soy agar (TSA) containing 1 μg ml-1 anhydrotetracycline for counter-selection, thus resulting in a UAMS-1 mutant unable to produce any of the 10 primary extracellular proteases. This mutant was used as a recipient strain to transduce regulatory mutations into this derivative of UAMS-1 that was unable to produce any extracellular protease.
Table 3. Primers used for constructing mutants
Existing sarA and codY mutants [Citation24,Citation34] were used as transduction donors to move these regulatory mutations into this protease-deficient derivative of UAMS-1. To generate protease-deficient rot and sigB mutants, we used the pKOR1 mutagenesis system [Citation39] to knock out each gene in this same protease-deficient strain. Specifically, upstream and downstream primer pairs () were used to amplify fragments upstream and downstream of rot and sigB, respectively. For each set of PCR products, the upstream and downstream amplification products were digested with BamHI and ligated prior to PCR amplification using the furthest upstream and furthest downstream primers used to generate the original PCR products for each gene. For each set of PCR products, upstream and downstream fragments for each gene were digested with BamHI and ligated prior to PCR amplification using the external upstream and downstream primers used to generate the original PCR products (). The resulting PCR products were then cloned into the temperature-sensitive plasmid pKOR1 using GatewayTM LR Clonase II (Thermo Fisher Scientific, Cat. # 11,791,020) according to the manufacturer’s recommendations. Plasmid transfer and allelic exchange was then carried out as described above.
All strains were maintained at −80°C in tryptic soy broth (TSB) containing 25% (v/v) glycerol. For each experiment, the appropriate strains were recovered from cold storage by plating on TSA with appropriate antibiotic selection. Antibiotics were used in the following concentrations: erythromycin, 5 or 10 μg ml−1; chloramphenicol, 10 μg ml−1; tetracycline, 5 μg ml−1; kanamycin, 50 μg ml−1; neomycin, 50 μg ml−1; spectinomycin, 1 mg ml−1. Kanamycin and neomycin were always used together to avoid spontaneously resistant strains.
Murine bacteremia model
Bacterial strains were recovered from cold storage at −80°C by plating on TSA with appropriate antibiotic selection. Multiple colonies recovered from these primary plates were inoculated into TSB with the appropriate antibiotic selection and grown to stationary phase (16 hrs) at 37°C. Each strain was then inoculated into fresh TSB without antibiotics to a standardized optical density (OD560) of 0.05. Cultures were then grown to an OD560 of 1.0. Bacterial cells were harvested by centrifugation, washed in sterile phosphate-buffered saline (PBS), and resuspended in an equal volume of PBS containing 10% DMSO and 5% bovine serum albumin (BSA). The number of colony-forming units (cfu) in this suspension was then confirmed by plate count on TSA. Aliquots of this suspension were then stored at −80°C.
For each in vivo experiment, bacteria were recovered from these standardized, frozen aliquots, washed with PBS, and standardized in PBS without additives to a cell density of 5 × 108 cfu per ml. 5–8 week-old female NIH-Swiss mice were infected via tail vein injection of 100 µl (5 × 107 cfu) of this standardized suspension. Mice were evaluated six times daily and the spleen, kidney, and heart harvested from any mice found dead or judged to require compassionate euthanasia; otherwise, tissues were harvested at 7 days post-infection. Organs were removed aseptically and homogenized using the Bullet Blender 5 Gold Bead Homogenizer (Next Advance). Serial dilutions were plated on TSA without antibiotic selection and the number of cfu per organ determined following overnight incubation at 37°C. All in vivo experiments were done using 10 mice per experimental group.
Preparation and analysis of stationary-phase conditioned medium (CM)
Bacterial strains were recovered from frozen stock cultures and grown overnight (16 hr). Cultures of each strain were then standardized to an optical density of 560 nm (OD560) of 10.0. CM was then prepared from each standardized culture by removing bacterial cells by centrifugation followed by filter sterilization.
Total protease activity was assessed using CM from stationary-phase bacterial cultures. Assays were done using the Protease Fluorescent Detection Kit, which uses casein as the protease substrate (Sigma Chemical Co., Cat. #PF0100), and the EnzChek Gelatinase/Collagenase Assay Kit (Thermo Fisher Scientific, Cat. #E12055). Protease assays included two biological replicates with six experimental replicates of each.
Exoprotein profiles were examined by SDS-PAGE using 4–12% gradient Novex Bis-Tris Plus gels (Life Technologies, Cat. #NW04125BOX). Proteins were visualized by staining with SimplyBlueTM SafeStain (Life Technologies, Cat. #LC6060) and imaged using Bio-Rad ChemiDocMP Imaging System (Bio-Rad Laboratories, Inc.). Western blots for extracellular protein A (eSpa), NucA, and NucB were done using commercially available antibodies (Sigma and Toxin Technologies, respectively) as previously described [Citation36,Citation40,Citation41]. Western blots included two biological replicates with two experimental replicates of each.
Assessment of biofilm formation
Biofilm formation was assessed in vitro using a microtiter plate assay [Citation23]. Assays were done in 96-well microtiter plates after coating overnight at 4°C with 100 μl of 20% human plasma in carbonate/bicarbonate buffer. Bacterial cultures grown overnight in TSB supplemented with 3% sodium chloride and 0.5% glucose (biofilm medium, BFM) were standardized to an OD560 of 0.05 in fresh BFM. Plasma was aspirated and each well of the microtiter plate inoculated with 200 μl of this standardized culture. Plates were then incubated statically overnight at 37°C. Wells were then washed, fixed, and stained with crystal violet as previously described [Citation23]. After staining, wells were washed three times with 250 μl PBS and left to dry overnight. The following day, the stain was eluted with 100 μl 100% EtOH for 10 min, and the eluent diluted in a new 96-well plate to achieve values within the linear range of the assay. Absorbance measurements were taken at 595 nm with a FLUOstar Omega microplate reader (BMG Labtech) and adjusted for the dilution factor as necessary. Biofilm assays included two biological replicates with six experimental replicates of each.
Capsule immunoblotting
Quantitative analysis of capsular polysaccharide production was done by western dot blot as previously described [Citation42]. Briefly, overnight cultures were grown for 16 hours and standardized to an OD660 of 5.0. Bacterial cells were harvested by centrifugation, washed with sterile PBS, and resuspended in 50 µl of sterile PBS. Cell suspensions were sequentially incubated with 0.5 µl of lysostaphin (10 mg/ml) and DNase I (2 units/µl), both at 30 min intervals at 37°C. This was followed by the addition of 0.5 µl of proteinase K (10 mg/ml) and a further incubation at 50°C for 30 min. This step was repeated before incubating at 75°C for an additional 15 min and clarification by centrifugation. Samples were serially diluted and 1.5 μl of each dilution spotted directly to a dry nitrocellulose membrane. The amount of capsule polysaccharide was determined using an anti-type 8 capsule antibody and the same immunoblotting protocol used for PIA detection as previously described [Citation24].
