
ABSTRACT
Surra, one of the most important animal diseases with economic consequences in Asia and South America, is caused by Trypanosoma evansi. However, the mechanism of immune evasion by T. evansi has not been extensively studied. In the present study, T. evansi extracellular vesicles (TeEVs) were characterized and the role of TeEVs in T. evansi infection were examined. The results showed that T. evansi and TeEVs could activate TLR2-AKT pathway to inhibit the secretions of IL-12p40, IL-6, and TNF-α in mouse BMDMs. TLR2−/- mice and mice with a blocked AKT pathway were more resistant to T. evansi infection than wild type (WT) mice, with a significantly lower infection rate, longer survival time and less parasite load, as well as an increased secretion level of IL-12p40 and IFN-γ. Kinetoplastid membrane protein-11 (KMP-11) of TeEVs could activate AKT pathway and inhibit the productions of IL-12p40, TNF-α, and IL-6 in vitro. TeEVs and KMP-11 could inhibit the productions of IL-12p40 and IFN-γ, promote T. evansi proliferation and shorten the survival time of infected mice in vivo. In conclusion, T. evansi could escape host immune response through inhibiting the productions of inflammatory cytokines via secreting TeEVs to activate TLR2-AKT pathway. KMP-11 in TeEVs was involved in promoting T. evansi infection.
Extracellular vesicles (EVs) secreted by Trypanosoma evansi (T. evansi) activate the TLR2-AKT signaling pathway to inhibit the production of inflammatory cytokines, thereby escaping the host’s immune response. Kinetoplastid membrane protein-11 (KMP-11) in EVs is related to the promotion of T.evansi infection via AKT pathway.
Graphical abstract
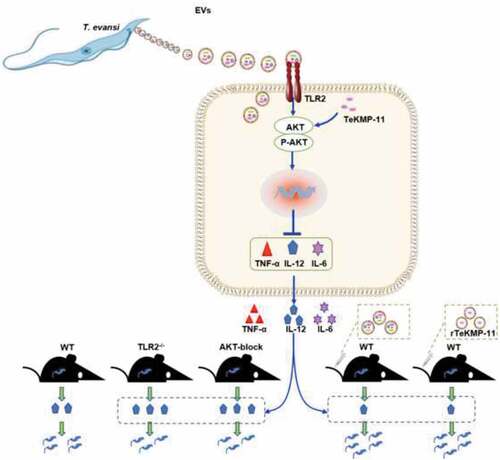
Introduction
Trypanosoma evansi is bloodborne protozoa that cause disease in several species and transmitted by hematophagous flies and vampire bats to cause surra, a serious disease of economic importance throughout the world [Citation1]. Trypanosoma evansi could infect most mammals exhibiting different clinical characteristics [Citation2,Citation3]. In camels, horses, and dogs, the parasite causes acute infection causing high fever, weight loss, anemia, genital inflammation, neurological symptoms, and eventually death [Citation4]. On the contrary, in cattle and buffalo, the parasite causes chronic infection and produces symptoms of emaciation, anorexia, anemia, and decreased productivity [Citation1,Citation3]. Wild mammals, such as puma and feral pigs, can also be infected with T. evansi [Citation5,Citation6]. T. evansi caused diseases are widely distributed in South America, Asia, Africa, and other regions, causing significant economic losses [Citation7]. Since 2005, several cases of human infection by T. evansi have been reported [Citation8–10], suggesting the potential of this parasite to be considered as zoonotic [Citation11]. T. evansi parasitizes in the host’s blood and is fully exposed to the host’s immune surveillance. However, T. evansi could evade host-specific immune response by constantly changing variable surface glycoproteins (VSGs) [Citation3]. Suramin is the drug that has most frequently been used for the treatment of surra in horses [Citation12]. There are no vaccines for the prevention of surra. Further understanding of the mechanism of host immune evasion by T. evansi will help us to explore new strategies for T. evansi control.
Innate immune responses are the first line of defense system developed early in animal evolution [Citation13]. Pathogen-associated molecular patterns (PAMPs) were recognized by Toll-like receptors (TLRs) and activate a series of signaling pathways to produce effector molecules, which ultimately regulate the host immune response [Citation14,Citation15]. TLRs are expressed by immune cells, like macrophages, dendritic cells (DCs), and neutrophils [Citation16]. Macrophages resist parasitic infection through phagocytosis, antigen presentation, and activation of other immune cells [Citation17]. TLRs play a double-edged sword role in parasitic infections. On the one hand, several parasitic protozoa could trigger TLR signaling pathway to protect host from parasite infection. TLR4 could recognize glycoinositolphospholipids (GIPLs) of T. cruzi and promote inflammatory response to resist T. cruzi infection [Citation18]. TLR2 of human NK cells could be activated by Liposphoglycan of Leishmania to release different mediators, such as ROS, inflammatory cytokines, and NO, which in turn help to resist Leishmania infection [Citation19]. On the other hand, TLRs could also play a negative role to facilitate parasitic infections. The lipophosphoglycan of Leishmania could interact with TLR2 to reduce the expression of TLR9 to inhibit the host immune response [Citation20]. TLR2−/- mice infected with T. cruzi could produce more inflammatory cytokines [Citation21]. Leishmania RNA virus 1 was recognized by mouse TLR3 to induce the production of inflammatory cytokines, which, however, made mice more susceptible to Leishmania infection [Citation22]. Nevertheless, the role of TLRs in T. evansi infection was still poorly understood.
EVs are vesicles with membrane structure released by cells [Citation23], which carried a large number of active molecular substances, including protein, lipid, RNA, miRNA, and other regulatory molecules [Citation24]. After pathogen infection, the host could secrete extracellular vesicles containing pathogen components to transmit signals to other cells [Citation25]. At the same time, pathogens such as bacteria [Citation26], fungi [Citation27] and parasites [Citation28–30] could also secrete EVs to regulate the host’s immune response. The reticulocyte-derived exosomes infected with Plasmodium yoelii carry P. yoelii antigen and participate in immune regulation to protect mice from lethal infections [Citation31]. EVs secreted by Leishmania contain a variety of virulence factors and non-coding RNAs. These virulence factors play a vital role in promoting the growth of Leishmania [Citation24]. Trichomonas vaginalis with high adhesion could increase the adhesion of low adhesion parasites to host epithelial cells by transferring exosomes to low adhesion parasites. T. vaginalis exosomes could down regulate IL-8 production in Ect cells, which is conducive to the establishment of chronic infection by T. vaginalis [Citation32]. Neospora caninum EVs could activate the TLR2/MAPK signaling pathway of mouse macrophages to regulate the production of inflammatory cytokines [Citation33]. However, whether T. evansi could release EVs to regulate host immune responses in T. evansi infection has not been reported.
To investigate the roles of TLRs and TeEVs in T. evansi infection, TLR activation and downstream signal pathways induced by T. evansi were determined. TeEVs were isolated and characterized. TeKMP-11 was screened by proteomic analysis from TeEVs. In vitro, the activation of TLR2-ERK/AKT pathways and the cytokines secretion induced by T. evansi, TeEVs, and rTeKMP-11 were examined. In T. evansi infected WT, TLR2−/-, TLR3−/-, TLR4−/- and AKT-blocked mice, the infection rate, survival time, parasite load, cytokines secretion, and pathological changes were analyzed. Furthermore, the functions of EVs and rTeKMP-11 in T. evansi infection were examined in vivo. Our results revealed that T. evansi evaded the host immune system by releasing EVs to activate TLR2-AKT signaling pathways.
Materials and methods
Animals
TLR2−/-, TLR4−/-, and TLR3−/- mice were purchased from the Model Animal Research Center of Nanjing University. WT C57BL/6 J mice were purchased from Changsheng Experimental Animal Center in China.
Parasites and cells
Trypanosoma evansi were kept frozen in our laboratory. Mice were infected with 2 × 106 T. evansi by intraperitoneal injection. When the number of T. evansi in the 1 ml peripheral blood reached 108, anticoagulated blood was collected and added an equal volume of phosphate buffered solution containing 1% glucose (PBSG) and centrifuged at 1,000 g for 5 min. The parasites were distributed at the junction of plasma and blood cells. Finally, T. evansi was purified by DE52 resin (Solarbio, China) [Citation34]. Briefly, the collected liquid sample containing blood cells and T. evansi was loaded onto a column containing DE52 resin and eluted with PBSG. The effluent was collected and centrifuged at 1,000 g for 5 min, then the cell pellet was resuspended with 2 mL RPIM-1640 and counted via a hemocytometer.
