
ABSTRACT
PE_PGRS proteins of Mycobacterium tuberculosis (Mtb) constitute a large family of complex modular proteins whose role is still unclear. Among those, we have previously shown, using the heterologous expression in Mycobacterium smegmatis, that PE_PGRS3 containing a unique arginine-rich C-terminal domain, promotes adhesion to host cells. In this study, we investigate the role of PE_PGRS3 and its C-terminal domain directly in Mtb using functional deletion mutants. The results obtained here show that PE_PGRS3 is localized on the mycobacterial cell wall and its arginine-rich C-terminal region protrudes from the mycobacterial membrane and mediates Mtb entry into epithelial cells. Most importantly, this positively charged helical domain specifically binds phosphorylated phosphatidylinositols and cardiolipin, whereas it is unable to bind other phospholipids. Interestingly, administration of cardiolipin and phosphatidylinositol but no other phospholipids was able to turn-off expression of pe_pgrs3 activated by phosphate starvation conditions. These findings suggest that PE_PGRS3 has the key role to serve as a bridge between mycobacteria and host cells by interacting with specific host phospholipids and extracting them from host cells, for their direct integration or as a source of phosphate, during phases of TB pathogenesis when Mtb is short of phosphate supply.
Introduction
Mycobacterium tuberculosis (Mtb) is one of the most successful human pathogens, that co-evolved with humans for more than 100.000 years [Citation1–3]. The most common outcome following Mtb infection is latent tuberculosis (TB), a benign coexistence between the human host and the tubercle bacilli that usually last for a lifetime, with no signs or symptoms of disease [Citation4,Citation5]. In 2–8% of cases, Mtb infection leads to overt disease, which most often involves the lung tissue, with inflammation, caseous necrosis, cavitation, and extensive tissue damage, that allows for the aerogenic transmission of Mtb [Citation6,Citation7]. Hence, Mtb usually survives and resists in host tissues for a lifetime despite the presence of a robust innate and adaptive immune responses. We lack a clear understanding of the immunological mechanisms involved in this process and molecular determinants responsible for these unique features of Mtb have been only partially identified [Citation8].
Proteins and other molecules available on the mycobacterial surface and mycomembrane are known to play a key role in host–pathogen interaction [Citation9]. Among these, some of the ESX type 7 secretion systems (T7SS) are essential for Mtb virulence, with the secretion of effector proteins such as those of the EsxA/B family and PE and PPE family [Citation10]. The expansion and diversification of PE and PPE proteins was a key event in the evolution of the virulent Mtb, where these genes occupy almost 10% of the genome coding capacity [Citation11–13]. Some of these proteins, as in the case of the PE_PGRS subfamily, are found only in the Mtb complex and few other pathogenic mycobacteria affecting mammals, where they have been implicated in mycobacterial pathogenesis [Citation14].
In Mtb, there are more than 50 functional pe_pgrs genes which encode proteins with a shared structure: a highly conserved PE domain of ≈ 100 amino acids in length; a linker domain with a conserved GRPLI motif that is essential for proper protein translocation on the mycobacterial outer membrane; a polymorphic glycine-rich domain of variable size with repetitive gly-gly-X motif [Citation15,Citation16]. Some of the PE_PGRS proteins have a unique C-terminal domain that can be up to 300 amino acids in length as for PE_PGRS30 [Citation17]. A mounting body of experimental evidence is supporting the role of PE_PGRS proteins in TB pathogenesis [Citation14,Citation18]. We have recently shown that PE_PGRS3 of Mtb contains, downstream of the PGRS domain, a unique arginine-rich C-terminal domain, with 30 arginine units out of the distal 77 amino acids [Citation19]. In a series of experiments involving the heterologous expression of the pe_pgrs3 gene in Mycobacterium smegmatis (Ms), we have shown that PE_PGRS3 is specifically expressed under low phosphate concentrations and mediates adhesion to host cells through the arginine-rich domain [Citation19]. In this study, we provide clues of the functional role of the arginine-rich domain of PE_PGRS3 in experimental models involving the use of Mtb. Our findings suggest that PE_PGRS3 plays an essential role in grabbing essential components of Mtb, whose provision needs to be ensured for mycobacterial survival.
Results
The arginine-rich C-terminal domain of PE_PGRS3 enhances mycobacteria entry in host cells
To investigate the role of the arginine-rich C-terminal domain of the PE_PGRS3 (xR-3Ct), the sequence corresponding to the 240 nucleotides at the 3ʹ of the Rv0278c gene, coding the xR-3Ct domain of 80 amino acids, was cloned in pET-SUMO vector and expressed in E. coli using standard procedures [Citation20] (Supplementary –d). Following purification, dialyzed and endotoxin-free recombinant xR-3Ct was used in a series of experiments to assess its functional role. Murine macrophages (J774) and human alveolar epithelial cells (A549) were infected with a panel of Ms recombinant strains with and without the addition in the culture medium of the recombinant xR-3Ct and adhesion and cell entry assessed by intracellular bacterial counting 4 h post-infection.
Figure 1. Recombinant arginine-rich C-terminal domain of the PE_PGRS3 promotes Mycobacterium smegmatis adhesion to macrophages and pneumocytes
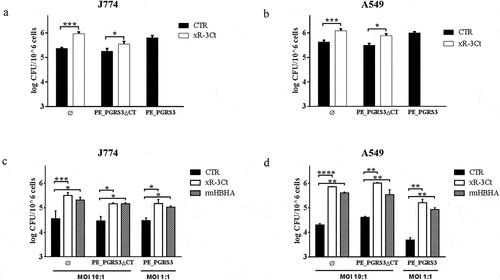
The addition of the recombinant xR-3Ct protein to a strain of Ms expressing the green fluorescent protein (MsGFP) enhanced adhesion to macrophages and pneumocytes, to achieve efficiency in cell entry similar to those observed for the recombinant Ms strain expressing PE_PGRS3 under the control of a strong promoter (MsGFPPE_PGRS3HA) [Citation19] ( and b). Interestingly, the addition of the recombinant xR-3Ct complemented the Ms expressing the functional mutant of the PE_PGRS3 lacking the C-terminal domain (MsGFPPE_PGRS3ΔCTHA) ( and b).
To better characterize these findings, J774 and A549 cells were infected using a lower MOI (1:1) of the MsGFPPE_PGRS3HA and compared with the infection carried out with the other functional deletion mutant strain and the parental strain administered at a higher MOI (MOI = 10:1) ( and d). As expected, since the MsGFPPE_PGRS3HA was administered at a ten times lower dose, the intracellular CFUs measured following infection were similar in J774 and in A549 with the CFUs of the other strains in non-conditioned media. However, when the recombinant xR-3Ct protein was added to the media, we observed enhanced mycobacterial entry in macrophages and pneumocytes for the three strains, regardless of the MOI used ( andd).
Moreover, we observed the same phenotype when we added another polybasic protein as the mycobacterial HBHA, which is known to mediate adhesion to epithelial cells but not to macrophages [Citation21,Citation22]. Again, no differences between MsGFP and recombinant Ms expressing PE_PGRS3 or its chimeras were observed, although, in contrast to previous findings [Citation22] addition of rmHBHA enhanced mycobacterial entry also in macrophages. These discrepancies may be attributable to the different experimental settings used and we cannot exclude that the addition of rmHBHA may activate macrophages to enhance bacterial uptake [Citation23,Citation24].
The PE_PGRS3 arginine-rich C-terminal domain specifically binds phosphatidylinositols and cardiolipin.
