
ABSTRACT
The fungal pathogen Candida auris has emerged as a new threat to human health. We previously reported the first isolate of C. auris (BJCA001) in China, which belongs to the South Asian clade (I) and was susceptible to all antifungals tested. In this study, we report the isolation of a drug-resistant C. auris strain (BJCA002) from the same city (Beijing). Strain BJCA002 belongs to the South African clade (III) and is resistant to fluconazole and amphotericin B based on the tentative MIC breakpoints. Taking advantage of the two isolates with distinct antifungal susceptibility and genetic origins, we performed a biological and genomic comparative study. Besides antifungal susceptibility, strains BJCA001 and BJCA002 showed differences in multiple aspects including morphologies, expression of virulence factors, virulence, mating type, and genomic sequence and organization. Notably, strain BJCA002 was less virulent than BJCA001 in both the Galleria mellonella and mouse systemic infection models. Genomic analysis demonstrated that strain BJCA002 but not BJCA001 had multiple mutations in drug resistance-associated genes, including a hot-spot mutation of ERG11 (VF125AL, namely V125A and F126L) and some missense mutations in CDR1, MDR1, and TAC1. Notably, strain BJCA001 carried 64 copies of the Zorro3 retrotransposon, whereas BJCA002 had only 3 copies in the genome. Taken together, our findings not only reveal the genetic and phenotypic diversities of the two isolates from Beijing, China, but also shed new light on the genetic basis of the antifungal resistance and virulence of C. auris.
Introduction
Candida auris was first reported in Japan in 2009 and is becoming a global threat [Citation1–4]. As of December 2020, C. auris cases have been reported in at least 44 countries across six continents based on data from the Centers for Disease Control and Prevention (CDC) (https://www.cdc.gov/). C. auris has also caused several outbreaks in hospitals across the globe [Citation5–8]. In 2016 and 2017, the CDC released a couple of clinical alerts to healthcare facilities throughout the United States warning of the emergence of C. auris infections.
In addition to its rapid emergence worldwide, the multidrug resistance properties of C. auris make it particular problem in clinical settings. Four major genetic clades of C. auris have been reported [Citation1,Citation9]. Distinct genetic clades correspond to different geographic regions. Most clinical isolates of C. auris are resistant to fluconazole, and some are resistant to two or more classes of antifungal drugs [Citation9,Citation10]. Mutations in the ERG11 gene (e.g., Y132F, K143R, V125A, and F126L, the V125A and F126L mutations also commonly referred to as VF125AL or F126L), encoding lanosterol demethylase, have been found to be related with fluconazole resistance of C. auris. Moreover, these mutations were specifically associated with different genetic clades: V125A and F126L with the South Africa clade (III), Y132F with the South America clade (VI), and Y132F or K143R with the South Asia clade (I) [Citation9,Citation10]. We recently found that C. auris could develop fluconazole resistance through an adaptive mechanism of aneuploidy formation [Citation11].
In 2018, we reported the first isolate (BJCA001) of C. auris in Beijing, China from the bronchoalveolar lavage fluid (BALF) of a hospitalized woman [Citation12]. Interestingly, strain BJCA001 was found to be susceptible to all tested antifungals used in clinics. In this study, we report a fluconazole-resistant isolate of C. auris from a different hospital in Beijing. This strain was isolated from a newborn boy with blood infection. A comparative analysis demonstrated many biological and genomic differences between the newly isolated fluconazole-resistant strain and the previously reported fluconazole-susceptible strain BJCA001.
Materials and methods
Strains and culture conditions
C. auris strains were routinely grown in YPD medium (20 g/L peptone, 10 g/L yeast extract, 20 g/L glucose; for solid medium, 20 g/L agar was added). The red dye phloxine B (5 mg/L) was added to the solid medium for colony staining. For the morphological assays, approximately 40–80 cells were plated onto each plate (90 mm in diameter) and incubated at 25°C for seven days.
To evaluate the cell size of C. auris, cells were grown to the stationary phase in liquid YPD or YCB-BSA medium at 37°C and then inoculated into liquid YPD or YCB-BSA medium for 24 hours of growth at 25°C and 37°C. Both the cell width and length were examined.
Whole-genome sequencing
Yeast-from cells of C. auris were grown on YPD medium at 37°C for 16 hours. Genomic DNA was extracted using the TIANamp Yeast DNA Kit (TianGen Biotech, Beijing, China) according to the company’s protocol. Briefly, yeast-form cells (collected from 10 mL culture) was resuspended, treated with the lysis buffer, and incubated at 37°C for 40 min. The samples were gently mixed by brief vortexing and cell debris was spun down by centrifugation. The supernatant was transferred to the DNA binding column for centrifugation. Genomic DNA was then released with TE buffer. For genome sequencing, a paired-end library with an average insert size of 300 bp was prepared and sequenced using the Illumina NovaSeq platform. In addition, single-molecule real-time (SMRT) sequencing was performed by Beijing Novogene Bioinformatics Technology Co., Ltd. (Beijing, China) using the PacBio Biosciences (PacBio) Sequel small-molecule real-time sequencing system (Pacific Biosciences, Menlo Park, CA, USA). The sequence data generated from the Illumina platform were used to proofread the PacBio assembly sequence.