Bone marrow-derived macrophages (BMDMs) were isolated as antecedently described [Citation35]. Euthanized mice were immersed in 75% alcohol, the marrow was washed out with RPMI-1640 from the femurs and tibiae. Subsequently, the cells were centrifuged at 300 g for 5 min. Finally, the cells were cultured in RPMI-1640 containing 25% L929-cell conditioned medium, 10% FBS, 1% penicillin and streptomycin (P/S) at 37°C for 7 days. On the seventh day, the cells were resuspended with 200 μL APC anti-mouse/human CD11b (Biolegend, UK) diluted with 1% BSA (1:80) for 2 h, then washed three times and resuspended in 200-μL PBS. The cells were analyzed by flow cytometry.
EVs isolation from T. evansi
EVs were purified with a modified protocol [Citation36]. Briefly, the purified T. evansi (107 cells/mL) was cultured in RPMI-1640 without FBS or P/S for 6 h, then the whole culture was centrifuged at 2,000 g for 10 min to remove parasites and debris. The obtained supernatants were centrifuged at 10,000 g for 45 min and filtration using filter membrane (0.22 µm), then ultracentrifugation at 100,000 g for 70 min. The collected EVs were washed with ice-cold PBS and resuspended in 200 µL PBS. The collected EVs were added to equal volume of RIPA lysate and placed on ice for 40 min. The BCA assay kit (Thermo Scientific, USA) was used to detect the EVs protein concentration. The particle size of TeEVs was ensured by nanoparticle tracking analysis (NTA) with ZetaView PMX 110 (Particle Metrix, Germany).
Scanning and transmission electron microscopy observation for TeEVs
TeEVs were applied onto a copper grid with carbon and 3% phosphotungsticacid to negatively stain the grid. The grid was observed using TEM (HITACHI, Japan).
2 × 107 T. evansi were centrifuged at 1,000 g for 5 min and the precipitate was fixed overnight (0.1 M sodium cacodylate buffer contain 2.5% glutaraldehyde). The samples were then fixed with 1% OsO4, dehydrated with ethanol, and embedded in epoxy resin block. Ultrathin section was cut from the block and observed by TEM.
2 × 105 T. evansi were plated into 24-well plates loaded with poly-l-lysine-treated (1 mg/mL, Sigma-Aldrich, Germany) coverslips. The cells were fixed with 4% glutaraldehyde for 24 h then washed and fixed in 1.0% osmium tetroxide. Next, the fixed sample was dehydrated in gradient ethanol solutions, then placed in tertiary butyl alcohol, frozen at −20°C, and finally sputtered with gold. The samples were visualized by SEM (Hitachi S-3400 N, Japan).
DiO-labeled TeEVs
TeEVs were labeled and purified with DiO-Membrane EVs Labeling & Purification Kit (Rengen Biosciences, China) according to the manufacturer’s instructions. BMDMs were cultured with RPMI-1640 containing 10% FBS and 1% P/S in 24-well plates with glass coverslips and stimulated with the labeled EVs at 37°C. After 2 h treatment, the cells were washed thrice and fixed in 4% paraformaldehyde for 10 min. The cell nucleus was counterstained with DAPI stain, and samples were analyzed with a Laser Scanning Confocal microscope (Olympus FV1000, Japan).
Proteomic analysis of TeEVs
LC-MS/MS was used to analyze the protein composition of TeEVs (Shanghai Omicsspace Biotech Co., Ltd, Shanghai, China). The built-in software Mascot’s Proteome Discoverer 2.1 (Thermo Scientific, USA) was used for library search identification and quantitative analysis. The EVs proteome was analyzed using TriTryp DB and Uniprot. HSP 70, EF-1α, Aldolase, and KMP-11 were selected from EVs proteome to be expressed and purified, and the recombinant proteins were used as antigens for polyclonal antibody preparation. Proteomic results were verified by Western blot with aforementioned antibodies.
Cloning, expression, and purification of rTeKMP-11
RNA of T. evansi was extracted using the Trizol reagent (Thermo Fisher Scientific, USA). KMP-11 sequence of T. evansi was obtained from the TriTrypDB (http://tritrypdb.org/tritrypdb/app, TevSTIB805.9.9950) and amplified by PCR using the primers: KMP-11-F (5ʹ-CGCGGATCCGCGATGGCCACCACATACGAAGA-3ʹ) and KMP-11-R (5ʹ-CCCAAGCTTGGGTCATTTTCCGGGGAACTGGGC-3ʹ). The amplified KMP-11 was ligated into pET-32a vector and expressed in E. coli Rosetta DE3 strain (TIANGEN, Beijing, China). The His-Tagged Protein Purification Kit (CWBIO, China) was used to purify the rTeKMP-11 protein. The tag was cut with recombinant bovine enterokinase (Sangon Biotech, Shanghai, China) and removed with the His-Tagged Protein Purification Kit. The protein was dialyzed against PBS at 4°C for at least 5 h, and the buffer solution was changed twice. Endotoxin was removed using Triton X-114 [Citation37]. The endotoxin level was screened by LAL analysis (GenScript, Piscataway, NJ, United States).
Immunofluorescence
2 × 105 T. evansi were plated into 24-well cell culture plates loaded with poly-l-lysine-treated coverslips. BMDMs were stimulated with EVs and plated into 24-well cell culture plates. The samples were fixed in 4% glutaraldehyde, and permeabilized using 0.25% Triton X-100. Then, the samples were blocked in 3% BSA and incubated with the anti-KMP-11 antibody (1:100) overnight at 4°C, washed three times and incubated with goat anti-mouse Ig-G conjugated with FITC (Proteintech, Wuhan, China) for 1 h. Nuclear were stained with DAPI. The samples were visualized by a Laser Scanning Confocal microscope.
Mice infection
TLR2−/-, TLR3−/-, TLR4−/-, AKT-blocked WT and WT mice (n = 10 per group) were infected with 1 × 104 T. evansi by intraperitoneal injection. MK-2206 (120 mg/kg, every 2 days by p.o., Selleck, USA) was used to block the AKT pathway in vivo [Citation38]. Blood samples were collected from tail vein every day to detect the parasite load, and the serum was collected for cytokines detection. The time of death was recorded. The spleen and lung of mice were harvested on the fifth day post infection (dpi) for HE-staining.
To explore the role of EVs in T. evansi infection, mice (n = 10 per group) were infected with 1 × 104 T. evansi by intraperitoneal injection. Meanwhile, 50 μg/mL EVs were injected intravenously (tail vein) into the mice for three consecutive days. In control group, an equal volume of sterile PBS was injected in mice.
To understand the role of rTeKMP-11 protein in the pathogenesis of T. evansi, mice (n = 10 mice per group) were pretreated with 50 μg/mL rTeKMP-11 protein or the empty carrier protein (ECP) coated with 100 μg DOTAP liposome (final volume 120 μL) using DOTAP Liposomal Transfer Reagent (Roche, Indianapolis, IN) by intravenously injection into tail vein for three consecutive days. DOTAP was used as the negative control. Meanwhile, mice were infected with 1 × 104 T. evansi by intraperitoneal injection. Blood samples from all mice treated with EVs, rTeKMP-11 protein, ECP, and DOTAP control were collected from tail vein every day to detect the parasites load, and the serum was collected for cytokines detection. The mice were monitored after infection, and the times of death time for the mice were recorded.
Real-time quantitative PCR analysis
3 × 106 WT mouse BMDMs were stimulated with 3 × 106 T. evansi or 50 μg/mL TeEVs for 4 h, respectively. The RNA of cells was extracted and reverse-transcribed to cDNA. RT-qPCR was performed at 95°C for 10 min, followed by 40 cycles of 95°C for 30 s, 58°C for 30 s and 72°C for 30 s, with a final extension step at 72°C for 10 min. The data were normalized to GAPDH. Primers were designed using the NCBI Primer-BLAST tool (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) and synthesized by Kumei Biotechnology Co., Ltd (Changchun, China). The primers are listed in .
Table 1. Primers used for Real-time PCR
Western blot analysis
3 × 106 WT mouse BMDMs were stimulated with 3 × 106 T. evansi, 50 μg/mL EVs, or 50 μg/mL rTeKMP-11 protein, and the cells at different time points were collected (0, 2, 4, 6, and 8 h). The protein samples of BMDMs cells, T. evansi and EVs were prepared with radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime, Shanghai, China) with 1 mM phenylmethanesulfonyl fluoride (PMSF, Sigma-Aldrich, USA). The proteins (30 μg) was separated by SDS-PAGE (12%) electrophoresis and transferred to PVDF membranes (Millipore, Bedford, MA, USA). Membranes were blocked with 5% skim milk for 2 h and incubated overnight at 4°C with the following antibodies: rabbit anti-p-P38, anti-total P38, anti-p-P65, anti-total P65, anti-p-ERK1/2, anti-total ERK1/2, anti-p-AKT, anti-total AKT, anti-GAPDH (Cell Signaling Technology, USA) mouse anti-HSP70, anti-Aldolase, anti-EF-1α, and anti-KMP-11 (mouse polyclonal antibodies). After washing 3 times with TBS-0.05% Tween 20 for 15 min, the blots were incubated with secondary HRP-conjugated antibodies (Proteintech, Wuhan, China) for 1 h and washed as before. Finally, target proteins were visualized using ECL luminous reagent (Millipore, Bedford, MA, USA). Protein expressions were quantified using Image J 2.0 software.