Polybasic proteins may interact with host components through binding to negatively charged molecules such as phospholipids commonly found in host membranes. Indeed, a recent report indicates that the lysine-rich domain of the HBHA specifically binds to 4,5 di-phosphorylated phosphatidylinositol [Citation25]. To investigate the ability of the PE_PGRS3 to bind phospholipids, the purified recombinant xR-3Ct was used to probe a nitrocellulose membrane where a series of phospholipids were adsorbed as described in the material and methods section (Supplementary and b). Following incubation, the anti-His antibody was used to detect the xR-3Ct protein captured by adsorbed lipids. The recombinant methylated HBHA (rmHBHA) was used as a positive control [Citation25,Citation26]. As shown in , recombinant xR-3Ct was able to bind phosphorylated phosphatidylinositol-4 phosphate (PtdIns (4)P), phosphatidylinositol-4,5 phosphate (PtdIns 4,5(P)), and phosphatidylinositol-3,4,5 phosphate (PtdIns (3,4,5)P), with affinity two times higher than the rmHBHA ( and b). Despite the clear binding to phosphorylated phosphatidylinositol, neither xR-3Ct nor rmHBHA are able to bind phosphatidylinositol (PI). Surprisingly, recombinant xR-3Ct showed a specific interaction with cardiolipin (CL). Hence, these two polybasic mycobacterial proteins show differential ability to bind common phospholipids found in host membranes.
Figure 2. Arginine-rich C-terminal domain specifically binds cardiolipin and phosphorylated phosphatidylinositols
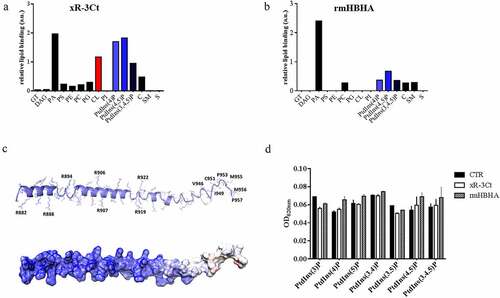
The analysis of chemical structures of all tested phospholipids allows rationalization of these results. Indeed, phosphatidic acid, phosphorylated phospholipids, and cardiolipin present a stronger overall negative charge (from −2 to −7), whereas all other phospholipids are either neutral or they present a single negative charge (Supplementary ). A strong electrostatic feature in the recognition of phospholipids by xR-3Ct well agrees with its predicted structural features. Structurally, xR-3Ct is predicted to adopt an extended α-helical conformation, followed at its C-terminus by a coiled-coil region (). Analysis of the electrostatic potential of this molecule shows a strongly positively charged surface for the helical region, whereas the coiled-coil region presents a neutral charge, with a hydrophobic character at its C-terminus (GISCSOQMMP) (). Therefore, it is not surprising that xR-3Ct recognizes amphipathic molecules, with the highest avidity for those with a higher negative charge.
Figure 3. Administration of cardiolipin, but not phosphorylated phosphatidylinositols, to phosphate-starved culture turns off pe_pgrs3 expression

To corroborate the hypothesis that xR-3Ct acts solely by sequestering phospholipids and has no phosphatase activity, the recombinant xR-3Ct was incubated with different phosphorylated PtdIns and the phosphatase activity was determined by measuring free phosphates (Pi) in solution with a malachite green assay. As shown in , no release in Pi was observed following incubation, thus showing that binding of phospholipids occurs without phosphatase activity.
Administration of free phosphate, cardiolipin and phosphatidylinositol to phosphate-starved culture turns off pe_pgrs3 transcriptional activity.
Since pe_pgrs3 is specifically expressed in phosphate starvation environment [Citation19], we investigated the consequences on the pe_pgrs3 transcriptional activity in a culture of phosphate starved Ms expressing PE_PGRS3-GFP under the control of the Mtb pe_pgrs3 promoter (MsPE_PGRS3GFP), following the administration of phosphorylated and non-phosphorylated host lipids. Phosphate starvation triggers the expression of PE_PGRS3-GFP and administration of inorganic phosphate turns off GFP expression, as previously described () [Citation19]. As expected, administration of non-phosphorylated lipids as cholesterol (C) and diacylglycerol (DAG) did not turn off pe_pgrs3 expression, but rather further induced fluorescence in the MsPE_PGRS3GFP phosphate starved culture (). Interestingly, administration of cardiolipin (CL) and phosphatidylinositol (PI) to MsPE_PGRS3GFP phosphate starved culture resulted in the turn off of fluorescence, suggesting that the mycobacterial cells do not any longer need the function of PE_PGRS3 in these conditions.
The highest level of fluorescence turn-off was observed upon the addition of free phosphate (). Conversely, PtdIns (4,5)P and PtdIns (3,4,5)P administration did not induce loss of fluorescence, suggesting that under these conditions (that is at least in Ms) these phosphorylated lipids cannot provide phosphate to the mycobacterial cells. This result prompted us to investigate whether binding by the C-terminal domain of the PE_PGRS3 to PtdIns (4)P, PtdIns 4,5(P), and PtdIns (3,4,5) is also observed with the whole bacterial cells. To this aim, we incubated Ms cells, cultured until mid-log phase, and resuspended in water, with the panel of selected PtdIns and then measured the Z-potential of mycobacteria. As shown in , no significant differences were observed between the surface charges of the MsGFPPE_PGRS3ΔCTHA and MsGFP strains, whereas MsGFPPE_PGRS3HA is characterized by a significantly lower Z potential (). Incubation of the MsGFPPE_PGRS3HA strain with the phosphorylated PtdIns enhanced the Z potential until the full retrieval of the net surface charge of the parental strain. Specifically, Zeta potential measurements showed that MsGFPPE_PGRS3HA net surface charge shifted from – 37.6 ± 1.9 mV to values of –43.7 ± 1.8 mV, –43.5 ± 2.8 mV, and – 44.2 ± 2.9 mV after the incubation with PtdIns(4)P, PtdIns 4,5(P) and PtdIns (3,4,5)P, respectively. Conversely, Z potential values of MsGFPPE_PGRS3ΔCTHA did not significantly change upon incubations with the phosphorylated PtdIns. These findings confirm our previous observation () that the xR-3Ct domain specifically interacts with phosphorylated PtdIns. Altogether, these data suggest that cardiolipin and PI may serve as a source of phosphate for mycobacteria, through the action of specific mycobacterial phosphatases. On the other hand, the inability of the phosphorylated PtdIns 4,5(P) and PtdIns (3,4,5) to turn off pe_pgrs3 transcriptional activity, albeit being able to bind the xR-3Ct domain, may be ascribed to the possible lack in Ms of specific phosphatases able to hydrolyze and free the phosphate ions from these more complex molecules.
Over-expression of the full-length PE_PGRS3 improves adhesion of the Mtb to pneumocytes, but not to phagocytic cells.
To investigate the functional role of the PE_PGRS3 in TB, we over-expressed various functional variants in Mtb, including (i) the full-length PE_PGRS3, (ii) its functional mutant lacking CT domain (residues 878–957, MtbGFPPE_PGRS3ΔCTHA) and (iii) its further truncated mutant, also lacking a part of the PGRS domain (residues 528–957, MtbGFPPE_PGRS3ΔGRPLIHA). Over-expression of the PE_PGRS3 in Mtb, or of its functional mutants, did not affect bacterial morphology (data not shown) nor growth rate in liquid media (). Proteinase K experiments show degradation of PE_PGRS3, thus suggesting that in Mtb the PE_PGRS3 full-length protein localizes on the mycobacterial surface (). Conversely, the two functional mutants were protected from proteinase K treatment, as proteinase K induced no degradation (). This finding indicates that these protein regions are not completely available on the mycobacterial surface and that the observed degradation of full-length PE_PGRS was limited to the C-terminal domain, which carries the HA epitope for detection. Namely, these results suggest that the full-length PE_PGRS3 is likely embedded in the mycomembrane and exposes solely the C-terminal arginine-rich domain.