Genomic assembly
The PacBio sequence data were error-corrected, trimmed, assembled and scaffolded using Canu v1.8 software guided by a genome size of 12.5 Mb [Citation13]. Illumina PE reads were quality trimmed for random hexamer primers on the 5′ read end using Trimmomatic v0.22 [Citation14]. The high-quality reads were aligned to the Canu genome assembly using Bwa v0.7.17 [Citation15] and filtered for concordant PE read alignments using SAMTools v1.8 [Citation16]. The Canu genome assembly was additionally corrected with the high-quality Illumina alignments using Pilon v1.23 [Citation17] to generate a final polished sequence assembly with SNP and INDEL corrections.
Gene prediction
Based on the final assemblies, proteins and coding genes were predicted using the program AUGUSTUS v3.2.1 [Citation18]. Putative ORFs were searched against the NR database of NCBI (http://www.ftp.ncbi.nlm.nih.gov/blast/executable s/blast +/LATES T/) and Candida genome database (http://www.candidagenome.org/).
Whole genome synteny and divergence analysis
The final assemblies of strains BJCA001 and BJCA002 were aligned with the open-source MAUVE aligner version 2.3.1 using a progressive algorithm [Citation19]. The alignment was performed using a skip-refinement and seed-weight of 15 to avoid spurious alignments while other mandatory arguments were run on the program default.
To calculate the genomic divergence of different regions between the two isolates, we broke the assembly of BJCA001 into 5-kbp sliding windows with 1000-bp step. The SNPs of each window compared with the BJCA002 assembly were extracted with the open source software package MUMmer, version 4.4 [Citation20]. The average divergence for each chromosome and the whole genome were calculated based on the SNP density.
The duplication-removed BAM datasets were used to estimate the copy number variation (CNV) for each isolate. Genomic regions with CNV were identified with the Splint script to avoid the “smiley pattern” bias [Citation21]. The read depth by 1000-bp nonoverlapping windows across the genome was generated with default internal parameters.
Reference-based alignment, variant calling, and Phylogenetic analysis
A total of 24 samples (including BJCA001 and BJCA002) representing the five major clades of C. auris were analyzed. The DNA sequence information was retrieved from the NCBI SRA database and previous publications [Citation9,Citation22]. Raw reads were trimmed to remove low-quality (Phred score ≤10), ambiguous, and adaptor bases using the FASTX-Toolkit v0.0.14 (http://hannonlab.cshl.edu/fastx_toolkit/index.html). The clean reads obtained were mapped to the CDC_B8441 genome (https://www.ncbi.nlm.nih.gov/assembly/GCA_002759435.2/) using Bwa v0.7.17 [Citation15] with default settings. SAMTools v1.8 [Citation16] was employed to convert the alignment results into BAM format and Picard Tools v1.56 (http://picard.source-forge.net) was used to remove duplicated sequences. The SAMTools and Genome Analysis Toolkit (GATK v2.7.2) program [Citation23] were used to detect the SNPs and INDELs. For GATK, HaplotypeCaller was used and the ploidy was set as “1”. The parameters “stand_call_conf” (thresholds for low – and high-quality variation loci) and stand_emit_conf (minimum Phred-scaled confidence threshold) were set to 50.0 and 20.0, respectively. The variation sites with a coverage depth ≥20 were retained for the subsequent analyses and final extraction.
Phylogenetic trees were constructed based on the whole genome SNPs of the samples. RAxML v8.1.6 (1000 bootstrap replicates) with the GTR model of nucleotide substitution and γ-distributed rates among sites was used for the analysis [Citation24].
Minimal inhibitory concentration (MIC) assay
MIC assays were performed according to the CCLS document M27-A3 with slight modifications [Citation11,Citation25]. Briefly, C. auris cells of each strain were initially patched on nutrient agar plates at 35°C for 24 hours. Approximately 500 cells were inoculated into 0.2 mL RPMI 1640 medium (w/v, 1.04% RPMI 1640, 3.45% MOPs, using NaOH for pH adjustment to 7.0) in a 96-well plate for MIC testing. Different antifungals in a series of concentrations (from 0.016 to 128 μg/mL) were used. Cells were incubated at 35°C in air for 24 hours. Candida krusei ATCC 6258 and Candida parapsilosis ATCC 22,019 served as quality controls.
Secreted aspartyl proteinase (SAP) activity assay
SAP activity was determined using the YCB-BSA method as described previously [Citation26]. Briefly, cells of C. auris were initially grown on YPD medium at 30°C for 48 hours. Approximately 5 × 106 cells of each strain in 5 μL ddH2O were spotted onto YCB-BSA medium plates and cultured at 25°C, 30°C, or 37°C for seven days. The width of the BSA precipitation rings (halos), which reflect the level of SAP activity, was measured. Three biological repeats were evaluated.