PIA immunoblotting
Production of the polysaccharide intercellular adhesion (PIA) was also assessed as previously described [Citation24]. Briefly, cultures were grown overnight (16 hrs) in BFM and standardized to an OD660 of 5.0. Bacterial cells were harvested by centrifugation and resuspended in 60 μl 0.5 M EDTA. Cell suspensions were boiled for 5 min followed by centrifugation at 14,000 x g for 2 minutes. 20 μl of Tris-buffered saline (20 mM Tris-HCl, 150 mM NaCl [pH 7.4]) was added to each sample before adding 1 μl proteinase K (10 mg/ml) and incubating for 10 minutes at 37°C and then 10 minutes at 70°C. For analysis, 2 μl of each sample was spotted directly onto a dry nitrocellulose membrane and PIA detected using an anti-PIA antibody as previously described [Citation24].
Statistical analysis
All in vitro experiments were done with at least two biological replicates, each of which included at least three experimental replicates. In some cases, the results observed with the parent strain were averaged and set to a value of 1.0. The results observed with each isogenic mutant were then plotted and analyzed relative to this value. In other experiments, results were plotted as raw values, with these values used for statistical analysis. Statistical analysis was done using ANOVA methods with Dunnett’s procedure to compare the results observed with each mutant to that observed with the isogenic parent strain or by paired t-test as appropriate. For analysis of in vivo survival studies, log-rank (Mantell-Cox) tests were used to assess the statistical significance of the results observed with each mutant relative to the parent strain or between isogenic mutants and their protease-deficient derivatives. For analysis of bacterial burdens, ANOVA methods with Dunnett’s procedure were used to compare the results observed with each mutant to that observed with the isogenic parent strain. For this analysis, cfu data was log10-transformed prior to analysis and p values calculated using permutation methods. Statistical analyses were performed using GraphPad Prism 5.0 (La Jolla). P-values ≤ 0.05 were considered statistically significant.
Ethics statement
Experiments involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of the University of Arkansas for Medical Sciences and performed according to NIH guidelines, the Animal Welfare Act, and US Federal law.
Results
The impact of different regulatory loci on virulence in UAMS-1
To better understand how regulatory mutations that affect biofilm formation also impact the pathogenesis of diverse clinical isolates of S. aureus in a murine sepsis model, we generated isogenic mutants of the USA200 strain UAMS-1 in sarA, agr, codY, fur, sigB, rot, and mgrA. This strain was chosen because we carried out the same study previously with the USA300 strain LAC, and while LAC and UAMS-1 are both clinical isolates, they are very different by comparison to each other as detailed above. The potential importance of such differences is illustrated by the results we report. Specifically, in our previous report focusing on the impact of these mutations on the virulence of LAC in the same sepsis model, we found that mutation of codY, sarA, and sigB resulted in a comparable decrease in virulence, while mutation of mgrA and rot increased virulence by comparison to the isogenic parent strain [Citation26]. In contrast, mutation of all of the loci examined in UAMS-1 other than fur and codY was found to attenuate virulence in UAMS-1 as assessed by lethality in our sepsis model (). These results were generally consistent with bacterial burdens observed in the spleen, kidney and heart. Specifically, statistically significant reductions in bacterial burden in the spleen were observed with UAMS-1 agr, sarA, and rot mutants, while in the heart they were observed with agr, mgrA, rot, sarA and sigB mutants (). Bacterial burdens in the kidney were less discriminatory in that only mutation of mgrA and sarA resulted in statistically significant reduction. Thus, the only mutant that exhibited reduced lethality and reduced bacterial burdens in all three organs examined was the UAMS-1 sarA mutant. Conversely, the only mutants that did not exhibit reduced bacterial burdens in any organ were the fur and codY mutants ().
Figure 1. Relative virulence of S. aureus regulatory mutants in acute sepsis. Top: Kaplan-Meier survival curves are shown for the USA200 strain UAMS-1 and the indicated isogenic mutants. Numbers in parenthesis indicate p values for each mutant by comparison to the results observed with LAC. NS = not significant. Bottom: The number of colony-forming units (CFU) in the spleen, kidney, and heart are shown by scatter plot. Asterisks above each plot statistically significant values by comparison to the parent strain UAMS-1 (U1). Error bars indicate standard error of the mean of log10 transformed values. Asterisk indicates statistical significance relative to the parent strain. NS = not significant
Correlation between virulence, protease production, the accumulation of extracellular proteins, and biofilm formation
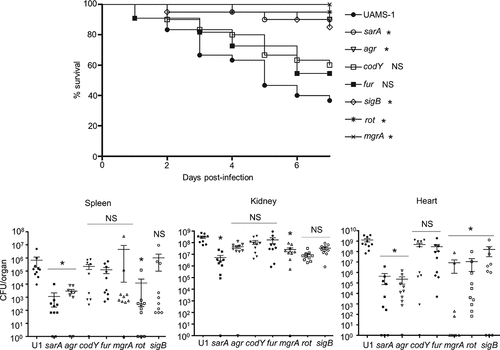
In our previous report focusing on LAC regulatory mutants in our sepsis model, we found a potentially important correlation between increased protease production and reduced virulence [Citation26]. To examine this correlation in UAMS-1, we assessed protease production in each of these regulatory mutants. As in LAC, protease production was significantly increased in codY, sigB, and sarA mutants (). The greatest increase was observed with the UAMS-1 sarA mutant followed by the sigB and codY mutants, respectively. These same relative relationships were also observed in the corresponding LAC mutants [Citation26]. Mutation of rot resulted in a modest increase in protease production (). We did not observe any increase in protease production with UAMS-1 agr, fur, or mgrA mutants.