Figure 4. Over-expression of the PE_PGRS3 enhances Mycobacterium tuberculosis cell entry in pneumocytes, but not in murine macrophages or human peripheral blood mononuclear cells (PBMCs)
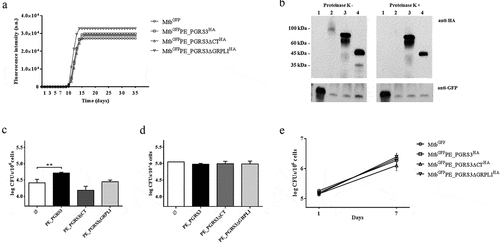
To investigate the effect of this exposed xR-3Ct domain on Mtb adhesion and entry in host cells, we infected pneumocytes (A549) and macrophages with the recombinant Mtb strains and evaluated the intracellular CFUs 1-h post infection. The MtbGFPPE_PGRS3HA strain showed a superior entry ability in A549 () but not in J774 macrophages () compared to the parental strain MtbGFP. Conversely, the MtbGFPPE_PGRS3ΔCTHA and the MtbGFPPE_PGRS3ΔGRPLIHA did not show any significant difference in their ability to gain entry in pneumocytes and macrophages. These results show that PE_PGRS3 enhances Mtb entry in epithelial cells but not in macrophages and that the enhanced cell entry is specifically due to the arginine-rich C-terminal domain. It is likely that Mtb entry in macrophages, which is mediated by multiple receptors, is already highly efficient and cannot be improved by PE_PGRS3. Indeed, infection of human peripheral blood mononuclear cells (PBMCs) with the Mtb strains did not result in any difference in total CFUs at day 1 and 7 post-infection among the strains, suggesting that PE_PGRS3 is not involved in the mycobacterial replication in the phagocytic cells ().
PE_PGRS3 is post-translationally cleaved in Mtb
Prompted by the observation that the PE_PGRS3 C-terminal domain is exposed to proteolysis, we performed a subcellular fractionation of the Mtb strains under study to assess the role of the arginine-rich domain in the association of the PE_PGRS3 with the mycobacterial cell wall. Indeed, localization of PE_PGRS proteins in M. smegmatis is only partially informative as a model of Mtb [Citation27], as Ms lacks the ESX-5 secretion system that is involved in the translocation/secretion of PE_PGRS proteins [Citation28, Citation29].
In the fractionation experiment, whole-cell lysate, the genapol extract, the cytosolic fraction following genapol extraction, and the culture supernatant (to measure secreted proteins) were obtained from the recombinant Mtb strains under study and cultured in Sauton media with or without Tween80 and in standard Pi or low Pi concentrations. As tested by western blot analysis, full-length PE_PGRS3HA (≈ 81 kDa) was detected only in the whole cell lysate fraction of the MtbGFPPE_PGRS3HA (), but not in any of the other fractions, regardless of the presence in the culture media of the mild detergent Tween80 or of low or standard Pi concentrations (–d). Differently, clear bands corresponding to their expected molecular weights were observed for MtbGFPPE_PGRS3ΔCTHA and MtbGFPPE_PGRS3ΔGRPLIHA, which were grown in the same conditions as the full-length PE_PGRS3. Consistent with its sensitivity to proteinase K, these findings suggest that the C-terminal domain of PE_PGRS3 is readily degraded.
Figure 5. PE_PGRS3 is cleaved in Mycobacterium tuberculosis.
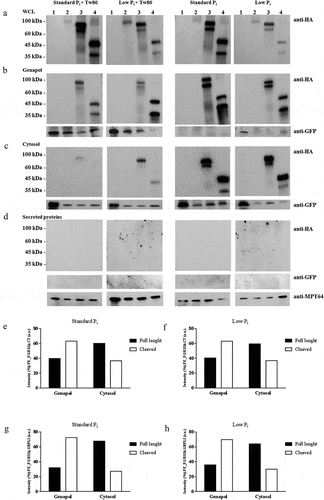
However, the results obtained with the other two functional deletion mutants provide relevant information. Subcellular fractionation of MtbGFP PE_PGRS3ΔCTHA clearly indicates that culture in Tween80 dramatically reduces the PE_PGRS3ΔCTHA extracted by Genapol or remaining in the cytosol following Genapol extraction, yet no signal was detected in the secreted fraction. Similar results were obtained with the MtbGFP PE_PGRS3ΔGRPLIHA strain, suggesting that the second GRPLI domain within the PGRS region of PE_PGRS3 does not significantly contribute to protein localization. These results indicate that growth in Tween80 containing medium significantly perturbs the localization of PE_PGRS protein on the mycobacterial surface and impacts cell permeability.
Interestingly, both PE_PGRS3ΔCTHA and PE_PGRS3ΔGRPLIHA were detected as two bands, with the lower band corresponding to the protein cleaved at N-terminus approximately 10 kDa downstream the amino acid in position 1. The relative amount of the lower band was higher in the genapol extract compared with cytosol obtained following genapol extraction, suggesting that cleaved PE_PGRS3 is more abundant on the mycobacterial surface (–h). Cleavage of the two proteins was consistent across the experimental conditions tested in and accounted for a loss of ≈ 120–150 amino acids, with the N-terminus truncated protein stable and more abundant in the outer membrane.
PE_PGRS33 is differently cleaved in Mtb and in M. bovis BCG
A recent report indicates that in M. marinum PE_PGRS proteins are cleaved at their N-terminus by the protease PecA, which is also present in other pathogenic mycobacteria species including Mtb [Citation30]. Since we could observe the cleavage for the truncated PE_PGRS3ΔCT but not for the full-length PE_PGRS3, we decided to investigate the same process for the well-characterized PE_PGRS33 by performing the cellular fractionation of the Mtb and M. bovis BCG expressing PE_PGRS33 HA. The recombinant pe_pgrs33 gene was expressed in Mtb and BCG under the control of its own promoter as previously described [Citation31,Citation32], which warrants constitutive expression of the protein.
As shown in –c, PE_PGRS33 was detected at the expected molecular weight (≈45 kDa) and at a lower molecular weight (≈35 kDa), suggesting that in Mtb and BCG PE_PGRS33 is cleaved similarly to what observed for the PE_PGRS3-truncated proteins. Again, a much stronger signal was detected in the genapol extract of MtbPE_PGRS33 HA cultured in the absence of Tween80 compared with that grown in the presence of Tween80, confirming that the presence of this mild detergent significantly alters mycobacterial permeability. The cleaved PE_PGRS33 was more abundant in the genapol extract than in the cytosolic fraction obtained following genapol extraction, suggesting that the cleaved form is most abundant in the mycobacterial cell wall. Moreover, PE_PGRS33 was poorly expressed in BCG compared with Mtb, despite both strains were transformed with the same plasmid, suggesting that protein stability or localization in BCG may be impaired.
Figure 6. PE_PGRS33 is mainly cleaved in Mycobacterium tuberculosis compared to Mycobacterium bovis BCG
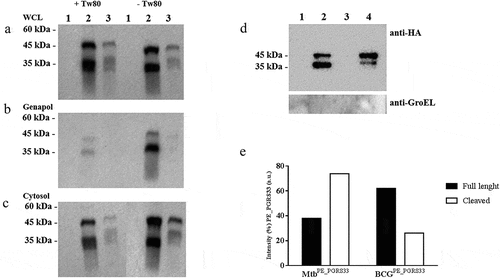
To further corroborate this hypothesis, the genapol fractions of Mtb and BCG strains expressing PE_PGRS33 HA were mixed with anti-HA antibody carried on magnetic beads to capture the protein available on the mycobacterial surface. Immunoblot of the eluted fractions clearly shows that in Mtb most of PE_PGRS33 is cleaved, while in BCG PE_PGRS33 is not efficiently cleaved ( ande). These results support the findings that in BCG, PE_PGRS translocation on the mycobacterial cell wall is impaired [Citation33].
Discussion
PE_PGRS3 is a protein belonging to the PE_PGRS family endowed with a peculiar and unique arginine-rich C-terminal domain [Citation19]. The pe_pgrs3 gene was shown to be specifically expressed under low inorganic phosphate (Pi) concentrations in Mtb and when heterologously expressed under the control of its own promoter in Ms. In this study, we aimed at rationalizing the role of PE_PGRS3 in connection with Mtb needs when in short supply of phosphate.