Galleria mellonella infection model
Galleria mellonella in the final instar larval stage were purchased from Tianjin Huiyu biological technology Co. LTD. (Tianjin, China). Larvae with a similar size (0.3 g-0.4 g) were used for infection assays. C. auris cells were incubated at 30°C with shaking to an OD600 = 1.8. C. auris cells (1 x 106 in 10 µL 1 x PBS) were washed twice with 1 x PBS and injected into each larva. After injection, the larvae were placed in plastic culture dishes and incubated at 37°C or 25°C in the dark. The survival rates were determined based on the daily records.
Mouse systemic infection
All animal experiments were performed according to the guidelines approved by the Animal Care and Use Committee of the School of Life Sciences, Fudan University. Mouse systemic infection experiments were performed as described in our previous report [Citation12]. Briefly, five BALB/c female mice (5 weeks old, 13–16 g) were used for intravenous infection of each C. auris sample. Each mouse was injected with approximately 2 × 107 fungal cells in 250 μL 1 x PBS via the lateral tail vein. The mice were humanely killed at 24 hours post-infection. The brain, kidney, spleen, liver, and lung of each infected mouse were removed, weighed, and homogenized for determination of colony-forming unit (CFU) on YPD medium.
Results
Isolation of the fluconazole-resistant strain BJCA002 of C. auris in Beijing, China
A male infant was born premature at 33 weeks (gestational age) by natural labor at Beijing Hospital. He was admitted to the neonatal intensive care unit (NICU) and monitored for vital signs. Meropenem and vancomycin were prescribed to prevent bacterial infection. On day 2 after birth, fluconazole was used to prevent fungal infection with intermittent usage of 12 mg per 72 hours. After 12 days of admission, yeast species were isolated from blood cultures, although the β-D-glucan test was negative. The isolates were identified as C. auris by the MALDI-TOF MS platform (4.15 version, Bruker, Germany) and verified by molecular identification methods. The sequences of the internal transcribed spacers (ITS) showed 100% identity to some reported C. auris isolates [Citation9,Citation27]. This isolate was referred to as BJCA002.
Phylogeny analysis demonstrated that strain BJCA002 belonged to the South African clade (III, ), whereas the isolate from Beijing, China previously reported by our group belongs to the South Asian clade (I). Therefore, there are at least two distinct genetic clades of C. auris in Beijing. We then tested the antifungal susceptibility of BJCA002 to nine different drugs (). The minimum inhibitory concentrations (MICs) of fluconazole and amphotericin B of strain BJCA002 were 64 mg/L and 4 mg/L, respectively. These values were higher than the MIC breakpoints determined for other Candida species, implying that the strain is multidrug-resistant. The MICs of itraconazole, posaconazole, voriconazole, 5-flucytosine, caspofungin, micafungin, and anidulafungin of strain BJCA002 were equal to or less than 2 mg/L (). Taken together, compared with strain BJCA001, BJCA002 had relatively higher MICs for all the tested drugs, suggesting a reduced susceptibility to antifungals.
Table 1. Antifungal susceptibility testing of C. auris strains BJCA001 and BJCA002
Figure 1. Phylogenic analysis of two strains isolated from Beijing, China and representative isolates of major clades of C. auris. The two strains (BJCA001 and BJCA002) isolated from Beijing are highlighted in bold. The phylogenic tree was generated using the program RAxML v7.3.2 with 381,150 SNPs. The generalized time-reversible (GTR) model, a Gamma distribution, and 1000 bootstraps were used to construct the support of the phylogenetic relationships. Countries from which the isolates originated are indicated in brackets. CHN, China; USA, United States of America; GBR, United Kingdom; JPN, Japan; KOR, Korea (South); SGP, Singapore; ZAF, South Africa; DEU, Germany; PAK, Pakistan; IND, India; RUS, Russia; NLD, Netherlands; IRN, Iran; COL, Colombia; VEN, Venezuela. The scale bar represents the expected number of substitutions per nucleotide position
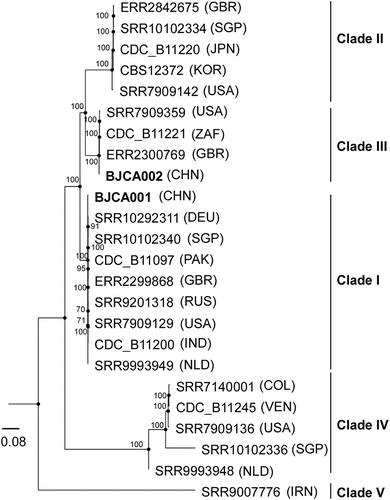
Comparison of the morphologies of strains BJCA002 and BJCA001
Given the differences in the genetic background and antifungal susceptibility between strains BJCA002 and BJCA001, we predicted that the two isolates could also differ in other biological features. Next, we examined the morphologies of the two isolates. As shown in Figure S1, the cell size of BJCA002 was much smaller than that of BJCA001 in both YCB-BSA and YPD media at 25°C and 37°C.
Filamentous growth is a key feature of virulence [Citation28]. We have previously demonstrated that BJCA001 is able to undergo filamentation [Citation29]. Here we found that this strain was also able to develop filaments on YPD medium at 25°C, although the morphologies of filamentous colonies of BJCA002 and BJCA001 appeared different in the presence of the red dye phloxine B (). Similar to BJCA001, the filamentation ability in BJCA002 was heritable. Once the cells became filamentous-competent, this ability was maintained for many generations.