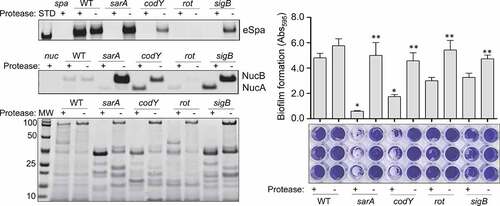
Figure 2. Impact of regulatory mutations on protease production in UAMS-1. Top: Total protease activity in conditioned medium (CM) was assessed with UAMS-1 (U1) and the indicated regulatory mutants using a commercially available casein-based FRET assay (left) or a gelatin-based FRET assay (right). Results obtained with U1 with each protease substrate were averaged and set to a value of 1.0. Results observed with all other strains are shown relative to this value. Bar charts are representative of results from at least two biological replicates for each of which included three experimental replicates. Results for each mutant are reported as mean fluorescence values (MFIs) ± the standard error of the means. Asterisks indicates statistical significance relative to the parent strain. NS = not significant. Middle: Abundance of extracellular protein A (eSpa) and the alternative forms of Nuc1 (NucA and NucB) as assessed by western blot. Bottom: SDS-PAGE profiles of CM from UAMS-1 and the indicated isogenic regulatory mutants
To assess the phenotypic impact of these changes in protease production, we examined the impact of these mutations on extracellular protein profiles. With the exception of the fur mutant, essentially all of these mutants were associated with a visible change in exoprotein profiles, but the most striking changes were a decrease in the abundance of high molecular weight extracellular proteins, and a corresponding increase in the abundance of lower molecular weight proteins, in sarA, codY, rot and sigB mutants (). In contrast, there was an apparent increase in the abundance of high molecular weight proteins in the UAMS-1 mgrA mutant. These results must be interpreted with caution in that all of the regulatory loci targeted in these studies are known to impact the production of exoproteins, but the correlation between these results and overall protease activity in sarA, codY, sigB and even rot mutants is suggestive of a cause-and-effect relationship between increased protease production and decreased accumulation of full-length high molecular weight exoproteins.
S. aureus produces protein A (Spa) in both surface-associated and extracellular forms [Citation43,Citation44], and in a previous report, we demonstrated that both forms are present in reduced amounts in CM from LAC codY, sarA and sigB mutants [Citation26]. In a subsequent report we demonstrated that this was also true in a UAMS-1 sarA mutant and that the abundance of extracellular protein A (eSpa) was fully restored in both LAC and UAMS-1 sarA mutants by limiting the ability of these mutants to produce extracellular proteases [Citation41]. Based on this, we examined the abundance of eSpa in other UAMS-1 regulatory mutants. The results demonstrated that eSpa is absent in CM prepared from UAMS-1 sarA, codY, rot and sigB mutants (). With the exception of codY, all of these regulatory loci have been implicated in expression from the spa promoter and the production of Spa [Citation45,Citation46], but this nevertheless suggests that the increased production of extracellular proteases is likely to play some role in defining the abundance of eSpa in these mutants in both LAC and UAMS-1.
Similarly, S. aureus produces an extracellular nuclease (Nuc1) as a larger protein (NucB) that is proteolytically cleaved to a smaller active form designated NucA [Citation47]. As assessed by western blot, both forms were detected in CM from UAMS-1, but only the NucA form was detected in CM from UAMS-1 sarA, codY, rot, and sigB mutants (). Neither form of nuclease was detected in CM from the mgrA mutant, and protease production was not increased in this mutant, thus making it likely that the absence of NucA and NucB in the UAMS-1 mgrA mutant is due to a lack of production rather than protease-mediated degradation. Biofilm formation was also reduced in all of these mutants, although in the case of rot the reduction was not statistically significant as assessed using our methods (). S. aureus has the capacity to produce at least 10 extracellular proteases (aureolysin, ScpA, SspA, SspB and the six spl-encoded proteases) [Citation48]. Eliminating the ability to produce these proteases enhanced biofilm formation in all four mutants including the rot mutant. It also restored the accumulation of high molecular weight proteins, at least partially restored the accumulation of eSpa, and resulted in a decrease in NucB degradation as evidenced by the reduced abundance of NucA ().
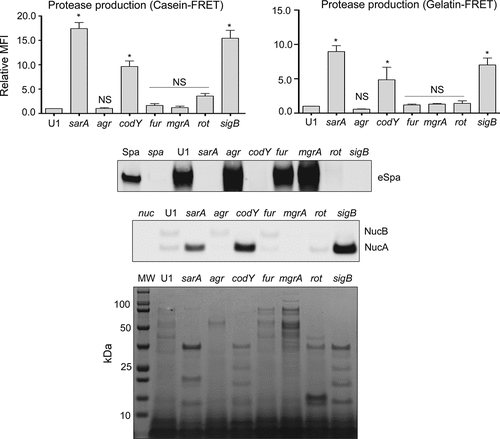
Figure 3. Impact of regulatory mutations on biofilm formation in UAMS-1. The relative capacity of UAMS-1 (U1) and its isogenic regulatory mutants to form a biofilm was assessed using a microtiter plate assay. Bottom image is a representative assay. Upper bar graph indicates cumulative absorbance values obtained from three biological replicates. Error bars indicate standard error of the mean. Asterisk indicates statistical significance (p ≤ 0.05) relative to the isogenic parent strain
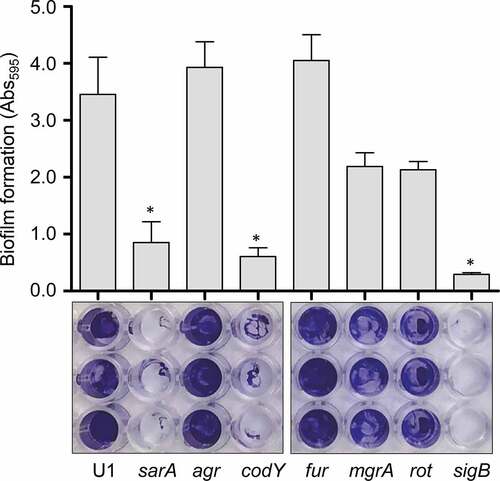
Figure 4. Impact of extracellular proteases in UAMS-1. Top left: Abundance of extracellular protein A (eSpa) was assessed by western blot using conditioned medium (CM) and an anti-Spa monoclonal antibody. Strains included were UAMS-1 (U1, a protease-deficient derivative of U1 (p), isogenic sarA (SA), codY (c), rot (r), and sigB (SB) mutants, and protease-deficient derivatives of each regulatory mutant (SAP, CP, and SBP, respectively). Purified protein A (Spa) and CM from a protein A mutant (spa) were included as positive and negative controls, respectively. Middle left: Abundance of extracellular nuclease (Nuc1) was assessed by western blot in the same strains. CM from a nuclease mutant (nuc) was included as a negative control. Bottom left: SDS-PAGE analysis of CM from the same S. aureus strains. MW: Molecular size markers. Right: The relative capacity of the same strains to form a biofilm was assessed using a microtiter plate assay. Graph indicates cumulative absorbance values obtained from three biological replicates. Error bars indicate standard error of the mean. Single asterisk indicates statistical significance relative to the isogenic parent strain. Double asterisks indicate significance relative to the isogenic regulatory mutant
Impact of protease production in specific UAMS-1 and LAC regulatory mutants
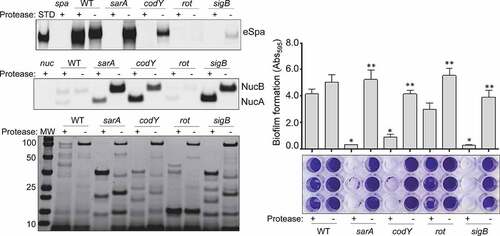
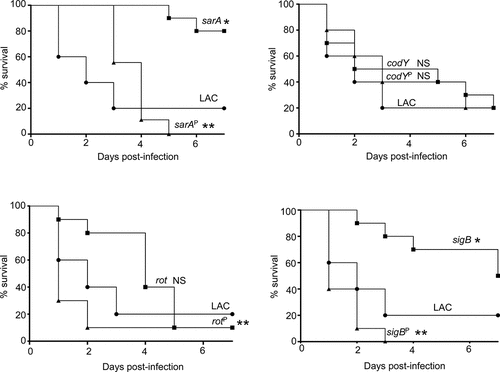
In our previous report, we demonstrated a correlation between reduced virulence and increased protease production in LAC codY, sarA, and sigB mutants [Citation26]. Based on the results described above for our UAMS-1 mutants, we also directly examined the impact of eliminating protease production in LAC codY, rot, sarA, and sigB mutants. All of the phenotypes discussed above were also evident in the LAC mutants, although with respect to biofilm formation, the reductions we observed with rot and sigB mutants did not reach statistical significance (). Nevertheless, eliminating protease production in LAC also enhanced biofilm formation, restored the accumulation of eSpa, limited the degradation of NucB, and generally restored overall exoprotein profiles in all four LAC mutants ().