Using protease protection assays, we show that PE_PGRS3 is available on the Mtb surface, as its arginine-rich C-terminal domain is readily degraded. Consistently, subcellular fractionation studies show that PE_PGRS3 is found in the genapol extract, as expected for a protein that localizes in the mycomembrane [Citation27,Citation34,Citation35b]. Importantly, the arginine-rich C-terminal domain of PE_PGRS3 protruding from the mycomembrane can promote adhesion to host cells, even when loosely associated or released by the mycobacterial cell. These results indicate that the adhesion properties of PE_PGRS3 [Citation19] may not require a mycomembrane – bound protein as expected for an adhesin, but rather suggest that the xR-3Ct domain serves as a “bridge” between mycobacteria and host cells to promote adhesion through electrostatic interactions mediated by its arginine residues.
The role of PE_PGRS3 in mediating Mtb interactions with the host cells does not explain the need of Mtb to overexpress this protein only when phosphate is lacking. To attempt an explanation for this finding, we screened the binding of PE_PGRS3 with several phospholipids with different complexities. As a result, we found that xR-3Ct of PE_PGRS3 binds phosphorylated phosphatidylinositol (PtdIns), cardiolipin (CL), and phosphatidic acid (PA) but no other phospholipids, with a higher affinity than observed for HBHA [Citation25]. We observed the binding of these PtdIns also to mycobacterial cells expressing PE_PGRS3, as demonstrated by the significant alterations of the Z potential of the mycobacterial surface in the presence of PtdIns. Therefore, xR-3Ct domains protruding from the mycobacterial surface mediate binding to the negatively charged lipids.
Structural features of xR-3Ct, displaying a positive electrostatic potential and a hydrophobic C-terminal arm, well account for the recognition of phosphatidylinositol, which carry both negatively charged phosphates and a lipidic arm. Similar considerations apply to cardiolipin, also able to bind xR-3Ct and to induce the turning off of the pe_pgrs3 transcriptional activity. These experimental evidences suggest that phosphate-starved mycobacteria start expressing PE_PGRS3 protein that then translocates to the mycobacterial surface to expose the arginine-rich domain and becomes available to bind host-derived cardiolipin and phosphatidylinositol. Lipids are known to be a major source of energy for Mtb in several steps of TB pathogenesis. In the caseating debris of human TB granulomas, there is an enrichment of cholesterol, cholesteryl esters, and triacylglycerides (TAGs), with the latter being the main components of the large lipid droplets commonly found in foamy macrophages, another important niche for Mtb [Citation36,Citation37]. Phagosome-containing Mtb can be found tightly associated with lipid droplets [Citation38] and Mtb can translocate to these droplets where it utilizes lipids as a carbon source for survival [Citation39,Citation40]. Interestingly, elevated levels of TAGs are found during rapid disease development caused by modern Mtb Beijing strains compared to ancient Mtb Beijing strains [Citation41], highlighting the importance of lipid metabolism in TB pathogenesis. Since these lipids lack phosphorous, the growth and persistence of Mtb under these nutrient conditions occurs in an environment where phosphate is a limiting factor [Citation42]. Hence, Mtb must be able to deploy mechanisms aimed at stealing phosphorylated molecules from the host to maintain adequate levels of this essential component.
Cardiolipin is a phosphorylated lipid that constitutes 10–20% of the mitochondrion inner membrane [Citation43] and damaged mitochondria release cardiolipin in the outer membrane and then in the cytoplasm. Mtb-infected and damaged foamy macrophages and necrotic cells that abound in the caseous granuloma may accumulate higher levels of cardiolipin released by damaged mitochondria. We hypothesize that PE_PGRS3 on the Mtb surface can serve to grab cardiolipin through the arginine-rich domain, protruding from the mycomembrane; cardiolipin can then be imported or degraded with the help of other proteins, to obtain phosphate. A similar scenario may be proposed for the phosphatidylinositol commonly found in the eukaryotic membranes. It is possible that Mtb-specific proteins are required to obtain phosphates from these phosphatidylinositols or that Mtb can simply accumulate these phospholipids on the surface and that host enzymes as PTEN [Citation44] may free phosphate molecules that can then be imported by Mtb. The role of PE_PGRS3 as a “molecular thief” well explains its overexpression in the absence of phosphate, and by grabbing these molecules, PE_PGRS3 ensures proper provisions of phosphate ions. Further studies are needed to support this hypothesis, though the unique presence of the arginine-rich domain in Mtb but not in other mycobacteria highlights further interest in the role of this protein in TB pathogenesis.
Moreover, PtdIns has been demonstrated to be of vital importance for the growth and viability of mycobacteria since it acts as a lipid anchor for key components of the cell wall, like the glycolipids phosphatidylinositol mannoside (PIM), lipomannan (LM), and lipoarabinomannan (LAM). In Mtb, PtdIns synthesis is catalyzed by the CDP-alcohol phosphotransferase phosphatidylinositol-phosphate synthase (PIPS), an essential enzyme for mycobacterial viability [Citation45]. Catalyzed by PIPS, PtdIns is obtained by the chemical reaction of CDP-diacylglycerol with inositol-phosphate to form phosphatidylinositol-phosphate, the precursor to PtdIns. PtdIns is unique to mycobacteria and few other bacterial species whereas it is widely distributed in eukaryotes, where PtdIns(4)P and PtdIns(4,5)P are commonly recognized to mediate anchoring of eukaryotic polybasic proteins [Citation46] and are involved in several pathways, including key steps in the intracellular lifestyle of Mtb in host cells as phagosome/lysosome fusion and autophagosome formation [Citation18,Citation47]. The essential role of PtdIns in Mtb, the availability of this compound in host cells, and the avidity of xR-3Ct for PtdIns point to the hypothesis that PE_PGRS3 exerts the precise role to enhance host cell interaction and, even more important, to tear PtdIns molecules from the host cells.
Another interesting feature of PE_PGRS3, arisen from subcellular fractionation studies of PE_PGRS3ΔCTHA and PE_PGRS3ΔGRPLIHA, is that a cleavage of the protein occurs that removes the N-terminal 120–150 residues, immediately upstream the PGRS domain. Densitometric analysis of these immunoblots clearly indicates that the cleaved protein is more abundant in the genapol extract than in the cytosolic fraction. Similar results were obtained when the same experiment was carried out in Mtb expressing the well-characterized PE_PGRS33. The cleavage we observed for PE_PGRS3 and PE_PGRS33 is in line with recent findings showing that in M. marinum LipY and PE_PGRS proteins are processed by another PE_PGRS protein, PecA [Citation30]. Cleavage of the PE protein LipY by PecA in M. marinum occurs at positions 136 and 149 [Citation48], whereas cleavage of PE_PGRS proteins results in the loss of ≈11 kDa, corresponding to the PE domain. The observed cleavage in Mtb is located within the position 139– 152 of PE_PGRSs, which includes, also in PE_PGRS3, the characteristic Ser-Gly–Gly–Ala motif that is the putative target of the protease PecA. Despite several efforts, we could not purify enough cleaved PE_PGRS33 or PE_PGRS3 to identify the exact position of the cleavage site.
In M. marinum and Mtb, translocation of PE_PGRS proteins, including PecA, occurs through the ESX5 type 7 secretion system [Citation29,Citation49,Citation50] and cleavage of the PE domain by the protease PecA occurs on the mycobacterial surface [Citation30]. Our findings that the cleaved forms of PE_PGRS3 and PE_PGRS33 are more abundant in the genapol extract than in the cytosolic fraction support this hypothesis, and the model we recently proposed [Citation14]. Moreover, the observation that expression of PE_PGRS33 in BCG results in poorly cleaved protein is in line with the recent findings by Ates et al. [Citation33,Citation51], indicating impaired translocation of PE_PGRS proteins in BCG.