Cytokines detection by ELISA
WT/TLR2−/- mouse BMDMs (3 × 106) were seeded into 6-well plates and incubated with 3 × 106 T. evansi, 50 μg/mL EVs or 50 μg/mL rTeKMP-11 protein. For the kinase inhibition test, BMDMs were pretreated with inhibitors of P38 (SB203580, 30 µM, Sigma-Aldrich), ERK (PD98059, 40 µM, Sigma-Aldrich), or AKT (AKT inhibitor IV, 5 µM, Santa Cruz; MK-2206, 1 μM, Selleck), respectively, and then stimulated with T. evansi, EVs, or rTeKMP-11 protein. The culture supernatants were harvested for ELISA assay at 8, 12, 18, and 24 h post stimulation.
The blood of infected or uninfected T. evansi mice was collected and the serum was collected for ELISA measurement. IL-1β, IL-2, IL-4, IL-6, IL-12p40, TNF-α, IL-17A, and IFN-γ were measured by Cytokine ELISA Ready-SET-Go kits (Thermo Scientific, USA) according to the instructions.
Determination of T. evansi viability
3 × 106 T. evansi were incubated with different concentrations of MK-2206 (2 nM, 50 nM, 500 nM and 1 μM) for 12 h to detect the effect of MK-2206 on T. evansi viability. 3 × 106 BMDMs were pretreated with MK-2206 (1 µM) for 60 min, then removed the supernatants and incubated with 3 × 106 T. evansi for 12 h to detect the effect of MK-2206 treated BMDMs on T. evansi viability. 3 × 106 T. evansi were co-cultured with IL-12p40 (100 pg/mL, Abcom, USA) treated BMDMs to detect the effect of IL-12p40 treated BMDMs on T. evansi viability. All cell supernatants were collected and stained with 10% trypan blue (Sigma-Aldrich, USA) for 2 min. The dead T. evansi were stained blue, while the living T. evansi were unstained. Subsequently, all alive T. evansi were counted via a hemocytometer.
Statistical analysis
The data were analyzed by One-Way ANOVA using SPSS version 19.0 (SPSS, Inc., Chicago, IL, USA) and were expressed by mean ± standard error of the mean (SEM) of triplicate experiments. All charts were generated by GraphPad prism 7.00 (GraphPad InStat Software, San Diego, CA). The asterisk indicated the statistically significant difference: *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
Results
TLR2−/- mice were more resistant to T. evansi infection compared with WT mice.
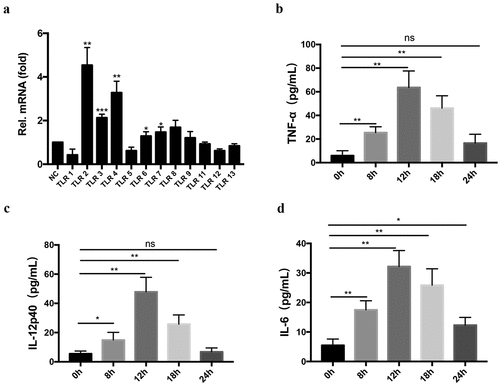
The isolated BMDMs were identified by flow cytometry and the purity was 96.62% (Figure S1). 3 × 106 WT mouse BMDMs were stimulated with T. evansi. After co-incubation for 4 h, compared with control, the transcriptional levels of TLR2, TLR3, and TLR4 were significantly up-regulated ().
Figure 1. T. evansi activated TLRs and induced inflammatory cytokines secretion in BMDMs. (a) 3 × 106 WT mouse BMDMs incubated with 3 × 106 T. evansi or medium alone, respectively. The relative mRNA levels of TLRs in total RNAs were analyzed by RT-qPCR. (b-d) WT mouse BMDMs were incubated with T. evansi, cytokines secretion in the supernatants were detected using ELISA assay. The data are presented as the mean ± SEM from at least three independent experiments. ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001 VS the negative control group
A pilot experiment had determined that 1 × 104 T. evansi could result in 100% parasitemia in WT mice via intraperitoneal injection (Figure S2A), which was set as the baseline to describe the parasitemia in TLR2−/-, TLR3−/-, and TLR4−/- mice. For parasite infection, 1 × 104 T. evansi was intraperitoneally injected into WT, TLR2−/-, TLR3−/-, and TLR4−/- mice, respectively. Results indicated that TLR2−/- mice showed higher resistance to T. evansi infection compared with WT, TLR3−/-, and TLR4−/- mice. The parasitemia in WT, TLR3−/-, and TLR4−/- mice were 100%, while the parasitemia in TLR2−/- mice was 83.3% (). The longest survival times of WT, TLR2−/-, TLR3−/-, and TLR4−/- mice infected with T. evansi were 6, 9, 6, and 6 days, respectively (). The parasitemia of WT, TLR3−/-, and TLR4−/- -infected mice reached the peak on the sixth day and died (3 × 108/mL), while the parasites load of TLR2−/–infected group was significantly lower than that of WT-infected group, and the number of parasites in the TLR2−/–infected group decreased significantly on the seventh day, increased again on the eighth day, and reached the peak of parasitemia on the ninth day (, S2B).
Figure 2. The TLR2−/- mice were more resistant to T. evansi infection than the WT mice. (a) WT, TLR2−/-, TLR3−/-, and TLR4−/- mice were infected with 1 × 104 T. evansi by intraperitoneal injection, respectively (n = 10 mice per group). The infection rate of mice was detected. (b) The death times of WT, TLR2−/-, TLR3−/-, and TLR4−/- mice-infected were recorded. (c) Parasite load was measured in peripheral blood of WT and TLR2−/- mice. (d) Hematoxylin-and-eosin staining of lung and spleen sections. (e, f) ELISA analysis of IL-12p40 and IFN-γ levels in the serum of WT and TLR2−/- mice infected with T. evansi. The values are shown as the mean ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01 for the WT group versus the TLR2−/- group
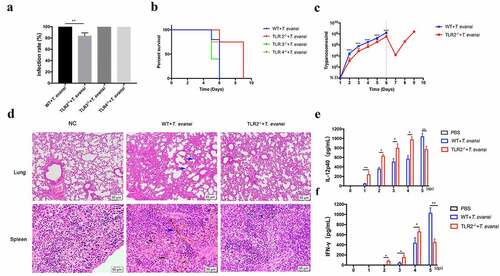
WT and TLR2−/- mice were euthanized at 5 dpi, and the pathological sections of the spleen and lung were analyzed. In WT-infected group, the spleens were hyperemic and a large amount of iron swallowing cells were observed. Interstitial widening and inflammatory exudate in the alveoli were observed. The TLR2−/–infected mice had significantly milder pathological changes than that in the WT-infected mice (). These results indicated that TLR2−/- mice were more resistant to T. evansi infection.
IL-12 [Citation39], IFN-γ [Citation40], and TNF-α [Citation41] play vital roles against Trypanosoma infection. The cytokine levels (IL-1β, IL-2, IL-4, IL-6, IL-12p40, IL-17A, TNF-α, and IFN-γ) in the serum at different times (0, 1, 2, 3, 4, and 5 dpi) were measured. The IFN-γ and IL-12p40 levels in TLR2−/–infected mice were significantly higher than that of the WT-infected mice up to 4 dpi. However, the IFN-γ and IL-12p40 secretions in the WT-infected mice was significantly higher than that of the TLR2−/–infected mice at 5 dpi ( e, f). T. evansi infection induced the production of IL-6 in both WT and TLR2−/- mice, no significant difference was observed between the two infected groups (Figure S3). There was no significant difference in the secretion of IL-1β, IL-2, IL-4, IL-17 and TNF-α in all infected mice.