Figure 2. Colony and cellular morphologies of strains BJCA002 (a) and BJCA001 (b). Yeast-form or filamentous cells of C. auris were plated on YPD medium and incubated at 25°C for 7 days. Scale bar, 10 μm
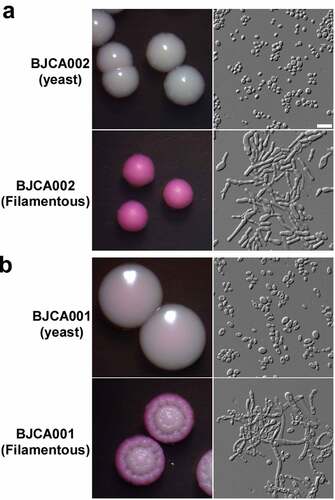
Strains BJCA002 and BJCA001 differ in the expression of SAPs
Secreted aspartic proteases (SAPs) are critical for virulence in pathogenic Candida species [Citation30]. We next examined SAP expression in the two C. auris strains using YCB-BSA medium. As shown in , the SAP levels of BJCA001 were comparable at all culture temperatures (25°C, 30°C, and 37°C), whereas the levels of BJCA002 were temperature-dependent. The latter strain secreted higher levels of SAPs at higher temperatures.
Figure 3. Comparison of SAP activities of C. auris strains BJCA001 and BJCA002. Approximately 5 × 106 yeast-form cells of BJCA002 (a) or BJCA001 (b) in 5 μL ddH2O were spotted onto YCB-BSA medium plates and then incubated at 25°C, 30°C, or 37°C for 7 days. The width of the white precipitation zones (halos) that indicate the level of SAP-mediated BSA hydrolysis activity are shown below the corresponding image (mm). Average values and standard deviations are presented (three repeats). Scale bar, 10 μm
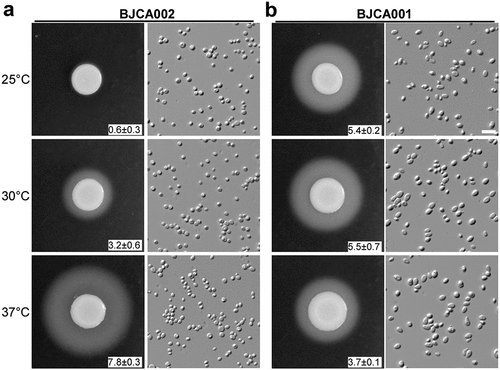
Strain BJCA002 is less virulent than BJCA001 in invertebrate animal and mouse infection models
Since the two C. auris strains differed in SAP secretion, we next compared their virulence using two animal models. In the G. mellonella invertebrate animal infection model, BJCA002 exhibited less virulence than BJCA001 at both 25°C and 37°C (). At 25°C and 37°C, strain BJCA001 (1 x 106 cells/insect) killed all the animals on days 5 and 3 post-infection, respectively. However, at both temperatures, 30% (25°C) or 40% (37°C) of the animals remained viable even at day 7 post-infection with strain BJCA002.
Figure 4. Virulence of C. auris strains BJCA001 and BJCA002 in a G. mellonella infection model. Yeast-form cells of C. auris were used. Approximately 1 × 106 C. auris cells of each strain were injected into each larva of G. mellonella. Ten larvae were injected for each strain. (a) Survival curves at 37°C. (b) Survival curves at 25°C
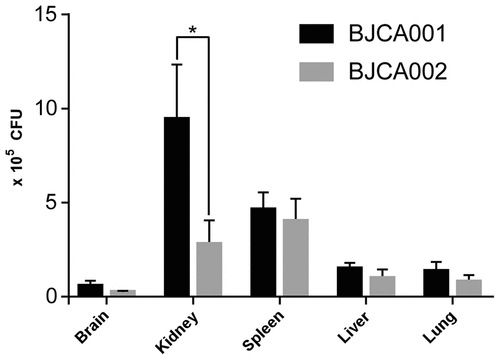
To further verify the virulence characteristics, we next performed fungal burden assays using the mouse systemic infection model (). The fungal burdens of the two C. auris strains were comparable in the brain, spleen, liver, and lung. However, the fungal burden of BJCA002 was significantly lower than that of BJCA001 in the mouse kidney. Taken together, strain BJCA002 was less virulent than BJCA001 in both animal models.
Figure 5. Virulence of C. auris strains BJCA001 and BJCA002 in a mouse systemic infection model. Fungal burden assays were performed. Five mice were used for infection of each strain. Each mouse was injected with approximately 2 × 107 fungal cells via the tail vein. Mice were killed for CFU assays at 24 h post-infection. *Indicates a significant difference (P value < 0.01, Student’s t-test, two-tailed)
Comparative analysis of the genomic characteristics of strains BJCA002 and BJCA001
To reveal the genetic basis of the biological differences between the two C. auris strains, we performed whole-genome sequencing assays using next-generation and third-generation sequencing instruments. Highly complete genome assemblies were generated for strains BJCA002 and BJCA001 ( and access numbers: CP068451-CP068458 for BJCA001 JAERMV000000000 for BJCA002; https://www.ncbi.nlm.nih.gov/bioproject/PRJNA691446). A summary of the genomic features of the two strains is presented in . The genome sizes of BJCA002 and BJCA001 were 12.39 Mb and 12.96 Mb, respectively. The genome assemblies were organized in 8 or 7 scaffolds based on a previous study [Citation17]. The divergence values of each corresponding chromosome of the two strains ranged from 0.260 to 0.789, and the average value was 0.394 for the whole genome (). As expected, the numbers of genes and introns, GC%, and sizes of intergenic regions were comparable between the two genomes. The SNPs and mutations are presented in supplementary Dataset S1.