Another note that we think is relevant to briefly discuss involves the impact that the use of a nonionic detergent in the culture medium has on the mycobacterial cell. In fact, culturing Mtb in liquid media containing Tween80 had a relevant impact on protein localization, indicating that this mild detergent may extract PE_PGRS3, PE_PGRS33, and potentially all other PE_PGRS proteins from the mycomembrane. This is in our opinion very important since it indicates that culturing Mtb in the presence of Tween80 could perturb the mycomembrane, leading to a loss of surface protein. As a result, culturing mycobacteria and Mtb in media without Tween80 or other mild detergents is desirable when performing protein subcellular fractionation studies or other functional assays aimed at studying mycomembrane-associated proteins.
In conclusion, the results of this study suggest that PE_PGRS proteins, once translocated through the plasmatic membrane in an ESX5-dependent mechanism [Citation29,Citation30], remain associated with the mycomembrane in a cleaved state missing their PE domains. Once in the outer membrane PE_PGRS proteins may “float” on the mycomembrane outer leaflet and may possibly be released to exert their activity as seen for instance for PE_PGRS3 and its arginine-rich motif that can bind host membranes. Similarly, PE_PGRS33 that interacts with TLR2 may be released or freed from the outer leaflet of the mycomembrane to promote inflammation and tissue damage [Citation31,Citation32]. We expect a similar scenario for other PE_PGRS proteins known to play a role during the intracellular lifestyle of Mtb in macrophages as PE_PGRS30 [Citation17], −41 [Citation52], and −47 [Citation53]. Most importantly, our results provide an experimental evidence of the ability of PE_PGRS3 on the mycobacterial surface to bind PtdIns and cardiolipin molecules, for their direct integration or as a vital source of phosphate for Mtb in the lipid-rich environment of foamy macrophages and caseum. This finding provides a functional rationale for the overexpression of PE_PGRS3 under short supply of phosphate, a condition which limits the ability of Mtb to synthesize endogenous vital molecules and likely favors a PE_PGRS3-mediated mechanism to steal these molecules from host cells. The ability of PE_PGRS3 to grab essential molecules during key steps of TB pathogenesis may be considered an ace in the hole of Mtb, able to persist for years in caseous granulomas and foamy macrophages.
Materials and methods
Bacterial strains, media and growth conditions, and electroporation
Mycobacterium smegmatis (Ms) mc2 155 and Mycobacterium tuberculosis (Mtb) H37Rv were grown at 37°C in Middlebrook 7H9 broth medium (difco Becton-Dickinson), supplemented with 0.2% glycerol (Sigma-Aldrich), 10% ADC (Becton-Dickinson), and 0.05% Tween 80 (Sigma-Aldrich) using standard procedures [Citation54,Citation55]. Ms and Mtb recombinant strains were obtained by electroporating competent cells with above mentioned homemade pMV modified vector containing hbha promoter upstream and the sequence coding HA epitope downstream the full-length Rv0278 gene or its chimeras, respectively. Furthermore, the modified pMV vector carried also the GFP sequence under the control of mycobacterial antigen 85 promoter (Supplementary Table 1) [Citation19].
Briefly, Ms mc2 155 and Mtb H37Rv grown to mid-exponential (log) phase and pre-treated with 1% glycine were widely washed with cold and sterile 10% glycerol. Two hundred microliters of concentrated cells were mixed with 1 μg of DNA, incubated for 5 min (m) at room temperature (RT), and then transferred to 0.2 cm cuvettes (BioRad). Samples were electroporated using an electroporation system Bio-RAD GenePulser X Cell™ with the following parameters: voltage 2500, capacitance 25µF, and resistance 1000 Ω [Citation54]. After the pulse, cells were recovered in 1 ml of 7H9 liquid medium and incubated for 1 day at 37°C.
Colonies were selected on 7H11 agar media supplemented with 10% OADC (Microbiol) containing 40 µg/ml kanamycin (Sigma Aldrich). Single distinct antibiotic-resistant colonies were isolated and sub-cultured in 7H9 medium supplemented with 10% ADC (Microbiol), 0.05% Tween 80, and kanamycin as previously indicated and were incubated at 37°C until mid-log phase. Finally, mycobacteria were stocked at −80°C after adding 20% glycerol until use. Serial dilutions were then carried out to establish bacterial concentration.
Cloning, expression, and purification of the PE_PGRS3 C-terminal domain
Sequence codifying the last 80 amino acids of the PE_PGRS3 (xR-3Ct) was amplified by using the following primers: Forward: 5ʹ- GGTTGCCGGCGGGTTTGGCGC −3ʹ and Reverse: 5ʹ- CTACGGCATCATCTGCGGTGA −3ʹ. The purified amplicon was cloned in the expression vector pET-SUMO (Invitrogen) downstream to the SUMO – 6x Histidine tag (6xHis) domain and under the control of the lacO promoter [Citation20]. Ligation was directly used to transform E. coli MACH 1 (Life technologies) and then plated on Luria Bertani agar medium (Sigma Aldrich) supplemented with 50 μg/ml kanamycin (Sigma Aldrich) to select transformant single colonies. Antibiotic-resistant colonies were sub-cultured in Luria Bertani broth medium (Sigma Aldrich) and extracted final vector was controlled by enzymatic restriction analysis and by sequencing.
To express xR-3Ct, E. coli BL21-DE3 pLyss chemically competent cells (Life technologies) were transformed with pET-SUMOxR−3Ct and then plated on Luria Bertani agar medium (Sigma Aldrich) containing 50 μg/ml Kanamycin (Sigma Aldrich). Selected single colonies were inoculated in Luria Bertani broth medium (Sigma Aldrich) containing 50 μg/ml Kanamycin (Sigma Aldrich), 50 μg/ml chloramphenicol (Sigma Aldrich) supplemented with 1% glucose (Sigma Aldrich), and incubated overnight at 37 C°. 250 ml culture was prepared and incubated until OD600 reached 0.6 when 1 mM Isopropyl β-d-1-thiogalactopyranoside (IPTG) (3 V chimica) was added. Three hours post induction, the cell pellet was harvested, washed with sterile cold Phosphate buffer (PBS), and resuspended in lysis buffer (PBS, containing 350 mM NaCl and the protease inhibitor cocktail (Sigma Aldrich), pH: 7.4). Cells were then lysed by sonication. The soluble fraction was collected and processed to purify the xR-3Ct by using Ni-NTA agarose column and Fast Protein Liquid Chromatography (FPLC- ӒKTA, GE health care life sciences) [Citation20,Citation55]. Protein elution was obtained by using PBS containing 350 mM NaCl and 200 mM Imidazole (Sigma Aldrich). rmHBHA was obtained as described above [Citation55].
Purified xR-3Ct and rmHBHA were dialyzed in sterile LPS free PBS by using Float-A-Lyzer G2 columns at 4°C according to the manufacturer’s instruction. Endotoxin was measured by using Limulus Amebocyte Lysate assay (LONZA). An endotoxin-free batch was considered at LPS concentration <0.1 EU/ml.
Lipids/PE_PGRS3 C-terminal domain interaction
Purified xR-3Ct was used to probe a nitrocellulose strip (Echelon) where different phosphorylated lipids were adsorbed [Citation25]. Following lipids were imbedded on the membrane strip: triglyceride (GT), diacylglycerol (DAG), phosphatidic acid (PA), phosphatidylserine (PS), phosphatidylethanolamine (PE), phosphatidylcholine (PC), phosphatidylglycerol (PG), cardiolipin (CL), phosphatidylinositol (PtdIns), PtdIns(4)P, PtdIns(4,5)P2, PtdIns(3,4,5)P3, cholesterol, sphingomyelin (SM), or sulfatide. Nonspecific binding was blocked by incubation with PBS containing 0.1% Tween 20 (PBS-T) and 3% BSA for 1 hour at room temperature. xR-3Ct was added at a final concentration of 0.5 µg/ml and incubated for 1 hour as indicated by the manufacturer’s instruction. The membrane was washed with PBS-T and a monoclonal anti-His antibody (Sigma-Aldrich) was to detect the binding. Finally, an IgG-Peroxidase (Sigma-Aldrich) was used as the secondary antibody. Immunoblot was developed using Supersignal West Dura Extended Duration Substrate (Thermo scientific) and chemiluminescence detected by ChemiDoc TM XRS+ system (Biorad). The same experiment was performed with the rmHBHA as previously described.