T. evansi induced low-level secretion of cytokines via TLR2-mediated ERK pathway in BMDMs.
Cytokines secretion (IL-1β, IL-2, IL-4, IL-6, IL-12p40, IL-17A, INF-γ, and TNF-α) induced by T. evansi in the culture supernatant of BMDMs at different time points (8, 12, 18, and 24 h) were detected. The results showed that T. evansi could induce low-level secretion of TNF-α IL-6, and IL-12p40. TNF-α (63.67 ± 8.066 pg/mL), IL-12p40 (47.92 ± 5.717 pg/mL), and IL-6 (37.17 ± 3.118 pg/mL) secretions reached the maximum at 12 h (, c, d). Secretion of other five cytokines could not be detected in T. evansi stimulated mouse BMDMs. IL-6, IL-12p40, and TNF-α in BMDMs of WT or TLR2−/- mice stimulated with T. evansi were detected at 12 h. The results showed that IL-12p40 in the TLR2−/- group (186.4 ± 8.573 pg/mL) was significantly higher (P < 0.001) than that in the WT group (46.96 ± 6.286 pg/mL), while TNF-α and IL-6 were not significantly different ().
Figure 3. T. evansi promoted inflammatory cytokines secretion via TLR2-mediated ERK pathway. (a) WT/TLR2−/- mouse BMDMs were stimulated with T. evansi for 12 h. The cytokines secretion levels were analyzed by ELISA. (b, c) WT BMDMs were stimulated with T. evansi, the levels of phosphorylation of ERK were analyzed by Western blot. (d) WT mouse BMDMs were stimulated with T. evansi after pretreated with inhibitor of ERK (PD98059; 40 µM) for 60 min. The amount of cytokines secretion was detected by ELISA. (e, f) WT/TLR2−/- BMDMs were stimulated with T. evansi for 6 h, then the p-ERK expression was determined by immunoblot and densitometric analysis. Bars represent the mean ± SEM from three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant, for the WT group versus the TLR2−/- group, or negative control group versus experimental group, or ERK inhibitor stimulation group versus WT stimulation group
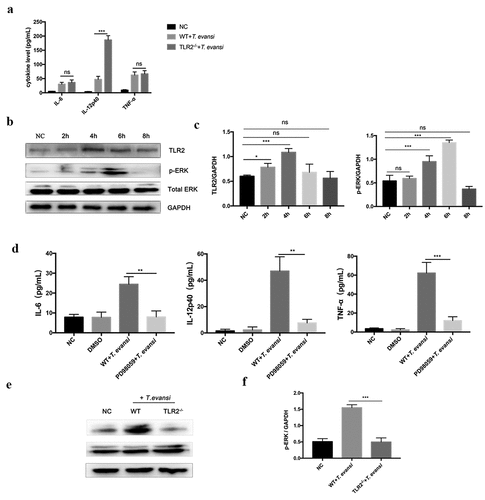
Subsequently, we examined several common inflammatory pathways, like NF-κB and MAPK. T. evansi could significantly activate TLR2 (P < 0.001) and ERK (P < 0.001) pathways after stimulation for 4 h and 6 h, respectively ( b, c). But no obvious changes were observed for p-P38 and p-P65 (Figure S4). In order to study whether TLR2 played a regulatory role, we used T. evansi to stimulate BMDMs of WT or TLR2−/- mouse and the results suggested that the phosphorylation level of ERK (P < 0.001) was significantly inhibited in TLR2−/- group (, f).
BMDMs were pre-treated with ERK inhibitors and stimulated with T. evansi. The results showed that T. evansi-induced the secretion of IL-6 (P < 0.01), IL-12p40 (P < 0.01), and TNF-α (P < 0.01) were significantly decreased in ERK-blocked BMDMs ().
T. evansi caused serious Trypanosomiasis by inhibiting the secretion of inflammatory cytokines via TLR2-mediated AKT pathway
Trypanosoma evansi could induce AKT activation after stimulation for 2 h and maintained at a high level (, b). To explore the role of the AKT pathway in regulating cytokines secretion, BMDMs were pre-treated with AKT inhibitor IV. In AKT inhibitor group, the production of IL-12p40 (117.8 ± 12.2 pg/mL), IL-6 (64.31 ± 9.138 pg/mL), and TNF-α (132.9 ± 19.01 pg/mL) induced by T. evansi were significantly increased (). BMDMs from TLR2−/- and WT mice were stimulated with T. evansi for 6 h, the phosphorylation level of AKT (P < 0.05) was significantly inhibited in BMDMs from TLR2−/- mice compared to that from WT mice (, e).
Figure 4. T. evansi inhibited the secretion of inflammatory cytokines via TLR2-mediated AKT pathway. (a, b) WT mouse BMDMs were stimulated with T. evansi, Western blot and densitometric analysis were used to detect the phosphorylation level of AKT. (c) WT mouse BMDMs stimulated with T. evansi after pretreated with AKT inhibitor IV for 60 min. ELISA analysis of TNF-α, IL-6, and IL-12p40 levels. (d, e) WT/TLR2−/- mouse BMDMs were stimulated with T. evansi for 4 h, and then the p-AKT expression was determined by immunoblot and densitometric analysis. (f) WT mice (n = 10 mice per group) were administered with MK-2206 to block AKT or without, then infected with 1 × 104 T. evansi by intraperitoneal injection. The death time was recorded. (g) The parasites load in peripheral blood was detected. (h) ELISA analysis of IL-12p40 and IFN-γ levels in the serum of AKT-blocked and WT mice infected with T. evansi. Bars represent the mean ± SEM for three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001, for negative control group versus experimental group, or AKT inhibitor group versus without AKT inhibitor group, or the WT group versus the TLR2−/- group
MK-2206 is another AKT inhibitor that often used in vivo assay. The effect of MK-2206 on T. evansi in vitro was performed. The result showed that MK-2206 did not affect parasite viability at concentrations of 0.02 μM to 1 μM in vitro (Figure S5A). Then T. evansi were co-incubated with WT mouse BMDMs pretreated with MK-2206 (1 µM) for 60 min. After 12 h co-incubation, parasites viability and the secretions of IL-12p40, IL-6 and TNF-α were examined. The results showed that the T. evansi viability was 27.23% in MK-2206-untreated group compared with 14.03% in the MK-2206-treated group (Figure S5B). In MK-2206-treated group, the production of IL-12p40 (104.9 ± 6.209 pg/mL), IL-6 (62.84 ± 5.485 pg/mL), and TNF-α (116.5 ± 7.517 pg/mL) induced by T. evansi were significantly increased (Figure S5C). We detected the effect of IL-12p40 on T. evansi in vitro. T. evansi were co-incubated with WT mouse BMDMs treated with IL-12p40 (100 pg/mL) for 12 h. The results showed that the T. evansi viability was 30.43% in IL-12p40-untreated group compared with 16.83% in the IL-12p40-treated group (Figure S5D).
To explore the role of AKT in T. evansi-infected mice, WT and AKT-blocked (MK-2206 treatment) mice were infected with T. evansi. WT-infected mice began to die on the fourth day and all died on the fifth day after infection, while the AKT-blocked mice began to die on the fifth day and all died on the seventh day after infection (). The parasites could be detected in the peripheral blood of WT mice on the second day after infection and reached the parasitemia on the fifth day (3 × 108/mL), while the parasites could be detected in the AKT-blocked mice on the third day and the number of parasites was significantly lower than WT mice (). IL-12p40 and IFN-γ secretions in the serum of AKT-blocked mice infected with T. evansi were significantly higher than that of WT mice ().
T. evansi could continuously secrete EVs.
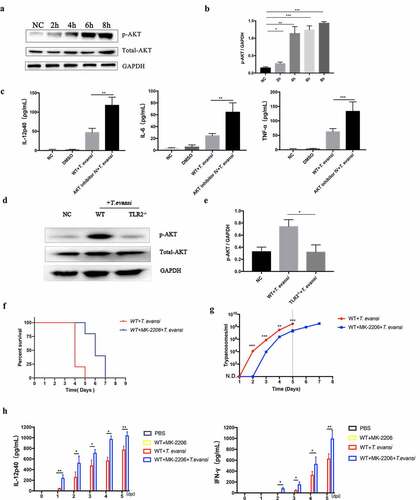
Negative staining observations using TEM showed that TeEVs were partially flat ball-shaped, with a double-layer membrane structure, and about 70–150 nm in diameter (). NTA showed that the mean diameter of the EVs was 110 nm (Figure S6). SEM results showed that the posterior end of T. evansi could extend out the nanotubes about 100 nm in diameter and TeEVs could be found on the surface of the parasite (). Ultrathin section observations revealed that free EVs and the nanotubes had similar membrane structures with the cell plasma membrane, and the nanotubes could dissociate to produce free EVs ().