Table 2. Genome assembly information of strains BJCA001 and BJCA002
Figure 6. Whole genome synteny and divergence. (a) Genome synteny between C. auris strains BJCA001 and BJCA002. Black blocks indicate shared syntenic regions of the two genomes, whereas gray blocks indicate large fragment recombination regions. Percentages of divergence for the two genomes and each corresponding pairs of chromosomes are also shown. Every point stands for the SNP density of a 5-kb window and 1-kb step size. (b) Examples of the Zorro3 retrotransposon cluster in strain BJCA001
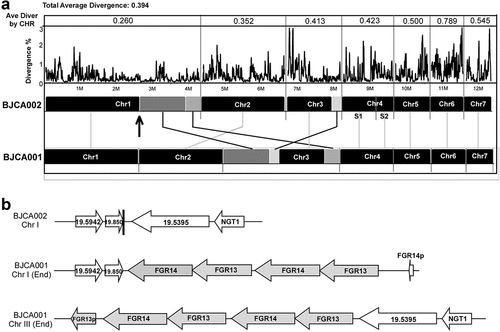
The assemblies of the BJCA002 and BJCA001 chromosomes showed a high similarity, and the two genomes were highly syntenic. However, there were some large chromosomal rearrangements (). For example, a large chromosomal rearrangement between chromosome 1 of strain BJCA002 and chromosome 3 of BJCA001 involved a translocation of two fragments of 1.26 Mb and 0.41 Mb, respectively. Moreover, a translocation of 139 kb was found in chromosome 3 of the two genomes (). This breaking region of strain BJCA001 could have resulted from the insert of a Zorro3 transposon in the chromosome.
Strains BJCA002 and BJCA001 differ in mating type
C. auris is a haploid species. Based on the genomic sequences, we analyzed the mating types of the two strains. BJCA002 was an MTLα strain, whereas BJCA001 was an MTLa strain. Consistently, BJCA002 was found to belong to the South African clade, among which all reported isolates carry an MTLα locus; strain BJCA001 belongs to the South Asian clade, among which all reported isolates carry an MTLa locus [Citation31].
Strains BJCA002 and BJCA001 differ in copy numbers of the non-LTR retrotransposon Zorro3
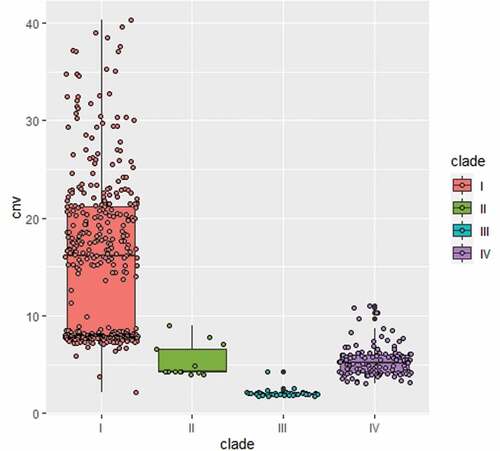
A conserved active non-LTR retrotransposon, Zorro3, was identified in C. auris. In fungi, the Zorro3 elements often include two genes, FGR14 and FGR13. FGR14 encodes a bifunctional enzyme with the activity of reverse transcriptase and endonuclease, whereas FGR13 encodes a zinc-finger protein with unknown function. Fgr14 has the predicted role in RNA-dependent DNA biosynthetic process, RNA binding, and RNA-directed DNA polymerase activity. Interestingly, we found that the genome of strain BJCA001 contained 64 copies of Zorro3, whereas BJCA002 had only 3 copies (). The increased genome size of strain BJCA001 could be due to the expansion of ZORRO3 retrotransposon elements. To further confirm the variation in copy numbers among the different clinical strains of C. auris, we analyzed 545 genomes available in the NCBI database. As shown in , isolates of the South Asian clade (I) generally had higher numbers of Zorro3, whereas isolates of the South African clade (III) had the lowest numbers. The mechanism and function of the expansion of this retrotransposon remains to be investigated.