FACS analysis
Recombinant Ms, expressing PE_PGRS3 under control of its own promoter and fused at the C-terminal with the GFP (MsPE_PGRS3GFP), Ms expressing GFP (MsGFP) and Ms mc2 155 non-fluorescent wild type strain were grown in standard and low phosphate Sauton medium (~50 μM Pi) as described above. Fluorescence was measured by using FACSCantoII flow cytometer (BD Bioscience), as described elsewhere [Citation19]. When fluorescence of MsPE_PGRS3GFP grown in low Pi environment switched on, the liquid culture was split into different tubes. Inorganic phosphate (Pi), diacylglycerol (DAG), phosphatidic acid (PA), phosphatidylserine (PS), cardiolipin (CL), phosphatidylinositol (PtdIns), PtdIns(4,5)P2, PtdIns(3,4,5)P3, and cholesterol were added at the final concentration of 100 µM. Fluorescence was measured at different time points following phosphate or lipid restoring.
Careful cytometry analysis provided for a gating strategy composed of different and serial steps. Initial Forward scatter (FSC-A) versus Side scatter (SSC-A) was carried out to identify bacteria, based on size and complexity, and to exclude debris. Progressively, two serial gating were carried out to get out doublets or inappropriate heterogeneities. Forward scatter height (FSC-H) versus FSC-A density plot and a Side scatter height (SSC-H) versus SSC-A plot were opportunely performed before measuring fluorescence. The fluorescence intensity of at least 50,000 ungated events was measured as previously described [Citation19]. The excitation laser line was at 488 nm (Excitation max value was 494 nm and emission max value was 520 nm). The data files were analyzed using FACSDiva Software (BD Bioscience).
Malachite green assay
Phosphatase activity of the xR-3Ct was assessed by using a malachite green based phosphatase assay (Echelon). Free Pi in solution forms a colored complex with molybdate/malachite green that is quantified by reading absorbance at 620 nm. The following phosphorylated phosphatidylinositols (PtdIns) were assayed: PtdIns(3)P, PtdIns(4)P, PtdIns(5)P, PtdIns(3,4)P, PtdIns(3,5)P, PtdIns(4,5)P, PtdIns(4,4,5)P.
Briefly, 25 μl containing 5 μg/ml of the xR-3Ct and rmHBHA were mixed with 25 μl of the PtdIns and incubated 1 h at 37°C in agitation. After incubation, 100 μl of room temperature malachite green solution was added to each well and the plate was incubated for 30 minutes without shaking. Absorbance was measured as above indicated.
Phosphatase activity of the xR-3Ct was also assessed on recombinant Ms strains expressing full-length PE_PGRS3 (MsGFPPE_PGRS3HA) or its chimera lacking CT domain (MsGFPPE_PGRS3ΔCTHA) and MsGFP. In the culture supernatant was measured free Pi (data not shown).
Zeta potential measurements
Recombinant Ms overexpressing PE_PGRS3 (MsGFPPE_PGRS3HA), its functional mutant lacking C-terminal domain (MsGFPPE_PGRS3ΔCTHA) and wild type (MsGFP) strains were grown in 7H9 completed medium until mid-log phase and then sub-inoculated in Sauton standard medium supplemented with 40 µg/ml kanamycin. Each specimen was diluted in water (1:100) and incubated with PtdIns(4)P, PtdIns(4,5)P2, PtdIns(3,4,5)P3 for 1 h. Finally, solutions were characterized by Zetasizer Nano ZS (Malvern, Herrenberg, Germany) equipped with a 633 nm He–Ne laser. Universal zeta dip cell (ZEN1002, Malvern, Herrenberg, Germany) was used for experiments with a sample volume of 1 ml. For each sample, three measurements were averaged. The Z-potential was calculated from the electrophoretic mobility using the Henry correction to Smoluchowski’s equation as reported previously [Citation19].
Proteinase K treatment
Recombinant Mtb strains were cultured in 7H9 medium supplemented with 10% ADC and 0.05% Tween 80, before sub-inoculating in Sauton medium until mid-log phase. Cells were harvested by centrifugation and washed two times with cold sterile PBS. Each pellet was re-suspended in the Buffer G2 (Qiagen) and divided into two aliquots. Proteinase K (Qiagen) was added to one of the samples according to the manufacturer’s instruction. Both samples were incubated in agitation for 30 minutes at RT. Finally, each aliquot was washed three times with PBS and resuspended in lysis buffer as previously described [Citation19].
Mycobacterial cell lysis, SDS-PAGE, western blotting, and immunoblotting
Recombinant Mtb strains were cultured as previously described and then sub-inoculated in diverse Sauton based medium until mid-log phase when cells were harvested by centrifugation (3500 rpm for 15 minutes, 4 C°). Growth conditions account for Sauton medium combinations supplemented or not with inorganic phosphate (Pi) or with 0.05% Tween 80. Each culture was divided into two aliquots. To obtain whole cell lysate (WCL), the cell pellet was washed with cold sterile PBS and re-suspended in lysis buffer (20 mM Tris, 150 mM NaCl, 1:100 protease inhibitor cocktail (Sigma Aldrich), pH: 7.5). Finally, the lysate was obtained using a mini bead beater (BioSpec) instrument [Citation34,Citation54]. To separate mycomembrane associated proteins, the cell pellet of the second aliquot was treated with lysis buffer containing 0.5% Genapol X-80 (Fluka) for 30 minutes at room temperature. The supernatant (Genapol fraction) was collected by centrifugation and pellet (Cytosolic fraction) was washed with cold PBS before lysis as previously described. Finally, secreted proteins were obtained by TCA precipitation of the culture medium. Proteins were separated on 4– 15% precast polyacrylamide gel (Smobio) by SDS-PAGE and then transferred to a nitrocellulose membrane (Bio-Rad) by western blotting. Nonspecific protein binding was blocked by treatment with PBS containing 0,05% Tween 20 (Sigma Aldrich) and 5% Skim Milk (Oxoid) for 1 h at room temperature.
Membranes were probed with monoclonal anti-HA (1:1000) (Covance), monoclonal anti-GFP (1:4000) (Sigma Aldrich), polyclonal anti-GroEL (1:5000) (GeneTex) antibodies, and with pooled sera (1:2000) obtained from mice immunized with the purified MPT64 antigen. IgG-Peroxidase (Sigma Aldrich) was used as a secondary antibody. Immunoblot was developed using Supersignal West Dura Extended Duration Substrate (Thermo scientific), and finally, chemiluminescence was detected by ChemiDoc TM XRS+ system (Biorad).
Capture of the PE_PGRS33 HA in the mycobacterial cell wall
Mtb and Mycobacterium bovis (Mbov) BCG were expressing the PE_PGRS33. Recombinant strains were obtained by cloning the Rv1818c gene, with its putative promoter, upstream the HA epitope sequence in a pMV206-based vector [Citation31,Citation32]. Recombinant MtbPE_PGRS33 HA and MbovPE_PGRS33 HA and the parental strains Mtb and Mbov BCG were grown as previously described and then sub-inoculated in Sauton medium, supplemented or not with 0.05% Tween 80, until mid-log phase (OD600 nm: 0.8 ± 0.2). Cultures were harvested and processed to obtain the Genapol fraction as indicated above. Genapol fractions of all strains were incubated with anti-HA magnetic beads (Miltenyi Biotec) following the manufacturer’s instruction. Beads binding PE_PGRS33 HA were separated from the solution by using a magnetic plate, washed three times before eluting the captured protein. Eluates were TCA precipitated before protein separation by SDS page electrophoresis and immunoblot as previously described. The membrane was probed with the anti-HA (1:1000) monoclonal antibody and anti-GroEL polyclonal antibody (1:5000).