Figure 5. Physical characterizations of TeEVs. (a) The partially flat ball-shaped vesicles were observed using negative staining by TEM. Scale bar = 100 nm. (b) EVs on the surface of T. evansi (left, scale bas = 100 nm) and the nanotubes protruded from the posterior end of T. evansi (right, scale bar = 200 nm) were visualized by SEM. (c) Thin section TEM analysis of EVs (left) and nanotubes (right). The membranes of EVs and nanotubes were similar to the cell plasma membrane and the nanotubes were dissociated to produce free EVs. Scale bars = 100 nm
TeEVs regulated inflammatory cytokines secretion via TLR2-mediated AKT/ERK pathways in BMDMs
Labeled EVs were co-incubated with BMDMs for 2 h and EVs were observed to be internalized into BMDMs (). WT mouse BMDMs were stimulated with TeEVs for 4 h. RT-qPCR results showed that EVs could significantly activate TLR2, TLR3, and TLR4 in BMDMs (). The low-level secretion of IL-12p40, IL-6, and TNF-α in the culture supernatants of BMDMs stimulated with EVs were detected (). The EVs-induced IL-12p40 secretion (P < 0.001) in TLR2−/- BMDMs was significantly higher than that in WT BMDMs. However, no significant differences were detected in IL-6 and TNF-α secretions ().
Figure 6. TeEVs could activate TLR2 and induce cytokines secretion in BMDMs. (a) After incubating the labeled EVs with BMDMs for 2 h, the internalization of EVs by BMDM was observed by confocal laser scanning. Green: labeled EVs. Blue: nuclei. Scale bar = 5 μm. (b) WT mouse BMDMs were incubated with TeEVs or medium alone, respectively. RT-qPCR was used to analyze the relative mRNA level of TLRs in total RNA. (c-e) After WT mouse BMDMs and EVs were co-incubated, the secretion of cytokines was detected by ELISA assays. Bars represent the mean ± SEM of triplicate experiments. ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001 VS the negative control group
Figure 7. TeEVs regulated inflammatory cytokines secretion via TLR2-mediated AKT/ERK pathways. (a, b) After WT mouse BMDMs and TeEVs were co-incubated, Western blot and densitometric analysis were used to measure the phosphorylation levels of ERK and AKT. (c, d) The BMDMs of WT/TLR2−/- mouse were stimulated with TeEVs for 6 h, the p-ERK and p-AKT expression were determined by immunoblot and densitometric analysis. (e) WT mouse BMDMs were stimulated with EVs after pretreated with PD98059 or AKT inhibitor IV for 60 min. The secretion levels of cytokines were measured by ELISA. (f) BMDMs of WT/TLR2−/- mouse with EVs were co-incubated for 12 h. The cytokine levels were measured by ELISA. Bars represent the mean ± SEM from three independent experiments. ns, no significance; **, P < 0.01; ***, P < 0.001 for the stimulated group versus the negative control group, or AKT/ERK inhibitor stimulation group versus WT stimulation group, or the WT group versus the TLR2−/- group
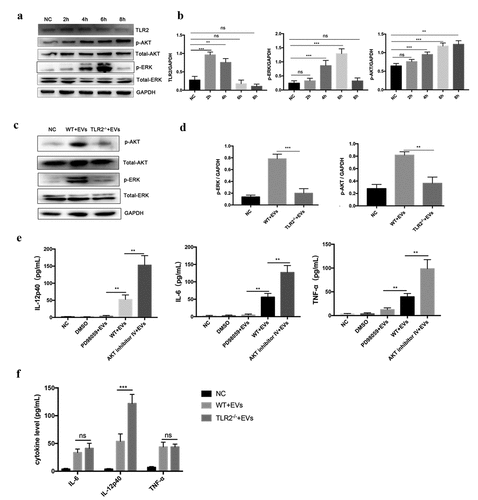
EVs could activate ERK and AKT pathways (, b), and the phosphorylation levels of ERK and AKT were significantly decreased in TLR2−/- mice BMDMs (, d). The secretion levels of IL-12p40, IL-6, and TNF-α decreased significantly after ERK inhibitor pretreatment, while the secretion levels of IL-12p40 (P < 0.01), IL-6 (P < 0.01), and TNF-α (P < 0.01) was increased significantly after AKT inhibitor pretreatment (). These results suggested that TeEVs played an important role in TLR2-mediated ERK and AKT activation, which ultimately regulated the secretion of inflammatory cytokines.
TeEVs promoted T. evansi proliferation and accelerated mice’s death in vivo
Trypanosoma evansi-infected mice in the EV group began to die on the fourth day, and all died on the fifth day, while T. evansi-infected mice in the PBS group began to die on the fifth day, and all died on the seventh day (). The parasite load of T. evansi-infected mice with EVs treatment was higher than that of PBS treated mice (). IL-12p40 and IFN-γ secretions in the EVs treated mice were significantly lower than that in the PBS treated mice ().
Figure 8. TeEVs could promote T. evansi proliferation in vivo. WT mice (n = 10 mice per group) were infected with 1 × 104 T. evansi by intraperitoneal injection. 50 μg EVs were injected intravenously (tail vein) into the mice for three consecutive days. Control mice were injected with an equal volume of PBS. (a) The death time was recorded daily. (b) Parasites load in peripheral blood was detected daily. (c) The production levels of IL-12p40 and IFN-γ in the serum were detected by ELISA. Bars represent the mean ± SEM of three independent experiments. *, P < 0.05; **, P < 0.01 for PBS group versus EVs group
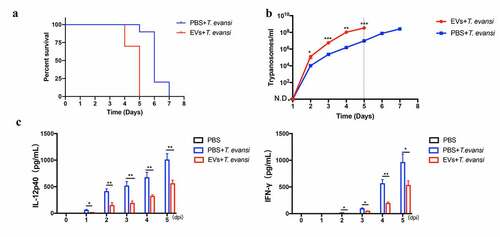
Proteomic analysis of TeEVs
Proteomics results showed that 746 proteins were identified in TeEVs, of which at least two unique peptides accounted for 57.5% (429/746). The proteins identified in TeEVs were similar to those reported in T. brucei [Citation36] (). The ribosomal proteins in the TeEVs proteome were detected. HSP-70, EF-1α, KMP-11, and Aldolase were confirmed by Western blot (). The proportion of biological process [Citation42], cell component (CC) and molecular function (MF) of the proteins were shown in . Among all of the identified proteins, 595 proteins were involved in 78 signal pathways, the top five of which were Proteasome, Aminoacyl-tRNA biosynthesis, RNA transport, Ribosome, and mRNA surveillance pathway (). In addition, these proteins were also related to PI3K-AKT, MAPK, NOD-like, and toll-like signaling pathways.
Table 2. Top 50 proteins enriched in extracellular vesicles of T. evansi.
Figure 9. Analysis of proteins in TeEVs. (a) Western blot analysis of the purified TeEVs with anti-HSP70, anti-Aldolase, anti-KMP-11, and anti-EF-1α polyclonal antibodies. (b) Gene ontology analysis of proteins in EVs. (c) KEGG analysis of differential expressed genes
T. evansi KMP-11 recombinant protein activated AKT pathway in vitro and promoted T. evansi proliferation in vivo
First, rTeKMP-11 protein was expressed and purified (). TeKMP-11 protein was detected in BMDMs stimulated with TeEVs and rTeKMP-11 for 2 h, indicating that EVs could release their cargo into BMDMs and rTeKMP-11 could enter the cytoplasm of BMDMs (). Then we detected the location of KMP-11 protein in T. evansi. The results showed that TeKMP-11 protein mainly existed in parasite coat, the flagellum, and the flagella pocket (). After stimulating BMDMs with different concentrations of rTeKMP-11, we analyzed the cell viability and selected the concentration of 50 μg/mL for subsequent experiments (Figure S7). Cytokines secretion and MAPK, AKT, and NF-κB pathways in BMDMs stimulated with rTeKMP-11 were examined. The secretion levels of IL-12p40 (64.24 ± 4.214 pg/mL), IL-6 (65.95 ± 10.99 pg/mL), and TNF-α (112.6 ± 14.69 pg/mL) reached the maximum at 12 h after stimulation of BMDMs by rTeKMP-11 (), and the control carrier protein had no effect on the production of cytokines in BMDMs (). The phosphorylation level of P38 peaked at 2 h and returned to baseline at 6 h and the phosphorylation level of AKT maintained at a high level (, g). The production of IL-12p40, IL-6, and TNF-α increased significantly after treatment with AKT inhibitor and reduced significantly using P38 inhibitor ().