Figure 5. Impact of extracellular proteases in LAC. Top left: Abundance of extracellular protein A (eSpa) was assessed by western blot using conditioned medium (CM) and an anti-Spa monoclonal antibody. Strains included were LAC, a protease-deficient derivative of LAC (p), isogenic sarA (SA), codY (c), rot (r), and sigB (SB) mutants, and protease-deficient derivatives of each regulatory mutant (SAP, CP, RP and SBP, respectively). Purified protein A (Spa) and CM from a protein A mutant (spa) were included as positive and negative controls, respectively. Middle left: Abundance of extracellular nuclease (Nuc1) was assessed by western blot in the same strains. CM from a nuclease mutant (nuc) was included as a negative control. Bottom left: SDS-PAGE analysis of CM from the same S. aureus strains. MW: Molecular size markers. Right: The relative capacity of the same strains to form a biofilm was assessed using a microtiter plate assay. Graph indicates cumulative absorbance values obtained from three biological replicates. Error bars indicate standard error of the mean. Single asterisk indicates statistical significance relative to the isogenic parent strain. Double asterisks indicate significance relative to the isogenic regulatory mutant
Impact of regulatory mutations and the role of increased protease production on virulence in LAC and UAMS-1
The results demonstrating a comparable impact of increased protease production in LAC and UAMS-1 rot mutants were somewhat surprising given that mutation of rot had relatively little impact in this regard by comparison to codY, sigB and sarA mutants in both UAMS-1 () and LAC [Citation26]. Based on this, we examined the impact of mutating rot on protease production using CM from early-exponential, post-exponential, and stationary-phase cultures of both LAC and UAMS-1 (OD560 of 1.5, 4.0, and 10.0, respectively). The results confirmed that protease production is significantly increased in both strains during the post-exponential and stationary growth phases (). Thus, these results suggest that even relatively modest increases in protease production can have biologically significant effects. To examine this further, we compared LAC and UAMS-1 to their isogenic regulatory mutants and protease-deficient regulatory mutants in our murine sepsis model. The results of these studies confirmed that mutation of rot, sarA, and sigB attenuated the virulence of UAMS-1, while mutation of codY had no significant effect (). They also confirmed that eliminating the ability to produce any of the 10 primary S. aureus extracellular proteases restored the virulence of rot, sarA, and sigB mutants, but had little impact on the virulence of the codY mutant. Although mutation of rot did not have a significant impact on virulence in LAC, it did in UAMS-1, and in both strains eliminating protease production enhanced virulence while mutation of codY had relatively little effect ().
Figure 6. Impact of rot on protease production in LAC and UAMS-1. Total protease activity was assessed with LAC, UAMS-1, and their isogenic rot mutants using conditioned medium (CM) harvested at the early exponential (EE), post-exponential (PE) and stationary growth phases (ST). Protease activity was assessed using a commercially available gelatin-based FRET assay. Bar charts are representative of results from at least two biological replicates for each of which included three experimental replicates. Results are reported as mean fluorescence values (MFIs) ± the standard error of the means. Asterisk indicates statistical significance by comparison to the isogenic parent strain at the same growth phase. Double asterisks indicate statistical significance by comparison to the corresponding UAMS-1 rot mutant at the same growth phase
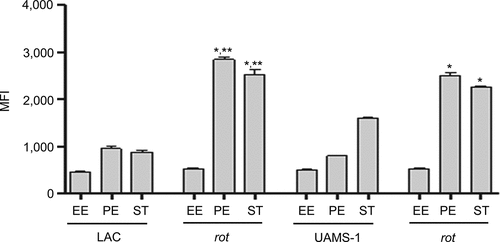
Figure 7. Impact of protease production on virulence in UAMS-1. A murine sepsis model was used to assess the relative virulence of UAMS-1, its sarA, codY, rot, and sigB mutants, and derivatives of each of these mutants unable to produce any extracellular protease (sarAP, codYP, rotP, and sigBP, respectively). Numbers indicate P values relative to the UAMS-1 parent strain. NS indicates no statistical significance by comparison to UAMS-1
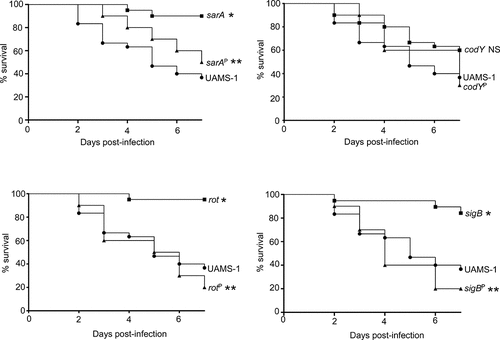
Figure 8. Impact of protease production on virulence in LAC. A murine sepsis model was used to assess the relative virulence of LAC, its sarA, codY, rot, and sigB mutants, and derivatives of each of these mutants unable to produce any extracellular protease (sarAP, codYP, rotP, and sigBP, respectively). Numbers indicate P values relative to the LAC parent strain. NS indicates no statistical significance by comparison to LAC
Impact of regulatory mutations on PIA and capsule production in UAMS-1
With respect to the impact of increased protease production on virulence, the biggest discrepancy we observed was associated with codY, mutation of which resulted in a significant increase in the production of extracellular proteases but had relatively little impact on virulence. With respect to LAC, this was in contrast to our previous report indicating that mutation of codY attenuated virulence in our sepsis model almost to the same degree as mutation of sigB and sarA [Citation26]. We have no explanation for this other than the inherent variability of in vivo experiments. Nevertheless, the discrepancy between protease production and virulence in a codY mutants suggests an important contribution of protease-independent phenotypes. One possibility is production of the polysaccharide intercellular adhesin (PIA) and/or capsular polysaccharides. Interestingly, the production of both of these was dramatically enhanced in a UAMS-1 codY mutant (). We have also confirmed that PIA production is also increased in a LAC codY mutant, albeit to a lesser extent than in a UAMS-1 codY mutant [Citation37]. Although we did not examine capsule production in a LAC codY mutant, this nevertheless suggests that the increased production of one or both of these polysaccharides could limit the attenuation of codY mutants despite their increased production of extracellular proteases.