Cell culture and mycobacteria infection
Murine macrophages (J774 – A1) and human type 2 pneumocytes (A549) were grown in Dulbecco’s modified eagle medium (DMEM) (Euroclone) enriched with 10% fetal bovine serum (FBS), 2 mM glutamine (Euroclone), 100 µg/ml streptomycin, and penicillin (Euroclone) and were kept in a humidified atmosphere containing 5% CO2 at 37°C. Before infection, cells were collected and suspended in the same medium without antibiotic and supplemented with 2% FBS. Cells were plated at the concentration of 1.2 × 106 cell/ml and were infected 24 h later [Citation19,Citation54].
Multiplicity of infection (MOI) 10:1 for 4 h was used for Ms infection for both J774 and A549; conversely an MOI of 1:1 (J774) and MOI of 10:1 (A549) for 1 h was used for Mtb infection and cells were maintained at standard atmosphere conditions (5% CO2 and 37°C) [Citation32]. Intracellular colony forming units (CFUs) were obtained at 4 h post infection for Ms and 1 h post infection for Mtb infected cells.
For the infection with Ms, xR-3Ct and rmHBHA were added to the infection solution at the final concentration of 1 μg/ml [Citation20].
Peripheral blood mononuclear cells (PBMCs) were isolated from human blood collected by healthy volunteers by using Ficoll lympholyte (Cederlane) following the manufacturer’s procedure. PBMCs were washed with sterile PBS and then resuspended in Roswell Park Memorial Institute (RPMI) 1640 medium (Euroclone) enriched with 10% FCS and 2 mM glutamine (Euroclone). Finally, PBMCs were seeded at a final concentration of 1.2 × 106 cell/ml and infected with Mtb recombinant strains at MOI = 1:10 [Citation56]. CFUs evaluated at 1 and 7 days post infection.
Statistical analysis
All data were generated from independent experiments with at least three technical replicates. Microsoft Excel (2010) and Graphpad Prism software version 6 (GraphPad software) were used to collect and to analyze the data. Data were expressed on a representative graph as mean ± SD and analyzed by one-way or two-way ANOVA comparison tests followed by the appropriate correction, as specified in the caption under each figure.
Modeling of xR-3Ct structure
Secondary structure prediction of xR-3Ct was performed using JPRED4 server [Citation57]. Homology modeling was performed using I-Tasser [Citation58]. A meta-threading approach, LOMETS, was used to retrieve template proteins of similar folds from the PDB library. The program SPICKER was used to cluster the decoys based on the pair-wise structure similarity. Best confidence model was selected based on the C-score. The electrostatic potential surface was computed with Chimera [Citation59].
Author contribution:
This study was designed by FDM and GD. Recombinant strains and protein purification and cellular localization were performed by FDM, AS, BB. The infection model was set up by FDM and BB and experiments were carried out by FDM, BB, IP, and MSali. VP and MP provided Zeta potential analysis. Protein modeling was performed by EK and RB. FDM, FM, and SB performed flow-cytometry assay. Data analysis and figure preparation were conducted by FDM, RB, and GD. FDM, RB, and GD wrote the paper. All authors contributed to revision and participated in the discussion and interpretation of the results. All authors contributed to the final revision of the project. All authors read, critically revised, and approved the final manuscript.
Supplemental Material
Download Zip (2.4 MB)Acknowledgments
This work has been supported by the intramural research grant of the Università Cattolica del Sacro Cuore Linea D3.2 2017 and Linea D1 awarded to GD. EK was supported by the Marie Skłodowska-Curie Action BactiVax, GA number 860325.
Data Availability Statement:
Raw data were generated at Università Cattolica del Sacro Cuore and Fondazione Policlinico Gemelli (Dipartimento di Scienze biotecnologiche di base, cliniche intensivologiche e perioperatorie – Sezione di Microbiologia; Dipartimento di Diagnostica per Immagini, Radioterapia Oncologica ed Ematologia; Dipartimento di Neuroscienze, Università Cattolica del Sacro Cuore) and Institute of Biostructures and Bioimaging. Derived data supporting the findings of this study are available from the corresponding author GD on request.
Supplementary Material
Supplementary data for this article can be accessed through the publisher’s website.
Disclosure statement
All authors declare no competing interests.
Additional information
Funding
Related Research Data
References
- Gagneux S. Host-pathogen coevolution in human tuberculosis. Philos Trans R Soc Lond B Biol Sci. 2012;367(1590):850–859.
- Gutierrez MC, Brisse S, Brosch R, et al. Ancient origin and gene mosaicism of the progenitor of Mycobacterium tuberculosis. PLoS Pathog. 2005;1(1):e5.
- Wirth T, Hildebrand F, Allix-Beguec C, et al. Origin, spread and demography of the Mycobacterium tuberculosis complex. PLoS Pathog. 2008;4(9):e1000160.
- Barry CE III, Boshoff HI, Dartois V, et al. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Microbiol. 2009;7(12):845–855.
- Delogu G, Goletti D. The spectrum of tuberculosis infection: new perspectives in the era of biologics. J Rheumatol Suppl. 2014;91(0):11–16.
- Dorhoi A, Reece ST, Kaufmann SH. For better or for worse: the immune response against mycobacterium tuberculosis balances pathology and protection. Immunol Rev. 2011;240(1):235–251.
- Orme IM. A new unifying theory of the pathogenesis of tuberculosis. Tuberculosis (Edinb). 2014;94(1):8–14.
- Delogu G, Provvedi R, Sali M, et al. Mycobacterium tuberculosis virulence: insights and impact on vaccine development. Future Microbiol. 2015;10(7):1177–1194.
- Dulberger CL, Rubin EJ, Boutte CC. The mycobacterial cell envelope – a moving target. Nat Rev Microbiol. 2020;18(1):47–59.
- Vaziri F, Brosch R. ESX/Type VII secretion systems – an important way out for mycobacterial proteins. Microbiol Spectr. 2019;4:7. 10.1128/microbiolspec.PSIB-0029-2019.
- Ates LS. New insights into the mycobacterial PE and PPE proteins provide a framework for future research. Mol Microbiol. 2020;113(1):4–21.
- Delogu G, Brennan MJ, Manganelli R. PE and PPE genes: a tale of conservation and diversity. Adv Exp Med Biol. 2017;1019:191–207.
- Fishbein S, Van WN, Warren RM, et al. Phylogeny to function: PE/PPE protein evolution and impact on Mycobacterium tuberculosis pathogenicity. Mol Microbiol. 2015;96(5):901–916.
- De Maio F, Berisio R, Manganelli R, et al. PE_PGRS proteins of Mycobacterium tuberculosis: a specialized molecular task force at the forefront of host–pathogen interaction. Virulence. 2020;11(1):898–915.
- Brennan MJ. The enigmatic PE/PPE multigene family of mycobacteria and tuberculosis vaccination. Infect Immun. 2017;85.
- Brennan MJ, Delogu G. The PE multigene family: a ‘molecular mantra’ for mycobacteria. Trends Microbiol. 2002;10(5):246–249.
- Iantomasi R, Sali M, Cascioferro A, et al. PE_PGRS30 is required for the full virulence of Mycobacterium tuberculosis. Cell Microbiol. 2012;14(3):356–367.
- De Matteis A, De Camilli P. Phosphoinositides. Biochim.Biophys.Acta. 1851;697-818:2015.
- De Maio F, Battah B, Palmieri V, et al. PE_PGRS3 of Mycobacterium tuberculosis is specifically expressed at low phosphate concentration, and its arginine-rich C-terminal domain mediates adhesion and persistence in host tissues when expressed in Mycobacterium smegmatis. Cell Microbiol. 2018;20(12):e12952.
- Minerva M, De MF, Camassa S, et al. Evaluation of PE_PGRS33 as a potential surface target for humoral responses against Mycobacterium tuberculosis. Pathog Dis. 2017; (8):75. doi:10.1093/femspd/ftx100.
- Delogu G, Brennan MJ. Functional domains present in the Mycobacterial hemagglutinin, HBHA. J Bacteriol. 1999;181(24):7464–7469.