Figure 10. rTeKMP-11 activated AKT pathway to inhibit the secretion of inflammatory cytokines in vitro and promoted T. evansi proliferation in vivo. (a) Purified rTeKMP-11 protein. M, protein molecular markers. Lane 1, Purified rTeKMP-11 protein with vector tagged protein. Lane 2, rTeKMP-11 protein after removal of vector tagged protein. (b) BMDMs were stimulated with EVs (left) and rTeKMP-11 (right) for 2 h, the TeKMP-11 protein in BMDMs was detected by IFA and observed under confocal laser scanning. Green: TeKMP-11. Blue: nuclei. Scale bar = 5 μm. (c) The localization of TeKMP-11 protein in T. evansi was observed by laser confocal microscopy. Green: TeKMP-11. Blue: nuclei. Scale bar = 5 μm. (d) Secretion levels of IL-12p40, IL-6, and TNF-α in BMDMs stimulated by 50 μg rTeKMP-11. (e) After stimulating with empty carrier protein (ECP), rTeKMP-11, and DOTAP for 12 h, the secretions of inflammatory cytokines were observed. (f, g) WT mouse BMDMs were stimulated with rTeKMP-11, Western blot and densitometric analysis were used to detect the phosphorylation levels of AKT and P38. (h) WT mouse BMDMs were stimulated with rTeKMP-11 after pretreated with SB203580 or AKT inhibitor IV for 1 h. The secretion levels of cytokines were measured by ELISA. (i-k) Mice (n = 10 mice per group) were injected intravenously (tail vein) with 50 μg rTeKMP-11 encapsulated in liposome, an equal volume of empty carrier protein or liposome for three consecutive days. (i) The death time was recorded daily. (j) Parasites load in peripheral blood was detected daily. (k) The production levels of IL-12p40, and IFN-γ in the serum were detected by ELISA. Bars represent the mean ± SEM for three experiments. ns, not significant; *, P < 0.05; **, P < 0.01; ***, P < 0.001, for the stimulated group versus the negative control group, or AKT/p38 inhibitor stimulation group versus WT stimulation group, or the rTeKMP-11 group versus the negative control group
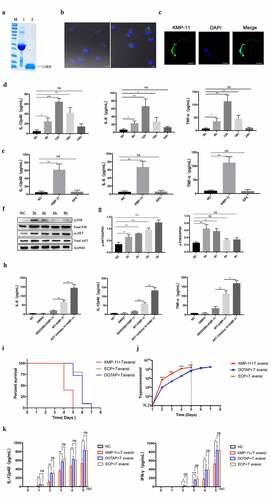
To study the role of KMP-11 protein in vivo, animal experiments were conducted. The results showed that T. evansi-infected mice in the rTeKMP-11 group began to die on the fourth day, and all died on the fifth day, while the mice in the ECP and DOTAP groups began to die on the fifth day, and all died on the seventh day (). The parasite load of T. evansi-infected mice injected with rTeKMP-11 was higher than that of ECP and DOTAP groups (). Finally, we tested the cytokines in the serum by ELISA at different times (0, 1, 2, 3, 4, and 5 dpi). IL-12p40 and IFN-γ secretions in the rTeKMP-11 group was significantly lower than that in the ECP and DOTAP groups ().
Discussion
Trypanosomas have developed a successful immune escape mechanism. Antigen variation of variant surface glycoproteins (VSGs) was the most important immune escape mechanism in Trypanosomes, which is to avoid the host’s humoral response by constantly converting VSGs [Citation43]. Trypanosomas could activate the polyclonal B cells to differentiate into short-lived plasmablasts, which ultimately lead to apoptosis/elimination of the B cell population [Citation41,Citation44–46]. As an important bloodborne protozoa, the immune evasion mechanism of T. evansi deserves more investigation. The results from the present study showed that T. evansi could secrete TeEVs to activate TLR2-AKT pathway, which in turn inhibited the productions of inflammatory cytokines to allow the survival of parasites. In addition, TeKMP-11 protein, a component of TeEVs, was shown to be involved in promoting T. evansi infection.
TLR2 plays a vital role in fighting parasite infections, including T. congolense, P. yoelii, T. gondii, and L. major [Citation47–50]. However, it also plays a negative role to promote parasite infection. TLR2−/- mice exhibited reduced severity of giardiasis through increased production of inflammatory cytokines [Citation51]. TLR2−/- mice had a lower parasite load than C57BL/6 wild-type mice in the first two weeks after L. amazonensis infection [Citation52]. In this study, we found that T. evansi could activate TLR2, TLR3 and TLR4 in mouse BMDMs. Compared with TLR3−/-, TLR4−/-, and WT mice, TLR2−/- mice were more resistant to T. evansi infection mainly due to decreased infection rate and the parasites load, delayed survival time and relieved pathological changes.
Proinflammatory cytokines IL-12 and IFN-γ played vital roles in Trypanosoma infections [Citation53,Citation54]. The secretion of IL-12-dependent IFN-γ was essential for fighting against T. brucei brucei and T. evansi infections [Citation55]. Studies have shown that IL-1, IFN-γ, IL-6, and TNF-α were increased in serum after T. evansi infection in rats [Citation56]. In this study, IFN-γ and IL-12p40 secretions were significantly higher in TLR2−/- mice than in WT mice at 2, 3, and 4 dpi, suggesting that IL-12p40 and IFN-γ might be critical in resisting T. evansi infection. BALB/c mice that were susceptible to African trypanosomes showed higher level of IL-6 in BMDMs than C57BL/6 J mice with relative resistance [Citation57]. The use of anti-IL-6 monoclonal antibody did not reduce the number of T. cruzi in blood or tissues [Citation58]. Our results showed that IL-6 secretion in serum of WT and TLR2−/- mice infected with T. evansi was increased, but there was no significant difference between the two groups. This result indicated that the secretion of IL-6 induced by T. evansi might not be regulated by TLR2 and did not play a significant role in resisting trypanosome infection. Interestingly, T. evansi could suppress multiple cytokine secretions that might help to maintain parasites survival in blood.
Macrophages play an important role in controlling parasite infections in many protozoa [Citation59,Citation60]. A complete monocyte system was essential to start and maintain the anti-Trypanosoma response [Citation61]. Macrophages are the major source of inflammatory cytokines in T. evansi infection [Citation62] However, the intracellular signaling pathway by which T. evansi induces BMDMs cytokines production remains unknown. In this study, we found that T. evansi could induce mouse BMDMs to secrete low-levels of IL-12p40, TNF-α and IL-6. Furthermore, T. evansi could induce the secretion of inflammatory cytokines via activating TLR2-ERK signaling pathway. However, no IFN-γ, IL-1β, IL-17A, IL-2, and IL-4 were detected. Previous studies have shown that IFN-γ can be rapidly produced by NK cells and NKT cells in the early stages during many pathogen infections [Citation63–66]. CD4+ and CD8+ T cells are major producers of IFN-γ during Plasmodium infection [Citation67]. In the present experiment, IFN-γ in serum might be secreted by other immune cells. There was no significant difference in IL-6 and TNF-α secretion between TLR2−/- mouse BMDMs and WT mouse BMDMs after T. evansi stimulation. Surprisingly, IL-12p40 secretion in BMDMs of TLR2−/- mice was significantly up-regulated compared with the BMDMs of WT mice. These results were consistent with the results of in vivo experiments. Furthermore, the phosphorylation of AKT induced by T. evansi was maintained for a long time and the secretions of IL-12p40, IL-6 and TNF-α were significantly enhanced in cells pre-treated with AKT inhibitor. AKT inhibitor induced increased IL-12p40 in serum and helped to resist T. evansi infection in mice. Similar studies found that Giardia inhibited the secretion of inflammatory cytokines through TLR2-AKT pathway [Citation51] and T. cruzi infection in TLR2−/- mice increased IL-12 secretion [Citation68]. These results suggested that T. evansi could inhibit IL-12p40 secretion through the TLR2-AKT signaling pathway to escape host immune response.
Many parasites could secrete EVs via the endosomal sorting complexes required for transport (ESCRT) dependent mechanism, such as T. cruzi and Neospora caninum [Citation33,Citation69]. In addition, a new mechanism of EVs formation was found in T. brucei, which could extend nanotubes from the posterior end of the parasite and form EVs through nanotubes dissociation [Citation36]. In this study, we isolated and identified TeEVs for the first time and found that the formation mechanism of TeEVs was consistent with that of T. brucei. Parasite-derived EVs could transfer their information to host cells by membrane fusion to play a vital role in the process of infecting the host and the host’s immune response [Citation28]. As an extracellular parasite, T. brucei could exchange information between the parasite and the host by secreting EVs, causing a series of immune responses in the host [Citation36]. The EVs secreted by N. caninum could activate TLR2 of host macrophages and regulate the production of a series of cytokines [Citation33]. In this experiment, we investigated whether T. evansi could secrete EVs to regulate host immune response. TeEVs could activate the TLR2-AKT signaling pathway and inhibit IL-12p40, IL-6, and TNF-α in vitro. In vivo, we proved that EVs could inhibit the secretion of IL-12p40 and IFN-γ and increase parasite load in peripheral blood.
