
ABSTRACT
The molecular mechanisms underlying Aeromonas hydrophila-pathogenesis are not well understood. Using head kidney macrophages (HKM) of Clarias gariepinus, we previously reported the role of ER-stress in A. hydrophila-induced pathogenesis. Here, we report that PI3K/PLC-induced cytosolic-Ca2+ imbalance induces the expression of pro-apoptotic ER-stress marker, CHOP in A. hydrophila-infected HKM. CHOP promotes HKM apoptosis by inhibiting AKT activation and enhancing JNK signaling. Elevated mitochondrial ROS (mtROS) was recorded which declined significantly by ameliorating ER-stress and in the presence of ER-Ca2+ release modulators (2-APB and dantrolene) and mitochondrial-Ca2+ uptake inhibitor, Ru360, together suggesting the role of ER-mitochondrial Ca2+ dynamics in mtROS generation. Inhibiting mtROS production reduced HKM death implicating the pro-apoptotic role of mtROS in A. hydrophila-pathogenesis. The expression of autophagic proteins (LC3B, beclin-1, and atg 5) was suppressed in the infected HKM. Our results with autophagy-inducer rapamycin demonstrated that impaired autophagy favored the cytosolic accumulation of mitochondrial DNA (mtDNA) and the process depended on mtROS levels. Enhanced caspase-1 activity and IL-1β production was detected and transfection studies coupled with pharmacological inhibitors implicated mtROS/mtDNA axis to be crucial for activating the caspase-1/IL-1β cascade in infected HKM. RNAi studies further suggested the involvement of IL-1β in generating pro-apoptotic NO in A. hydrophila-infected HKM. Our study suggests a novel role of ER-mitochondria cross-talk in regulating A. hydrophila pathogenesis. Based on our observations, we conclude that A. hydrophila induces ER-stress and inhibits mitophagy resulting in mitochondrial dysfunction which leads to mtROS production and translocation of mtDNA into cytosol triggering the activation of caspase-1/IL-1β-mediated NO production, culminating in HKM apoptosis.
Introduction
A. hydrophila, a Gram-negative bacterium is capable of infecting a wide range of organisms. In fish, it is known to cause ulcerative disease syndrome (UDS) while in humans it causes serious health complications [Citation1]. Although, the mechanism underlying A. hydrophila pathogenesis is not well understood; at the cellular level, the major outcome of this host-pathogen interaction is apoptosis of the host cells [Citation2,Citation3].
Pathogen-induced alterations in cytosolic Ca2+ [(Ca2+)C] levels have been found to be crucial for apoptosis of the host macrophages [Citation4]. ER is the chief location for synthesis and correct folding of cellular proteins which is highly dependent on (Ca2+)C homeostasis [Citation5]. Perturbation in (Ca2+)C homeostasis interferes with normal ER protein load and functioning of the organelle, leading to stress condition termed as ER-stress [Citation6,Citation7]. To overcome ER-stress, eukaryotic cells have evolved the unfolded protein response (UPR), characterized by induction of IRE1 driven BiP and PERK-eIF2 mediated expression of CHOP [Citation6,Citation8]. ER-stress is essential for the survival of cells; however, prolonged ER-stress can instigate apoptosis [Citation5,Citation6,Citation9,Citation10]. CHOP is considered a well-known marker for ER-stress induced apoptosis [Citation11].
Among the different intracellular signaling molecules that trigger (Ca2+)C release, PI3K/PLC is important [Citation12]. Different bacterial ligands have been reported to activate PI3K/PLC mediated release of Ca2+ from intracellular sources thus modulating several downstream signaling molecules [Citation13]. The pro-apoptotic role of PI3K-PLC-mediated (Ca2+)C alteration has recently been established in A. hydrophila-pathogenesis [Citation14]. Though, A. hydrophila-induced ER-stress has been testified earlier [Citation3]; the involvement of PI3K-PLC axis in initiating ER-stress induced apoptosis has not been reported in A. hydrophila-pathogenesis.
In order to overcome stress, ER releases Ca2+ [(Ca2+)ER] via ER membrane receptors [Citation15]. The (Ca2+)ER is propelled out through specific channels [Citation16] and enters into the mitochondria via mitochondrial calcium uniporters (MCU) and voltage-dependent anion channels (VDAC), and the process is aided by the juxtaposition of the two organelles [Citation17–19]. The mechanism of ER-stress-induced apoptosis is not well known, but there are reports implicating the cross-talk between ER and mitochondria are critical in different model systems including fish [Citation3,Citation5].