- Pethe K, Alonso S, Biet F, et al. The heparin-binding haemagglutinin of M. tuberculosis is required for extrapulmonary dissemination. Nature. 2001;412(6843):190–194.
- Jung ID, Jeong SK, Lee CM, et al. Enhanced efficacy of therapeutic cancer vaccines produced by co-treatment with Mycobacterium tuberculosis heparin-binding hemagglutinin, a novel TLR4 agonist. Cancer Res. 2011;71(8):2858–2870.
- Lei Y, Shao J, Ma F, et al. Enhanced efficacy of a multi-epitope vaccine for type A and O foot and-mouth disease virus by fusing multiple epitopes with mycobacterium tuberculosis heparin-binding hemagglutinin (HBHA), a novel TLR4 agonist. Mol Immunol. 2020;121:118–126.
- Raze D, Verwaerde C, Deloison G, et al. Heparin-binding hemagglutinin adhesin (HBHA) is involved in intracytosolic lipid inclusions formation in mycobacteria. Front Microbiol. 2018;9:2258.
- Delogu G, Bua A, Pusceddu C, et al. Expression and purification of recombinant methylated HBHA in Mycobacterium smegmatis. FEMS Microbiol Lett. 2004a;239(1):33–39 .
- Cascioferro A, Daleke MH, Ventura M, et al. Functional dissection of the PE domain responsible for translocation of PE_PGRS33 across the mycobacterial cell wall. PLoS ONE. 2011;6(11):e27713.
- Abdallah AM, Verboom T, Weerdenburg EM, et al. PPE and PE_PGRS proteins of Mycobacterium marinum are transported via the type VII secretion system ESX-5. Mol Microbiol. 2009;73(3):329–340.
- Bottai D, Di LM, Majlessi L, et al. Disruption of the ESX-5 system of Mycobacterium tuberculosis causes loss of PPE protein secretion, reduction of cell wall integrity and strong attenuation. Mol.Microbiol. 2021;83:1209. doi:10.1111/j.1365-2958.2012.08001.x
- Burggraaf MJ, Speer A, Meijers AS, et al. Type VII secretion substrates of pathogenic mycobacteria are processed by a surface protease. MBio. 2019;10.
- Camassa S, Palucci I, Iantomasi R, et al. Impact of pe_pgrs33 gene polymorphisms on Mycobacterium tuberculosis infection and pathogenesis. Front Cell Infect Microbiol. 2017;7:137.
- Palucci I, Camassa S, Cascioferro A, et al. PE_PGRS33 contributes to Mycobacterium tuberculosis entry in macrophages through Interaction with TLR2. PLoS ONE. 2016;11(3):e0150800.
- Ates LS, Sayes F, Frigui W, et al. RD5-mediated lack of PE_PGRS and PPE-MPTR export in BCG vaccine strains results in strong reduction of antigenic repertoire but little impact on protection. PLoS Pathog. 2018b;14(6):e1007139.
- Cascioferro A, Delogu G, Colone M, et al. PE is a functional domain responsible for protein translocation and localization on mycobacterial cell wall. Mol Microbiol. 2007;66(6):1536–1547.
- Delogu G, Pusceddu C, Bua A, et al. Rv1818c-encoded PE_PGRS protein of Mycobacterium tuberculosis is surface exposed and influences bacterial cell structure. Mol Microbiol. 2004b;(3): 725–733. 10.1111/j.1365-2958.2004.04007.x.
- Korb VC, Chuturgoon AA, Moodley D. Mycobacterium tuberculosis: manipulator of protective immunity. Int J Mol Sci. 2016;17(3):131.
- Russell DG, Cardona PJ, Kim MJ, et al. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat Immunol. 2009;10(9):943–948.
- Roque NR, Lage SL, Navarro R, et al. Rab7 controls lipid droplet-phagosome association during mycobacterial infection. Biochim Biophys Acta Mol Cell Biol Lipids. 2020;1865(8):158703.
- Shim D, Kim H, Shin SJ. Corrigendum: Mycobacterium tuberculosis infection-driven foamy macrophages and their implications in tuberculosis control as targets for host-directed therapy. Front Immunol. 2020a;11:1601.
- Shim D, Kim H, Shin SJ. Mycobacterium tuberculosis infection-driven foamy macrophages and their implications in tuberculosis control as targets for host-directed therapy. Front Immunol. 2020b;11:910.
- Tong J, Liu Q, Wu J, et al. Mycobacterium tuberculosis strains of the modern Beijing sublineage excessively accumulate triacylglycerols in vitro. Tuberculosis (Edinb). 2020;120:101892.
- Elliott SR, Tischler AD. Phosphate starvation: a novel signal that triggers ESX-5 secretion in Mycobacterium tuberculosis. Mol Microbiol. 2016;100(3):510–526 .
- Mileykovskaya E, Dowhan W. Cardiolipin membrane domains in prokaryotes and eukaryotes. Biochim Biophys Acta. 2009;1788(10):2084–2091.
- Rodgers SJ, Ferguson DT, Mitchell CA, et al. Regulation of PI3K effector signalling in cancer by the phosphoinositide phosphatases. Biosci Rep. 2017;37.
- Sciara G, Clarke OB, Tomasek D, et al. Structural basis for catalysis in a CDP-alcohol phosphotransferase. Nat Commun. 2014;5(1):4068.
- Choy CH, Han BK, Botelho RJ. Phosphoinositide diversity, distribution, and effector function: stepping out of the box. Bioessays. 2017;39.
- Palamiuc L, Ravi A, Emerling BM. Phosphoinositides in autophagy: current roles and future insights. Febs J. 2020;287(2):222–238.
- Daleke MH, Cascioferro A, De PK, et al. Conserved PE and PPE protein domains target LipY lipases of pathogenic mycobacteria to the cell surface via ESX-5. J Biol Chem. 2011(21). 10.1074/jbc.M110.204966.
- Bunduc CM, Ummels R, Bitter W, et al. Species-specific secretion of ESX-5 type VII substrates is determined by the linker 2 of EccC5. Mol Microbiol. 2020;(1). 10.1111/mmi.14496.
- Houben EN, Bestebroer J, Ummels R, et al. Composition of the type VII secretion system membrane complex. Mol Microbiol. 2012;86(2):472–484.
- Ates LS, Dippenaar A, Ummels R, et al. Mutations in ppe38 block PE_PGRS secretion and increase virulence of mycobacterium tuberculosis. Nat Microbiol. 2018a;3(2):181–188.
- Deng W, Long Q, Zeng J, et al. Mycobacterium tuberculosis PE_PGRS41 enhances the intracellular survival of M. smegmatis within macrophages via blocking innate immunity and inhibition of host defense. Sci Rep. 2017;7(1):46716.
- Saini NK, Baena A, Ng TW, et al. Suppression of autophagy and antigen presentation by Mycobacterium tuberculosis PE_PGRS47. Nat Microbiol. 2016;1(9):16133.
- De Maio F, Maulucci G, Minerva M, et al. Impact of protein domains on PE_PGRS30 polar localization in mycobacteria. PLoS ONE. 2014;9(11):e112482.
- Delogu G, Bua A, Pusceddu C, et al. Expression and purification of recombinant methylated HBHA in Mycobacterium smegmatis. FEMS Microbiol Lett. 2004;(1): 33–39. 10.1016/j.femsle.2004.08.016.
- Battah B, Chemi G, Butini S, et al. A repurposing approach for uncovering the anti-tubercular activity of FDA-approved drugs with potential multi-targeting profiles. Molecules. 2019; (23):24. 10.3390/molecules24234373.
- Drozdetskiy A, Cole C, Procter J, et al. JPred4: a protein secondary structure prediction server. Nucleic Acids Res. 2015;43(W1):W389–W394.
- Yang J, Yan R, Roy A, et al. The I-TASSER suite: protein structure and function prediction. Nat Methods. 2015;12(1):7–8.
- Huang CC, Meng EC, Morris JH, et al. Enhancing UCSF chimera through web services. Nucleic Acids Res. 2014;42(W1):W478–W484.