Figure 9. Impact of regulatory mutations on PIA and capsule production. Top: PIA production was assessed by dot blot using antibodies specific for PIA as previously described [Citation41]. An ica mutant was included as a negative control. Bottom: Capsule production was assessed by dot blot after serial dilution using antibodies specific for type-8 capsular polysaccharide as previously described [Citation42]. A cap mutant was included as a negative control
![Figure 9. Impact of regulatory mutations on PIA and capsule production. Top: PIA production was assessed by dot blot using antibodies specific for PIA as previously described [Citation41]. An ica mutant was included as a negative control. Bottom: Capsule production was assessed by dot blot after serial dilution using antibodies specific for type-8 capsular polysaccharide as previously described [Citation42]. A cap mutant was included as a negative control](/cms/asset/9f19bb9d-1803-49e4-af17-3272e611dc89/kvir_a_1879550_f0009_b.gif)
Discussion
There is an increasing need for alternative strategies to combat S. aureus infections, and increasing interest in anti-virulence approaches that target key regulatory loci [Citation18,Citation19]. The two best-studied regulatory loci in this regard are the accessory gene regulator (agr) and the staphylococcal accessory regulatory (sarA), and putative inhibitors of the function of these loci have been described [Citation20–22]. However, given that S. aureus causes diverse forms of infection, it is imperative to consider regulatory loci as therapeutic targets in the context of these diverse forms of infection. For instance, mutation of agr generally reduces toxin production, suggesting that inhibitors of agr may be helpful in the context of acute, toxin-mediated infections. However, mutation of agr also enhances biofilm formation [Citation23,Citation25]. Thus, such inhibitors could have the adverse consequence of increasing the likelihood of chronic, biofilm-associated infections. Indeed, it is not uncommon to isolate agr mutants from chronically infected patients [Citation49], and there is one report that examined sequential isolates from the same patient and concluded that mutations that limit the regulatory functions of agr can be directly correlated with phenotypic adaptations that favor chronic infections including osteomyelitis [Citation50].
This issue is further complicated by the complexity of S. aureus regulatory circuits and the genetic and phenotypic diversity among clinical isolates. For instance, one report used comparative genomics tools to construct a model of the regulatory network of S. aureus and six related staphylococcal species. This network included 46 transcription factors with more than 1,900 binding sites, 2,800 target genes, and approximately 320 regulatory interactions between these transcription factors, and the results suggested that only 20% of the interactions in S. aureus regulatory circuits are conserved among all S. aureus strains [Citation51].
We have begun to address these issues by making direct comparisons between the relative impact of different regulatory loci in diverse animal models and in clinical isolates that are demonstrably different from each other. Given our specific interest in orthopedic infection and the important role of biofilms in these infections, we initially focused on the impact of S. aureus regulatory loci on biofilm formation. This led us to place a primary focus on sarA, mutation of which limits biofilm formation in both LAC and UAMS-1 to a greater degree than any other regulatory locus we have examined [Citation24].
We also demonstrated that mutation of sarA limits virulence in murine models of implant-associated infection, sepsis, and osteomyelitis, and that this is due in all cases to the increased production of extracellular proteases [Citation24,Citation26,Citation36,Citation37,Citation41,Citation52–57]. In the USA300, methicillin-resistant strain LAC, we found that mutation of codY and sigB attenuated virulence in a murine sepsis model to a degree comparable to that observed in an isogenic sarA mutant [Citation26]. One phenotype that all of these mutants had in common was the increased production of extracellular proteases. We had previously evaluated the impact of these additional regulatory loci on biofilm formation [Citation24,Citation52]. However, our sepsis studies were limited to LAC, thus leaving open the question whether a similar degree of attenuation would be observed with these regulatory mutants in other clinical isolates of S. aureus. To address this, we generated mutations in the USA200, methicillin-sensitive strain UAMS-1, which was isolated originally from the bone of an osteomyelitis patient during surgical debridement [Citation28].
As in our previous study with LAC, mutations were generated in UAMS-1 in agr, codY, fur, mgrA, rot, sarA and sigB. With respect to the impact of these mutations on virulence in our sepsis model, we did observe some differences between LAC and UAMS-1. Specifically, in UAMS-1, mutation of agr, mgrA, and rot limited virulence, while mutation of codY did not, and this was not the case with the corresponding LAC mutants [Citation26]. Thus, only mutation of sarA and sigB resulted in reduced virulence in both LAC and UAMS-1.
Defining the mechanistic reasons for such differences is extremely complicated by the complexity and highly interactive nature of S. aureus regulatory circuits, but the results observed with the UAMS-1 codY mutant were particularly surprising in that mutation of codY resulted in a comparable increase in protease activity in both strains and the hypothesis that increased protease production attenuates virulence owing to the decreased accumulation of S. aureus virulence factors. However, mutation of codY has been shown to result in increased expression of agr in S. aureus [Citation58,Citation59], and while it would therefore be anticipated that mutation of codY would result in increased protease production as was observed in the UAMS-1 codY mutant, it would also be anticipated that it would result in increased expression of agr. To the extent that agr significantly enhances the production of multiple S. aureus virulence factors [Citation1], this could provide a counter-balance to the increased production of extracellular proteases, particularly in a strain like UAMS-1 that expresses agr at relatively low levels by comparison to a strain like LAC [Citation37]. Similarly, mutation of sigB results in increased expression of agr, but also decreased expression of sarA [Citation60]. With respect to the impact of sigB on sarA, there are reports to the contrary [Citation61], but we found that the amount of SarA present in whole cell lysates is reduced in LAC and UAMS-1 sigB mutants by comparison to the isogenic parent strains (data not shown). While mutation of sigB did not abolish the production of SarA, this suggests that the increased production of extracellular proteases in sigB mutants could be mediated by the impact of sigB on production of SarA as a repressor of protease production even despite its impact on the expression of agr. Addressing this issue will require additional experiments that are beyond the scope of this report, but this nevertheless emphasizes the complexity of S. aureus regulatory circuits, the need to assess the impact of critical elements within this circuit in diverse clinical isolates, and the potential importance of the balance between the production of S. aureus virulence factors and their protease-mediated degradation.