KMP-11 is a unique protein of kinetoplastid proteoa that is widely distributed on the surface of parasites. KMP-11 plays an important role in the cytoplasmic division of T. brucei [Citation70]. As a virulence factor, KMP-11 induced increased IL-10 production, thus enhancing the infection of macrophages by Leishmania [Citation71]. In this study, KMP-11 was identified as an important molecule of TeEVs to promote disease development in mice. rTeKMP-11 activated AKT and p38 signaling pathways to inhibit or promote the secretions of IL-12p40, IL-6, and TNF-α in vitro. EVs also carry a variety of proteins, including many virulence factors. The role of these EVs proteins in the interaction between T. evansi and host remains to be explored.
In conclusion, T. evansi could inhibit the productions of inflammatory cytokines via releasing TeEVs to activate TLR2-AKT signal pathways to survive successfully. TeKMP-11, a component of TeEVs, was involved in promoting T. evansi infection, which revealed a novel mechanism of T. evansi immune evasion.
CRediT author statement
Ran Wei: Conceptualization, Original draft preparation, Methodology. Xin Li: Conceptualization, Methodology. Xiaocen Wang: Conceptualization, Original draft preparation. Nan Zhang: Conceptualization, Software. Yuru Wang: Software, Writing - Review & Editing. Xichen Zhang: Conceptualization, Data Curation. Pengtao Gong: Conceptualization, Data Curation. Jianhua Li: Supervision, Project administration.
Animal welfare statement
All experimental procedures strictly followed the guidelines provided by the administration of experimental animal affairs approved by state council of people’s Republic of China and the animal welfare and research ethics committee of Jilin University (IACUC license No.: 201,701,009).
Supplemental Material
Download Zip (533.9 KB)Acknowledgments
Thanks to the colleagues for effective discussions on the experiment. Thanks to Xinrui Wang for the help with the Olympus FluoView FV1000 confocal microscope. We thank Yuanyuan Zhang and Yi Xin, technicians from the Instrument Development Center of Jilin University, for helping us with TEM and SEM analysis.
Disclosure statement
The authors declare that they have no competing interests.
Data availability statement
The data that support the findings of this study are available from the corresponding author, Jianhua Li, upon reasonable request.
Supplementary material
Supplemental data for this article can be accessed here
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
Related Research Data
References
- Aregawi WG, Agga GE, Abdi RD, et al. Systematic review and meta-analysis on the global distribution, host range, and prevalence of Trypanosoma evansi. Parasit Vectors. 2019;12:67.
- Birhanu H, Roge S, Simon T, et al. Surra Sero K-SeT, a new immunochromatographic test for serodiagnosis of Trypanosoma evansi infection in domestic animals. Vet Parasitol. 2015;211(3–4):153–157.
- Desquesnes M, Dargantes A, Lai D-H, et al. Trypanosoma evansi and Surra: a review and perspectives on transmission, epidemiology and control, impact, and zoonotic aspects. Biomed Res Int. 2013;2013:20.
- Elhaig MM, Sallam NH. Molecular survey and characterization of Trypanosoma evansi in naturally infected camels with suspicion of a Trypanozoon infection in horses by molecular detection in Egypt. Microb Pathog. 2018;123:201–205.
- Rashid I, Akbar H, Gharbi M, et al. First report of Trypanosoma Evansi Infection (SURRA) IN A PUMA (FELIS CONCOLOR) OF LAHORE ZOO, PAKISTAN. J Zoo Wildl Med. 2017;48(3):918–921.
- Herrera HM, Norek A, Freitas TP, et al. Domestic and wild mammals infection by Trypanosoma evansi in a pristine area of the Brazilian Pantanal region. Parasitol Res. 2005;96:121–126.
- Ereqat S, Nasereddin A, Al-Jawabreh A, et al. Prevalence of Trypanosoma evansi in livestock in Palestine. Parasit Vectors. 2020;13:21.
- Van Vinh Chau N, Buu Chau L, Desquesnes M, et al. A clinical and epidemiological investigation of the first reported human infection with the zoonotic parasite Trypanosoma evansi in Southeast Asia. Clin Infect Dis. 2016;62:1002–1008.
- Joshi PP, Shegokar VR, Powar RM, et al. Human trypanosomiasis caused by Trypanosoma evansi in India: the first case report. Am J Trop Med Hyg. 2005;73:491–495.
- Misra KK, Roy S, Choudhury A. Biology of Trypanosoma (Trypanozoon) evansi in experimental heterologous mammalian hosts. J Parasit Dis. 2016;40(3):1047–1061.
- Kurup S, Tewari A. Induction of protective immune response in mice by a DNA vaccine encoding Trypanosoma evansi beta tubulin gene. Vet Parasitol. 2012;187(1–2):9–16.
- Faccio L, Da Silva AS, Gressler LT, et al. Susceptibility of Brazilian isolates of Trypanosoma evansi to suramin sodium: test in experimentally infected mice. Exp Parasitol. 2013;134(3):309–312.
- Gourbal B, Pinaud S, Beckers GJM, et al. Innate immune memory: an evolutionary perspective. Immunol Rev. 2018;283(1):21–40.
- Kawai T, Akira S. Pathogen recognition with Toll-like receptors. Curr Opin Immunol. 2005;17(4):338–344.
- Janeway CA Jr., Medzhitov R. Innate Immune Recognition. Annu Rev Immunol. 2002;20(1):197–216.
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511.
- Souza COS, Gardinassi LG, Rodrigues V, et al. Monocyte and Macrophage-Mediated Pathology and protective immunity during schistosomiasis. Front Microbiol. 2020;11:1973.
- Oliveira AC, Peixoto JR, de Arruda LB, et al. Expression of functional TLR4 confers proinflammatory responsiveness to trypanosoma cruzi glycoinositolphospholipids and higher resistance to infection with T. cruzi. J Immunol. 2004;173(9):5688–5696.
- De Veer MJ, Curtis JM, Baldwin TM, et al. MyD88 is essential for clearance ofLeishmania major: possible role for lipophosphoglycan and Toll-like receptor 2 signaling. Eur J Immunol. 2003;33(10):2822–2831.
- Srivastava S, Pandey SP, Jha MK, et al. Leishmania expressed lipophosphoglycan interacts with Toll-like receptor (TLR)-2 to decrease TLR-9 expression and reduce anti-Leishmanial responses. Clin Exp Immunol. 2013;172(3):403–409.
- Campos MA, Closel M, Valente EP, et al. Impaired production of proinflammatory cytokines and host resistance to acute infection with Trypanosoma cruzi in mice lacking functional myeloid differentiation factor 88. J Immunol. 2004;172(3):1711–1718.
- Ives A, Ronet C, Prevel F, et al. Leishmania RNA virus controls the severity of mucocutaneous leishmaniasis. Science. 2011;331(6018):775–778.
- Zaborowski MP, Balaj L, Breakefield XO, et al. Extracellular vesicles: composition, biological relevance, and methods of study. Bioscience. 2015;65(8):783–797.
- Nowacki FC, Swain MT, Klychnikov OI, et al. Protein and small non-coding RNA-enriched extracellular vesicles are released by the pathogenic blood fluke Schistosoma mansoni. J Extracell Vesicles. 2015;4(1):28665.
- Schorey JS, Cheng Y, Singh PP, et al. Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO Rep. 2015;16(1):24–43.
- Bose S, Aggarwal S, Singh DV, et al. Extracellular vesicles: an emerging platform in gram-positive bacteria. Microb Cell. 2020;7(12):312–322.
- Rodrigues ML, Nakayasu ES, Oliveira DL, et al. Extracellular vesicles produced by Cryptococcus neoformans contain protein components associated with virulence. Eukaryot Cell. 2008;7(1):58–67.
- Szempruch AJ, Dennison L, Kieft R, et al. Sending a message: extracellular vesicles of pathogenic protozoan parasites. Nature Rev Microbiol. 2016;14(11):669–675.
- Coakley G, Maizels RM, Buck AH. Exosomes and other extracellular vesicles: the new communicators in parasite infections. Trends Parasitol. 2015;31(10):477–489.
- Atayde VD, da Silva Lira Filho A, Chaparro V, et al. Exploitation of the Leishmania exosomal pathway by Leishmania RNA virus 1. Nat Microbiol. 2019;4(4):714–723.
- Martin-Jaular L, Nakayasu ES, Ferrer M, et al. Exosomes from Plasmodium yoelii-infected reticulocytes protect mice from lethal infections. PLoS One. 2011;6(10):e26588.