Figure 7. Variation of the copy numbers of the Zorro3 retrotransposon element FGR14 in C. auris isolates of the four major clades. Each dot denotes an isolate, and 545 strains were analyzed. The genomic sequences used for analysis were from the NCBI database
Mutations of drug-resistance-associated genes
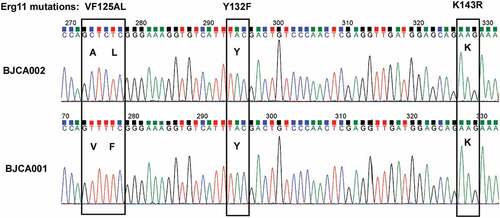
SNP analysis revealed several missense mutations in BJCA002 (Dataset S1). For example, four missense mutations (G71C, T46S, L279I, and L431W) were found in CDR4, which encodes a putative ABC transporter superfamily and could be related to fluconazole resistance in C. albicans and C. auris [Citation32,Citation33]. Two missense mutations (L286F and V428I) were found in MDR1 in BJCA002, which encodes an multidrug efflux pump [Citation34]. Mutations were also found in TAC1 and MRR1. Tac1 and Mrr1 could play a role in the transcriptional regulation of MDR1 and CDR genes. Most importantly, two missense mutations (V125A and F126L) were found in the lanosterol 14-α-demethylase-encoding gene ERG11 (), which could be directly attributed to the fluconazole resistance of strain BJCA002. Taken together, these mutations could provide an explanation for the different antifungal susceptibilities between strains BJCA001 and BJCA002.
Figure 8. Analysis of hot-spot mutations in ERG11. Strain BJCA001 carries no mutations in the three reported hot-spot regions, whereas BJCA002 has a VF125AL mutation (V125A and F126L)
Discussion
We have previously reported the first isolate of C. auris (BJCA001) in Beijing, China. Strain BJCA001 was susceptible to all the antifungals tested based on the tentative MIC breakpoints [Citation12]. In the current study, we report a C. auris isolate (BJCA002) from the same city that is resistant to fluconazole (MIC = 64 mg/L) and amphotericin B (MIC = 4 mg/L). To explore the diverse characteristics of natural isolates of C. auris, we performed a comparative study using the two strains (BJCA001 and BJCA002). We found several distinct biological and genetic features between the two strains, which could contribute to their differences in pathogenesis and antifungal resistance.
Strain BJCA001 belongs to the South Asian clade (I) and contains an MTLa locus, whereas strain BJCA002 belong to the South African clade (III) and contains an MTLα locus. C. auris isolates of these two distinct lineages have been shown to significantly differ in biological phenotypes and antifungal susceptibility [Citation35]. Here we provide new biological and genetic evidence to reveal the diversity of clinical isolates. For example, strains BJCA001 and BJCA002 differed in cell size and SAP expression, which could be directly associated with virulence. SAPs represent a major virulence factor of pathogenic Candida species [Citation30]. Strain BJCA002 expressed a relatively lower level of SAPs at 25°C and 30°C (), which might benefit it as a commensal on the skin of the human host. However, both strains were able to develop filaments under certain culture conditions. It remains to be investigated whether this morphological change is also related to pathogenesis in C. auris as in other Candida species.
To uncover the genetic basis of the difference in antifungal resistance of strains BJCA001 and BJCA002, we sequenced and assembled their genomes ( and Dataset S1). A couple of chromosome rearrangements were found in the two genomes, which could affect the expression of virulence – and antifungal resistance-associated genes. SNP analysis revealed that the fluconazole-resistant strain BJCA002 carried a number of mutations in potential antifungal resistance-associated genes, including ERG11, CDR1, MDR1, TAC1, and MRR1. Notably, a mutation of ERG11 (VF125AL, namely V125A and F126L) in BJCA002 has been previously reported as a hot-spot associated with fluconazole resistance [Citation9,Citation31]. MDR1 encodes a plasma membrane multidrug efflux pump of the major facilitator superfamily (MFS) that confers resistance to multiple antifungal drugs in C. albicans. In clinical isolates of C. albicans, a constitutive activation of the MDR1 gene was often associated with fluconazole resistance and disruption of this gene led to increased susceptibility to fluconazole [Citation36]. Given the close relationship between C. albicans and C. auris, one would expect that mutations of MDR1 in this study contribute to the fluconazole resistance of C. auris.
We identified an active Zorro3 retrotransposon in the two genomes. A striking difference between the two strains was the presence of 64 copies of the retrotransposon in BJCA001, in contrast to only 3 copies in BJCA002. An analysis of the data available in the NCBI database revealed that C. auris isolates of the South Asian clade contain much a greater higher copy number of the Zorro3 retrotransposon, whereas isolates of the South African and the other two clades often have a lower copy number. Some Zorro3 insertions are close to antifungal resistance-associated genes (Dataset S2), which may affect the expression of these genes and thus regulate antifungal susceptibility. Interestingly, the transposition of the Zorro2 retrotransposon is activated by miconazole, suggesting that these retrotransposons could be associated with antifungal resistance [Citation37]. In C. albicans, the Zorro3 element FGR14 also regulates filamentous growth [Citation38]. However, how the retrotransposon directly regulates antifungal resistance and morphological changes remains to be investigated. For example, what are the biological effects of the high level expression of the retrotransposon elements FGR14 and FGR13? Does the retrotransposon play a role in the rapid evolution and emergence of this fungal pathogen?