Mutation of rot also reduced the virulence of UAMS-1 in our sepsis model but enhanced virulence in LAC [Citation26]. As with codY and sigB, this difference is difficult to explain in that rot also impacts expression of both agr and sarA. Specifically, RNAIII, the primary effector molecule of the agr system, blocks translation of rot mRNA, and rot expression is repressed by both SarA and SigB [Citation1]. However, Rot also influences the production of S. aureus virulence factors directly, and this includes extracellular proteases. For instance, a recent report demonstrated that mutation of rot increases protease production via direct binding to the promoters of the corresponding genes and that this increase is sufficient to limit the capacity of LAC to form a biofilm [Citation62]. There is also a recent report confirming that mutation of rot results in increased expression of the gene encoding aureolysin (aur) and the sspABC operon and increased protease activity as assessed by gelatin-based zymography [Citation63]. We found that, by comparison to mutation of codY, sigB and sarA, mutation of rot had only a modest impact on protease production, and we were not able to demonstrate a significant decrease in biofilm formation in a LAC rot mutant [Citation24].
Nevertheless, this is consistent with our observation that mutation of rot in both LAC and UAMS-1 led to a decreased accumulation of high molecular weight proteins and increased accumulation of lower molecular weight proteins. Although rot is known to serve multiple regulatory roles that impact exoprotein production [Citation64], we believe this shift from high to lower molecular weight proteins is likely to reflect degradation of at least some S. aureus extracellular proteins resulting from an overall increase in protease activity. Further support for this comes from the absence of eSpa in CM from a UAMS-1 rot mutant. However, Rot has been shown to bind the spa promoter and stimulate transcription [Citation65], thus suggesting that the absence of Rot would result in reduced production of protein A irrespective of its impact on protease production. To the extent that protein A has been shown to promote biofilm formation [Citation66], its absence in a rot mutant might be expected to limit biofilm formation as previously indicated [Citation62],
Taken together, these results emphasize the complexity and interactive nature of S. aureus regulatory circuits, and by extension the need to address the impact of different regulatory loci in divergent clinical isolates in the context of phenotypes of potential clinical relevance and, more importantly, virulence in relevant animal models. One of these phenotypes that has been found to be increasingly important in recent years is protease production. Specifically, while extracellular proteases play important roles in tissue invasion, the acquisition of nutrients, and subversion of host defenses, it has also been shown that eliminating protease production increases the virulence of LAC owing to the increased abundance of important virulence factors [Citation67]. Conversely, mutation of sarA limits virulence in murine models of sepsis and osteomyelitis owing to the increased production of these proteases and corresponding decrease in the availability of these same virulence factors [Citation36,Citation54,Citation55,Citation57,Citation68].
Based on this, we focused in this report on directly assessing the impact of each regulatory mutation on protease production in UAMS-1 and its impact on these phenotypes, including virulence in our sepsis model. As with LAC, mutation of rot, codY, sarA and sigB all resulted in a significant increase in overall protease production. Also as in LAC, mutation of sarA had the greatest effect followed by mutation of sigB, codY, and rot, respectively. Biofilm formation was limited to a significant degree in UAMS-1 codY, sarA, and sigB mutants, which with the exception of codY, was consistent with the results previously generated using these same mutants in both LAC and UAMS-1 [Citation24]. Mutation of codY in UAMS-1 did result in a significant increase in protease production, and in this report we confirm that this contributes significantly to the reduced capacity of a UAMS-1 codY mutant to form a biofilm. However, the biofilm phenotype we report is in direct contrast with what has previously been reported, which indicated that mutation of codY in UAMS-1 had little impact on biofilm formation [Citation24,Citation69]. One possible explanation for this disparity is that the biofilm assays were sometimes compromised by what appeared to be intracellular aggregation of the codY mutant, which resulted in a tendency for the entire aggregate to be removed during the washing steps (data not shown). This is consistent with the dramatic increase in the production of PIA in a codY mutant [Citation24,Citation37]. Thus, the increased production of extracellular proteases in codY mutants may limit the abundance of proteins required for the initial adherence phase of biofilm formation but simultaneously result in increased intercellular aggregation. However, this does not preclude the possibility that the increased production of extracellular proteases in a UAMS-1 codY mutant impacts relevant phenotypes of S. aureus, and in fact we demonstrated that this is the case as evidenced by the reduced accumulation of high molecular weight proteins and changes in the abundance and/or form of eSpa and Nuc1.
This was also true with rot, sarA and sigB mutants, and in all three cases we confirmed that increased protease production makes a significant contribution to their reduced virulence in our sepsis model. Thus, we believe the results we report provide strong support for the hypothesis that a key but largely overlooked component of S. aureus regulatory circuits is the ability to limit the production of extracellular proteases such that they serve their intended purposes without compromising the abundance of critical S. aureus virulence factors. Moreover, they demonstrate that this is the case in diverse clinical isolates of S. aureus as represented by the USA300, methicillin-resistant strain LAC and the USA200, methicillin-sensitive strain UAMS.
Acknowledgments
This work was supported by NIH grant R01-AI119380 to MSS. Additional support was provided by a generous gift from the Texas Hip and Knee Center and research core facilities supported by the Center for Microbial Pathogenesis and Host Inflammatory Responses (P20-GM103450), the Translational Research Institute (UL1TR000039) and by the United States Army Congressionally Directed Medical Research Programs (W81X1H-14-PRORP-EA). The authors thank Horace J. Spencer for assistance with statistical analyses and Robin L. Mulkey for technical assistance with murine infections.
Disclosure statement
The authors report no conflict of interest.
Additional information
Funding
References
- Jenul C, Horswill AR. Regulation of Staphylococcus aureus virulence. Microbiol Spectr. 2018. DOI:10.1128/microbiolspec.GPP3-0031-2018
- Priest NK, Rudkin JK, Feil EJ, et al. From genotype to phenotype: can systems biology be used to predict Staphylococcus aureus virulence? Nat Rev Microbiol. 2012;10:791–797.
- Kobayashi SD, Malachowa N, DeLeo FR. Pathogenesis of Staphylococcus aureus abscesses. Am J Pathol. 2015;185:1518–1527.
- Lowy FD. Staphylococcus aureus infections. N Engl J Med. 1998;339:520–532.