- Twu O, de Miguel N, Lustig G, et al. Trichomonas vaginalis exosomes deliver cargo to host cells and mediate host∶parasite interactions. PLoS Pathog. 2013;9(7):e1003482.
- Li S, Gong PT, Tai LX, et al. Extracellular vesicles secreted by Neospora caninum are recognized by toll-like receptor 2 and modulate host cell innate immunity through the MAPK Signaling Pathway. Front Immunol. 2018;9:1633.
- Tavares KC, Da Silva AS, Wolkmer P, et al. Cryopreservation of Trypanosoma evansi after DEAE-cellulose purification: evaluation of infective parameters. Res Vet Sci. 2011;90(2):257–259.
- Pineda-Torra I, Gage M, De Juan A, et al. Isolation, culture, and polarization of murine bone marrow-derived and peritoneal macrophages. Methods Mol Biol. 2015;1339:101–109.
- Szempruch AJ, Sykes SE, Kieft R, et al. Extracellular vesicles from Trypanosoma brucei mediate virulence factor transfer and cause host anemia. Cell. 2016;164(1–2):246–257.
- Aida Y, Pabst MJ. Removal of endotoxin from protein solutions by phase separation using Triton X-114. J Immunol Methods. 1990;132(2):191–195.
- Cheng Y, Ren X, Zhang Y, et al. eEF-2 kinase dictates cross-talk between autophagy and apoptosis induced by Akt inhibition, thereby modulating cytotoxicity of novel Akt inhibitor MK-2206. Cancer Res. 2011;71(7):2654–2663.
- Aliberti JC, Cardoso MA, Martins GA, et al. Interleukin-12 mediates resistance to Trypanosoma cruzi in mice and is produced by murine macrophages in response to live trypomastigotes. Infect Immun. 1996;64(6):1961–1967.
- Wu H, Liu G, Shi M. Interferon Gamma in African trypanosome infections: friends or foes? Front Immunol. 2017;8:1105.
- Vincendeau P, Bouteille B. Immunology and immunopathology of African trypanosomiasis. An Acad Bras Cienc. 2006;78(4):645–665.
- Crawford AC, Lehtovirta-Morley LE, Alamir O, et al. Biphasic zinc compartmentalisation in a human fungal pathogen. PLoS Pathog. 2018;14(5):e1007013.
- Taylor JE, Rudenko G. Switching trypanosome coats: what’s in the wardrobe? Trends Genet. 2006;22(11):614–620.
- Tabel H, Wei G, Shi M. T cells and immunopathogenesis of experimental African trypanosomiasis. Immunol Rev. 2008;225(1):128–139.
- Stijlemans B, Caljon G, Van Den Abbeele J, et al. Immune Evasion Strategies of Trypanosoma brucei within the mammalian host: progression to pathogenicity. Front Immunol. 2016;7. DOI:10.3389/fimmu.2016.00233
- Magez S, Torres JEP, Obishakin E, et al. Infections with extracellular trypanosomes require control by efficient innate immune mechanisms and can result in the destruction of the mammalian humoral immune system. Front Immunol. 2020;11. DOI:10.3389/fimmu.2020.00382
- Kuriakose S, Onyilagha C, Singh R, et al. TLR-2 and MyD88-Dependent activation of MAPK and STAT proteins regulates proinflammatory cytokine response and immunity to experimental Trypanosoma congolense Infection. Front Immunol. 2019;10. DOI:10.3389/fimmu.2019.02673
- Zheng H, Tan ZP, Zhou TL, et al. The TLR2 is activated by sporozoites and suppresses intrahepatic rodent malaria parasite development. Sci Rep. 2015;5(1). DOI:10.1038/srep18239
- Del Rio L, Butcher BA, Bennouna S, et al. Toxoplasma gondii triggers myeloid differentiation factor 88-Dependent IL-12 and Chemokine Ligand 2 (Monocyte Chemoattractant Protein 1) responses using distinct parasite molecules and host receptors. J Iimmunol. 2004;172(11):6954–6960.
- Muraille E, De Trez C, Brait M, et al. Genetically resistant mice lacking MyD88-Adapter protein display a high susceptibility to Leishmania major Infection associated with a Polarized Th2 response. J Iimmunol. 2003;170(8):4237–4241.
- Li X, Zhang X, Gong P, et al. TLR2−/− mice display decreased severity of giardiasis via enhanced proinflammatory cytokines production dependent on AKT signal pathway. Front Immunol. 2017;8. DOI:10.3389/fimmu.2017.01186
- Guerra CS, Silva RM, Carvalho LO, et al. Histopathological analysis of initial cellular response in TLR-2 deficient mice experimentally infected by Leishmania (L.) amazonensis. Int J Exp Pathol. 2010;91(5):451–459.
- Tarleton RL. Immune system recognition of Trypanosoma cruzi. Curr Opin Immunol. 2007;19(4):430–434.
- Hunter CA, Slifer T, Araujo F. Interleukin-12-mediated resistance to Trypanosoma cruzi is dependent on tumor necrosis factor alpha and gamma interferon. Infect Immun. 1996;64(7):2381–2386.
- Barkhuizen M, Magez S, Atkinson RA, et al. Interleukin-12p70-dependent interferon- gamma production is crucial for resistance in African trypanosomiasis. J Infect Dis. 2007;196:1253–1260.
- Paim FC, Duarte MM, Costa MM, et al. Cytokines in rats experimentally infected with Trypanosoma evansi. Exp Parasitol. 2011;128:365–370.
- Kaushik RS, Uzonna JE, Zhang Y, et al. Innate resistance to experimental African trypanosomiasis: differences in cytokine (TNF-alpha, IL-6, IL-10 and IL-12) production by bone marrow-derived macrophages from resistant and susceptible mice. Cytokine. 2000;12:1024–1034.
- Truyens C, Torrico F, Angelo-Barrios A, et al. The cachexia associated with Trypanosoma cruzi acute infection in mice is attenuated by anti-TNF-alpha, but not by anti-IL-6 or anti-IFN-gamma antibodies. Parasite Immunol. 1995;17:561–568.
- Colineau L, Clos J, Moon KM, et al. Leishmania donovani chaperonin 10 regulates parasite internalization and intracellular survival in human macrophages. Med Microbiol Immunol. 2017;206:235–257.
- Dos-Santos AL, Carvalho-Kelly LF, Dick CF, et al. Innate immunomodulation to trypanosomatid parasite infections. Exp Parasitol. 2016;167:67–75.
- Stijlemans B, De Baetselier P, Magez S, et al. African trypanosomiasis-associated anemia: the contribution of the interplay between parasites and the mononuclear phagocyte system. Front Immunol. 2018;9:218.
- Mekata H, Konnai S, Mingala CN, et al. Kinetics of regulatory dendritic cells in inflammatory responses during Trypanosoma evansi infection. Parasite Immunol. 2012;34:318–329.
- Farrar MA, Schreiber RD. The molecular cell biology of interferon-gamma and its receptor. Annu Rev Immunol. 1993;11:571–611.
- Schroder K, Hertzog PJ, Ravasi T, et al. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–189.
- Bafica A, Santiago HC, Goldszmid R, et al. Cutting edge: TLR9 and TLR2 signaling together account for MyD88-dependent control of parasitemia in Trypanosoma cruzi infection. J Immunol. 2006;177:3515–3519.
- Da Silva HB, Fonseca R, Alvarez JM, et al. IFN-gamma priming effects on the maintenance of effector memory CD4(+) T Cells and on phagocyte function: evidences from infectious diseases. J Immunol Res. 2015. DOI:10.1155/2015/202816
- Inoue S, Niikura M, Mineo S, et al. Roles of IFN-gamma and gammadelta T Cells in Protective immunity against blood-stage malaria. Front Immunol. 2013;4:258.
- Ropert C, Gazzinelli RT. Regulatory role of Toll-like receptor 2 during infection with Trypanosoma cruzi. J Endotoxin Res. 2004;10:425–430.
- Bayer-Santos E, Aguilar-Bonavides C, Rodrigues SP, et al. Proteomic analysis of trypanosoma cruzi secretome: characterization of two populations of extracellular vesicles and soluble proteins. J Proteome Res. 2013;12:883–897.
- Li Z, Wang CC. KMP-11, a basal body and flagellar protein, is required for cell division in Trypanosoma brucei. Eukaryot Cell. 2008;7:1941–1950.
- De Mendonca SC, Cysne-Finkelstein L, Matos DC. Kinetoplastid membrane protein-11 as a vaccine candidate and a virulence factor in Leishmania. Front Immunol. 2015;6:524.