Several earlier reports have documented a correlation between innate immune signaling and mitochondrial functioning [Citation20,Citation21]. (Ca2+)ER on entering the mitochondria impairs the ETC causing excessive ROS production by the mitochondria [Citation22,Citation23]. There are few reports suggesting mitochondrial ROS (mtROS) as one of the major contributory factors in host cell bactericidal activity, although the exact mechanism remains obscure [Citation21]. Additionally, the role of mtROS in microbial pathogenesis is not well elucidated in fish.
Recent reports suggest that mitochondrial DNA (mtDNA) acts as a modulator of immune responses [Citation24,Citation25]. Under varied conditions of stress, mtDNA is translocated into the cytosol and is sensed as a damage-associated molecular pattern (DAMP) by immune receptors triggering downstream signaling cascade to produce immune-effector molecules [Citation25]. The involvement of mtDNA in regulating host immunity to microbial immunity is fairly well established [Citation24,Citation26]. It has also been observed that mtROS plays an important role in triggering the release of mtDNA in cytoplasm impacting host immune responses [Citation21,Citation27,Citation28]. Autophagy is a conserved cellular quality control mechanism which facilitates the selective turnover of damaged cell-organelles, including mitochondria [Citation29]. Suppression of autophagy machinery led to cytosolic translocation of mtDNA in macrophages [Citation27]. To the best of our knowledge, the role of mtROS/mtDNA axis in regulating microbial pathogenesis has not been reported in fish.
One of the major outcomes of ER-mitochondrial interaction is inflammasome formation, leading to caspase-1 activation [Citation30]. Though there is very little information on inflammasomes in fish, the presence of signature molecules like caspase-1 and IL-1β in several fish [Citation31–34] suggests similar mechanism helps them to respond to varying insults, like higher vertebrates [Citation35]. Caspase-1 has been reported to be conserved from teleosts to mammals in terms of specificity, processing, and function [Citation33]. During caspase-1 activation, pro-caspase-1 is processed into active caspase-1 [Citation30], but the existence of alternatively spliced isoforms of caspase-1 has also been reported in fish [Citation33]. There are several reports documenting the activation of caspase-1 in response to A. hydrophila [Citation34].
The pro-inflammatory cytokine IL-1β is produced by innate immune cells in response to viral [Citation36], bacterial [Citation37], and parasitic infection [Citation38]. Although IL-1β has been reported in fishes [Citation39–43], but the synthesis of mature IL-1β is debatable. There are reports suggesting IL-1β processing to be both caspase-1 dependent [Citation32,Citation33] and caspase-1 independent in fish [Citation34,Citation44].
Head kidney macrophages (HKMs) from Clarias gariepinus have been established as an alternate model to understand A. hydrophila pathogenesis at the cellular level [Citation3,Citation14]. A. hydrophila is reported to induce activation of an intricate network of signaling molecules ultimately leading to caspase-3-mediated HKM apoptosis [Citation3]. The role of both caspase-9 as well as caspase-8 has been observed in triggering A. hydrophila-induced HKM apoptosis [Citation3]. Pro-apoptotic role of (Ca2+)C in ER-stress mediated HKM apoptosis is evident in the A. hydrophila-pathogenesis [Citation3], the upstream events that initiate the process have not been reported. In the present study, we describe the primal role of PI3K-PLC axis on ER-stress and mtROS generation that triggers the mtDNA/caspase-1/IL-1β axis induced apoptosis of A. hydrophila-infected HKM.
Results
PI3K-PLC signaling is imperative in triggering A. hydrophila-induced ER stress
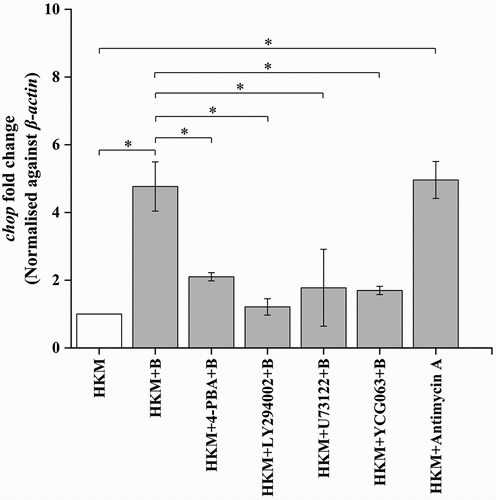
We had previously observed the role of ER-stress [Citation3] and PI3K-PLC induced (Ca2+)C surge in A. hydrophila-induced HKM apoptosis [Citation14]. Hence, our first step was to correlate the two molecular events in A. hydrophila-pathogenesis. HKMs were pre-incubated separately with PI3K specific inhibitor, LY294002 and PLC specific inhibitor, U73122, respectively, and chop expression monitored by RT-qPCR at 2 h p.i. We had observed chop expression to be the maximum at 2 h p.i. in A. hydrophila-infected HKM and selected this time point for the study (data not shown). We noted that pre-incubation with LY294002 and U73122 led to significant inhibition of chop-mRNA expression (). ER-stress inhibitor, 4-PBA was used as control for the study which effectively repressed chop mRNA () and protein expression in A. hydrophila-infected HKM (Fig. S1). We conclude that PI3K-PLC induced (Ca2+)C surge has a primal role in A. hydrophila-induced ER stress.