- Thomer L, Schneewind O, Missiakas D. Pathogenesis of Staphylococcus aureus bloodstream infections. Annu Rev Pathol. 2016;11:343–364.
- Otto M. Staphylococcal infections: mechanisms of biofilm maturation and detachment as critical determinants of pathogenicity. Annu Rev Med. 2013;64:175–188.
- De Oliveira DMP, Forde BM, Kidd TJ, et al. Antimicrobial resistance in ESKAPE pathogens. Clin Microbiol Rev. 2020;33:e00181–19.
- Brady RA, Leid JG, Calhoun JH, et al. Osteomyelitis and the role of biofilms in chronic infection. FEMS Immunol Med Micro. 2008;52:13–22.
- Ford CA, Cassat JE. Advances in the local and targeted delivery of anti-infective agents for management of osteomyelitis. Expert Rev Anti Infect Ther. 2017;15:851‐860.
- Urish KL, Cassat JE. Staphylococcus aureus osteomyelitis: bone, bugs, and surgery. Infect Immun accepted manuscript. 2020;88. DOI:10.1128/IAI.00932-19
- Ansari S, Jha RK, Mishra SK, et al. Recent advances in Staphylococcus aureus infection: focus on vaccine development. Infect Drug Resist. 2019;12:1243‐1255.
- Gutiérrez D, Fernández L, Rodríguez A, et al. Are phage lytic proteins the secret weapon to kill Staphylococcus aureus? mBio. 2018;9:e01923–17.
- Galanzha EI, Shashkov E, Sarimollaoglu M, et al. In vivo magnetic enrichment, photoacoustic diagnosis, and photothermal purging of infected blood using multifunctional gold and magnetic nanoparticles. PLoS One. 2012;7:e45557.
- Meeker DG, Wang T, Harrington WN, et al. Versatility of targeted antibiotic-loaded gold nanoconstructs for the treatment of biofilm-associated bacterial infections. Int J Hyperthermia. 2018;34:209–219.
- Meeker DG, Jenkins SV, Miller EK, et al. Synergistic photothermal and antibiotic killing of biofilm-associated Staphylococcus aureus using targeted antibiotic-loaded gold nanoconstructs. ACS Infect Dis. 2016;2:241–250.
- Nedosekin DA, Juratli MA, Sarimollaoglu M, et al. Photoacoustic and photothermal detection of circulating tumor cells, bacteria and nanoparticles in cerebrospinal fluid in vivo and ex vivo. J Biophotonics. 2013;6:523–533.
- Zharov VP, Mercer KE, Galitovskaya EN, et al. Photothermal nanotherapeutics and nanodiagnostics for selective killing of bacteria targeted with gold nanoparticles. Biophys J. 2006;90:619–627.
- Cai X, Zheng W, Li Z. High-throughput screening strategies for the development of anti-virulence inhibitors against Staphylococcus aureus. Curr Med Chem. 2019;26:2297–2312.
- Gordon CP, Williams P, Chan WC. Attenuating Staphylococcus aureus virulence gene regulation: a medicinal chemistry perspective. J Med Chem. 2013;56:1389–1404.
- Arya R, Princy SA. An insight into pleiotropic regulators agr and sar: molecular probes paving the new way for antivirulent therapy. Future Microbiol. 2013;8:1339–1353.
- Benzar IF, Mashruwala A, Boyd JM, et al. Drug-like fragments inhibit agr-mediated virulence expression in Staphylococcus aureus. Sci Rep. 2019;9:6786.
- Balamurugan P, Krishna VP, Bharath D, et al. Staphylococcus aureus quorum regulator SarA targeted compound, 2-[(Methylamino)methyl]phenol inhibits biofilm and down-regulates virulence genes. Front Microbiol. 2017;8:1290.
- Beenken KE, Blevins JS, Smeltzer MS. Mutation of sarA in Staphylococcus aureus limits biofilm formation. Infect Immun. 2003;71:4206–4211.
- Atwood DN, Loughran AJ, Courtney AP, et al. Comparative impact of diverse regulatory loci on Staphylococcus aureus biofilm formation. Microbiologyopen. 2015;4:436–451.
- Beenken KE, Mrak LN, Griffin LM, et al. Epistatic relationships between sarA and agr in Staphylococcus aureus biofilm formation. PLoS One. 2010;5:e10790.
- Rom JS, Atwood DN, Beenken KE, et al. Impact of Staphylococcus aureus regulatory mutations that modulate biofilm formation in the USA300 strain LAC on virulence in a murine bacteremia model. Virulence. 2017;8:1776–1790.
- Otto M. Basis of virulence in community-associated methicillin-resistant Staphylococcus aureus. Ann Rev Microbiol. 2010;64:143–162.
- Gillaspy AF, Hickmon SG, Skinner RA, et al. Role of the accessory gene regulator (agr) in pathogenesis of staphylococcal osteomyelitis. Infect Immun. 1995;63:3373–3380.
- Cassat JE, Dunman PM, McAleese F, et al. Comparative genomics of Staphylococcus aureus musculoskeletal isolates. J Bacteriol. 2005;187:576‐592.
- King JM, Kulhankova K, Stach CS, et al. Phenotypes and virulence among Staphylococcus aureus USA100, USA200, USA300, USA400, and USA600 clonal lineages. mSphere. 2016;1:e00071–16.
- Gillaspy AF, Lee CY, Sau S, et al. Factors affecting the collagen binding capacity of Staphylococcus aureus. Infect Immun. 1998;66:3170‐3178.
- Cassat J, Dunman PM, Murphy E, et al. Transcriptional profiling of a Staphylococcus aureus clinical isolate and its isogenic agr and sarA mutants reveals global differences in comparison to the laboratory strain RN6390. Microbiol. 2006;152:3075–3090.
- Tsang LH, Cassat JE, Shaw LN, et al. Factors contributing to the biofilm-deficient phenotype of Staphylococcus aureus sarA mutants. PLoS One. 2008;3:e3361.
- Blevins JS, Beenken KE, Elasri MO, et al. Strain-dependent differences in the regulatory roles of sarA and agr in Staphylococcus aureus. Infect Immun. 2002;70:470‐480.
- Fey PD, Endres JL, Yajjala VK, et al. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. MBio. 2013;4:e00537–12.
- Zielinska AK, Beenken KE, Mrak LN, et al. sarA-mediated repression of protease production plays a key role in the pathogenesis of Staphylococcus aureus USA300 isolates. Mol Microbiol. 2012;86:1183–1196.
- Atwood DN, Beenken KE, Loughran AJ, et al. XerC Contributes to diverse forms of Staphylococcus aureus Infection via agr-dependent and agr-independent pathways. Infect Immun. 2016a;84:1214–1225.