Taken together, our comparative study may not reflect the general differences between isolates of the South Asian and South African clades, but it provides new insights into the diversity of natural strains of C. auris. Our study demonstrated the existence of at least two different genetic clades of C. auris in Beijing, China. Biological and genomic analyses indicated that they differed in a number of aspects, including the cell phenotype, SAP secretion, virulence, antifungal resistance, and genetic basis. These variations may benefit the survival of the species in distinct ecological niches.
Supplemental Material
Download Zip (1.4 MB)Acknowledgments
We thank all the members of the Huang lab for insightful discussions concerning the topic of this manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- Du H, Bing J, Hu T, et al. Candida auris: epidemiology, biology, antifungal resistance, and virulence. PLoS Pathog. 2020;16(10):e1008921. Epub 2020/ 10/23. PubMed PMID: 33091071; PubMed Central PMCID: PMCPMC7581363 inhibitors and diagnostics of biofilms.
- Jeffery-Smith A, Taori SK, Schelenz S, et al. Candida auris: a review of the literature. Clin Microbiol Rev. 2018;31(1). PubMed PMID: 29142078; PubMed Central PMCID: PMCPMC5740969. DOI:10.1128/CMR.00029-17
- Chowdhary A, Sharma C, Meis JF. Candida auris: a rapidly emerging cause of hospital-acquired multidrug-resistant fungal infections globally. PubMed PMID: 28542486; PubMed Central PMCID: PMCPMC5436850 PLoS Pathog. 2017;13(5):e1006290.
- Satoh K, Makimura K, Hasumi Y, et al. Candida auris sp. nov., a novel ascomycetous yeast isolated from the external ear canal of an inpatient in a Japanese hospital. PubMed PMID: 19161556 Microbiol Immunol. 2009;53(1):41–44.
- Taori SK, Khonyongwa K, Hayden I, et al. Candida auris outbreak: mortality, interventions and cost of sustaining control. J Infection. 2019;79(6):601–611. PubMed PMID: WOS: 000500934300013.
- Ruiz-Gaitan A, Moret AM, Tasias-Pitarch M, et al. An outbreak due to Candida auris with prolonged colonisation and candidaemia in a tertiary care European hospital. Mycoses. 2018;61(7):498–505. PubMed PMID: WOS: 000436407500010.
- Eyre DW, Sheppard AE, Madder H, et al. A candida auris outbreak and its control in an intensive care setting. N Engl J Med. 2018;379(14):1322–1331. Epub 2018/ 10/04. PubMed PMID: 30281988.
- Schelenz S, Hagen F, Rhodes JL, et al. First hospital outbreak of the globally emerging Candida auris in a European hospital. Antimicrob Resist Infect Control. 2016;5(1):35. PubMed PMID: 27777756; PubMed Central PMCID: PMCPMC5069812.
- Lockhart SR, Etienne KA, Vallabhaneni S, et al. Simultaneous emergence of multidrug-resistant candida auris on 3 continents confirmed by whole-genome sequencing and epidemiological analyses. Clin Infect Dis. 2017;64(2):134–140. PubMed PMID: 27988485; PubMed Central PMCID: PMCPMC5215215.
- Chowdhary A, Anil Kumar V, Sharma C, et al. Multidrug-resistant endemic clonal strain of Candida auris in India. Eur J Clin Microbiol Infect Dis. 2014;33(6):919–926. PubMed PMID: 24357342.
- Bing J, Hu T, Zheng Q, et al. Experimental evolution identifies adaptive aneuploidy as a mechanism of fluconazole resistance in Candida auris. Antimicrob Agents Chemother. 2020;65(1):e01466–20. Epub 2020/ 10/21. PubMed PMID: 33077664.
- Wang X, Bing J, Zheng Q, et al. The first isolate of Candida auris in China: clinical and biological aspects. Emerg Microbes Infect. 2018;7(1):93. PubMed PMID: 29777096; PubMed Central PMCID: PMCPMC5959928.
- Koren S, Walenz BP, Berlin K, et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. PubMed PMID: 28298431; PubMed Central PMCID: PMC5411767 Genome Res. 2017;27(5):722–736.
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. PubMed PMID: 24695404; PubMed Central PMCID: PMC4103590 Bioinformatics. 2014;30(15):2114–2120.
- Li H, Durbin R. Fast and accurate short read alignment with burrows-wheeler transform. PubMed PMID: 19451168; PubMed Central PMCID: PMC2705234 Bioinformatics. 2009;25(14):1754–1760.
- Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. PubMed PMID: 19505943; PubMed Central PMCID: PMC2723002.
- Walker BJ, Abeel T, Shea T, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9(11):e112963. PubMed PMID: 25409509; PubMed Central PMCID: PMC4237348.
- Stanke M, Steinkamp R, Waack S, et al. AUGUSTUS: a web server for gene finding in eukaryotes. PubMed PMID: WOS: 000222273100062 Nucleic Acids Res. 2004;32(Web Server):W309–W12.
- Darling AC, Mau B, Blattner FR, et al. Mauve: multiple alignment of conserved genomic sequence with rearrangements. PubMed PMID: 15231754; PubMed Central PMCID: PMC442156 Genome Res. 2004;14(7):1394–1403.
- Kurtz S, Phillippy A, Delcher AL, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12. PubMed PMID: 14759262; PubMed Central PMCID: PMC395750.