- Bose JL, Fey PD, Bayles KW. Genetic tools to enhance the study of gene function and regulation in Staphylococcus aureus. Appl Environ Microbiol. 2013;79:2218–2224.
- Bae T, Schneewind O. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid. 2006;55:58–63.
- Beenken KE, Mrak LN, Zielinska AK, et al. Impact of the functional status of saeRS on in vivo phenotypes of Staphylococcus aureus sarA mutants. Mol Microbiol. 2014;92:1299–1312.
- Rom JS, Ramirez AM, Beenken KE, et al. The impacts of msaABCR on sarA-associated phenotypes are different in divergent clinical isolates of Staphylococcus aureus. Infect Immun. 2020;88:e00530–19.
- Luong TT, Sau K, Roux C, et al. Staphylococcus aureus ClpC divergently regulates capsule via sae and codY in strain Newman but activates capsule via codY in strain UAMS-1 and in strain Newman with repaired saeS. J Bacteriol. 2011;193:686–694.
- O’Halloran DP, Wynne K, Geoghegan JA. Protein A is released into the Staphylococcus aureus culture supernatant with an unprocessed sorting signal. Infect Immun. 2015;83:1598‐1609.
- Movitz J. Formation of extracellular protein A by Staphylococcus aureus. Eur J Biochem. 1976;68:291–299.
- Gao J, Stewart GC. Regulatory elements of the Staphylococcus aureus protein A (Spa) promoter. J Bacteriol. 2004;186:3738‐3748.
- Hsieh H-Y, Tseng CW, Stewart GC. Regulation of Rot expression in Staphylococcus aureus. J Bacteriol. 2008;190:546–554.
- Kiedrowski MR, Kavanaugh JS, Malone CL, et al. Nuclease modulates biofilm formation in community-associated methicillin-resistant Staphylococcus aureus. PLoS One. 2011;6:e26714.
- Shaw L, Golonka E, Potempa J, et al. The role and regulation of the extracellular proteases of Staphylococcus aureus. Microbiology. 2004;150:217–228.
- Altman DR, Sullivan MJ, Chacko KI, et al. Genome plasticity of agr-defective Staphylococcus aureus during clinical infection. Infect Immun. 2018;86:e00331–18.
- Suligoy CM, Lattar SM, Noto Llana M, et al. Mutation of agr is associated with the adaptation of Staphylococcus aureus to the host during chronic osteomyelitis. Front Cell Infect Microbiol. 2018;8:18.
- Ravcheev DA, Best AA, Tintle N, et al. Inference of the transcriptional regulatory network in Staphylococcus aureus by integration of experimental and genomics-based evidence. J Bacteriol. 2011;193:3228–3240.
- Atwood DN, Beenken KE, Lantz TL, et al. Regulatory mutations impacting antibiotic susceptibility in an established Staphylococcus aureus Biofilm. Antimicrob Agents Chemother. 2016b;60:1826–1829.
- Loughran AJ, Atwood DN, Anthony AC, et al. Impact of individual extracellular proteases on Staphylococcus aureus biofilm formation in diverse clinical isolates and their isogenic sarA mutants. Microbiologyopen. 2014;3:897–909.
- Loughran AJ, Gaddy D, Beenken KE, et al. Impact of sarA and phenol-soluble modulins on the pathogenesis of osteomyelitis in diverse clinical isolates of Staphylococcus aureus. Infect Immun. 2016;84:2586–2594.
- Ramirez AM, Byrum SD, Beenken KE, et al. Exploiting correlations between protein abundance and the functional status of saeRS and sarA to identify virulence factors of potential importance in the pathogenesis of Staphylococcus aureus osteomyelitis. ACS Infect Dis. 2020;6:237–249.
- Weiss EC, Zielinska A, Beenken KE, et al. Impact of sarA on daptomycin susceptibility of Staphylococcus aureus biofilms in vivo. Antimicrob Agents Chemother. 2009;53:4096‐4102.
- Zielinska AK, Beenken KE, Joo HS, et al. Defining the strain-dependent impact of the Staphylococcal accessory regulator (sarA) on the alpha-toxin phenotype of Staphylococcus aureus. J Bacteriol. 2011;193:2948–2958.
- Rivera FE, Miller HK, Kolar SL, et al. The impact of CodY on virulence determinant production in community-associated methicillin-resistant Staphylococcus aureus. Proteomics. 2012;12:263‐268.
- Roux A, Todd DA, Velázquez JV, et al. CodY-mediated regulation of the Staphylococcus aureus Agr system integrates nutritional and population density signals. J Bacteriol. 2014;196:1184‐1196.
- Bischoff M, Entenza JM, Giachino P. Influence of a functional sigB operon on the global regulators sar and agr in Staphylococcus aureus. J Bacteriol. 2001;183:5171–5179.
- Cheung AL, Chien Y, Bayer AS. Hyperproduction of alpha-hemolysin in a sigB mutant is associated with elevated SarA expression in Staphylococcus aureus. Infect Immun. 1999;67:1331–1337.
- Mootz JM, Benson MA, Heim CE, et al. Rot is a key regulator of Staphylococcus aureus biofilm formation. Mol Microbiol. 2015;96:388–404.
- Gimza BD, Larias MI, Budny BG, et al. Mapping the global network of extracellular protease regulation in Staphylococcus aureus. mSphere. 2019;4:e00676–19.
- Saïd-Salim B, Dunman PM, McAleese FM, et al. Global regulation of Staphylococcus aureus genes by Rot. J Bacteriol. 2003;185:610–619.
- Oscarsson J, Harlos C, Arvidson S. Regulatory role of proteins binding to the spa (protein A) and sarS [staphylococcal accessory regulator) promoter regions in Staphylococcus aureus NTCC 8325-4. Int J Med Microbiol. 2005;295:253–266.
- Merino N, Toledo-Arana A, Vergara-Irigaray M, et al. Protein A-mediated multicellular behavior in Staphylococcus aureus. J Bacteriol. 2009;191:832–843.
- Kolar SL, Ibarra JA, Rivera FE, et al. Extracellular proteases are key mediators of Staphylococcus aureus virulence via the global modulation of virulence-determinant stability. Microbiologyopen. 2013;2:18–34.
- Byrum SD, Loughran AJ, Beenken KE, et al. Label-free proteomic approach to characterize protease-dependent and independent effects of sarA inactivation on the Staphylococcus aureus exoproteome. ACS J Proteome Res. 2018;17:3384–3395.
- Majerczyk CD, Sadykov MR, Luong TT, et al. Staphylococcus aureus CodY negatively regulates virulence gene expression. J Bacteriol. 2008;190:2257–2265.