- Gallone B, Steensels J, Prahl T, et al. Domestication and divergence of saccharomyces cerevisiae beer yeasts. Cell. 2016;166(6):1397–410 e16. PubMed PMID: 27610566; PubMed Central PMCID: PMC5018251.
- Lee WG, Shin JH, Uh Y, et al. First three reported cases of nosocomial fungemia caused by Candida auris. J Clin Microbiol. 2011;49(9):3139–3142. PubMed PMID: 21715586; PubMed Central PMCID: PMCPMC3165631.
- McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. PubMed PMID: 20644199; PubMed Central PMCID: PMC2928508.
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. PubMed PMID: 16928733 Bioinformatics. 2006;22(21):2688–2690.
- Arendrup MC, Prakash A, Meletiadis J, et al. Comparison of EUCAST and CLSI reference microdilution MICs of eight antifungal compounds for candida auris and associated tentative epidemiological cutoff values. Antimicrob Agents Ch. 2017;61(6):e00485–17. ARTN PubMed PMID: WOS: 000403783400005.
- Tao L, Du H, Guan GB, et al. Discovery of a “white-gray-opaque” tristable phenotypic switching system in candida albicans: roles of non-genetic diversity in host adaptation. PLoS Biol. 2014;12(4):e1001830. ARTN . . PubMed PMID: WOS: 000335458800019.
- Tian S, Rong C, Nian H, et al. First cases and risk factors of super yeast Candida auris infection or colonization from Shenyang, China. Emerg Microbes Infect. 2018;7(1):128. Epub 2018/ 07/12. PubMed PMID: 29992959; PubMed Central PMCID: PMCPMC6041263.
- Whiteway M, Bachewich C. Morphogenesis in Candida albicans. Annu Rev Microbiol. 2007;61(1):529–553. Epub 2007/ 05/18. PubMed PMID: 17506678.
- Yue H, Bing J, Zheng Q, et al. Filamentation in Candida auris, an emerging fungal pathogen of humans: passage through the mammalian body induces a heritable phenotypic switch. Emerg Microbes Infect. 2018;7(1):188. Epub 2018/ 11/30. PubMed PMID: 30482894; PubMed Central PMCID: PMCPMC6258701.
- Naglik JR, Challacombe SJ, Hube B. Candida albicans secreted aspartyl proteinases in virulence and pathogenesis. Microbiol Mol Biol Rev. 2003;67(3): 400–428. table of contents. PubMed PMID: 12966142; PubMed Central PMCID: PMC193873.
- Munoz JF, Gade L, Chow NA, et al. Genomic insights into multidrug-resistance, mating and virulence in Candida auris and related emerging species. Nat Commun. 2018;9(1):5346. Epub 2018/ 12/19. PubMed PMID: 30559369; PubMed Central PMCID: PMCPMC6297351.
- Coste AT, Karababa M, Ischer F, et al. TAC1, transcriptional activator of CDR genes, is a new transcription factor involved in the regulation of candida albicans ABC transporters CDR1 and CDR2. PubMed PMID: WOS: 000225824400027 Eukaryot Cell. 2004;3(6):1639–1652.
- Rybak JM, Munoz JF, Barker KS, et al. Mutations in TAC1B: a novel genetic determinant of clinical fluconazole resistance in candida auris. mBio. 2020;11(3) Epub 2020/ 05/14. doi: 10.1128/mBio.00365-20. PubMed PMID: 32398311; PubMed Central PMCID: PMCPMC7218281. . DOI:
- Wirsching S, Michel S, Morschhauser J. Targeted gene disruption in Candida albicans wild-type strains: the role of the MDR1 gene in fluconazole resistance of clinical Candida albicans isolates. PubMed PMID: WOS: 000087647000008 Mol Microbiol. 2000;36(4):856–865.
- Szekely A, Borman AM, Johnson EM. Candida auris isolates of the southern asian and south african lineages exhibit different phenotypic and antifungal susceptibility profiles in vitro. J Clin Microbiol. 2019; Epub 2019/ 03/15. doi: 10.1128/JCM.02055-18. PubMed PMID: 30867237; PubMed Central PMCID: PMCPMC6498034; 57(5). doi: . .
- Wirsching S, Michel S, Morschhauser J. Targeted gene disruption in Candida albicans wild-type strains: the role of the MDR1 gene in fluconazole resistance of clinical Candida albicans isolates. Mol Microbiol. 2000;36(4):856–865. Epub 2000/ 06/09. PubMed PMID: 10844673.
- Zhu CX, Yan L, Wang XJ, et al. Transposition of the Zorro2 retrotransposon is activated by miconazole in Candida albicans. Biol Pharm Bull. 2014;37(1):37–43. Epub 2014/ 01/07. PubMed PMID: 24389479.
- Uhl MA, Biery M, Craig N, et al. Haploinsufficiency-based large-scale forward genetic analysis of filamentous growth in the diploid human fungal pathogen C.albicans. EMBO J. 2003;22(11):2668–2678. Epub 2003/ 05/30. PubMed PMID: 12773383; PubMed Central PMCID: PMCPMC156753.