Figure 1. A. hydrophila-induced PI3K/PLC axis activates ER-stress. HKM pre-incubated with 4-PBA, LY-294002, U73122, YCG063, and Antimycin A were infected with or without A. hydrophila and chop expression studied at 2 h p.i. Vertical bars represent mean ± SE (n = 3). Asterisk indicates significant difference between indicated groups (*p < 0.05). HKM, uninfected HKM; HKM+B, HKM infected with A. hydrophila; HKM+4-PBA+B, HKM pre-incubated with 4-PBA infected with A. hydrophila; HKM+LY-294002 + B, HKM pre-incubated with LY-294002 infected with A. hydrophila; HKM+U73122 + B, HKM pre-incubated with U73122 infected with A. hydrophila; HKM+YCG063 + B, HKM pre-incubated with YCG063 infected with A. hydrophila, HKM+Antimycin A, HKM pre-incubated with Antimycin A.
ER-stress promotes HKM apoptosis by regulating the AKT-JNK signaling cascade
Pathogen-induced activation of MAPK pathway has been reported in various cellular processes including apoptosis of host macrophages [Citation45]. Toward this direction, we studied the involvement of MAPK family member JNK, which is known to induce apoptosis in several model systems [Citation46]. We measured the levels of total and phosphorylated JNK, respectively, by immunoblotting from cell lysates of infected HKM collected at 24 h p.i. We could detect increased phosphorylation of JNK in the infected HKM ().
Figure 2. A. hydrophila-induced ER-stress induces HKM apoptosis via JNK-Akt signaling pathway. HKM pre-incubated with SP600125, 124,005 or transfected with sc-siRNA or chop-siRNA were infected with A. hydrophila and at 24 h p.i. (a) phosphorylation of JNK, (b) levels of total and phospho-Akt, and (c) HKM apoptosis was studied. Vertical bars represent mean ± SE (n = 3). Asterisk indicates significant difference between indicated groups (*p < 0.05). HKM, uninfected HKM; HKM+B, HKM infected with A. hydrophila; HKM+SP600125 + B, HKM pre-incubated with SP600125 infected with A. hydrophila; HKM+124005 + B, HKM pre-incubated with 124,005 infected with A. hydrophila; HKM+sc-siRNA, HKM transfected with sc-siRNA (scrambled siRNA); HKM+chop-siRNA+B, HKM transfected with chop-siRNA infected with A. hydrophila.
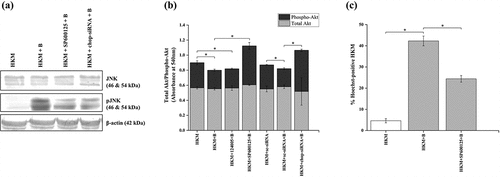
We tried to investigate whether ER-stress was possibly activating JNK. The HKMs transfected with chop-siRNA were infected and the changes in total and phosphorylated JNK levels studied at 24 h p.i. We observed that chop-siRNA effectively inhibited the phosphorylation of JNK in the infected HKM, though the total JNK levels remained unaltered (). The specific pharmacological inhibitor for JNK, SP600125 served as negative control in the study (). This reflects that ER-stress activates JNK in the A. hydrophila-infected HKM.
The role of PI3K in HKM apoptosis made us interested to study the role of PI3K-dependent kinase, Akt in the cascade of events. HKMs were transfected with chop-siRNA and then infected with A. hydrophila and the changes in total and phospho-Akt levels studied using specific ELISA kits. We observed significant reduction in active or phospho-Akt levels in the infected HKM (), though the total Akt levels remained unaltered (). HKM pre-incubated with Akt inhibitor, 124,005 served as positive control for the experiment.
Once we confirmed JNK activation in infected HKM, we set out to determine its consequences. HKM were pre-incubated with SP600125 prior to infection and apoptotic cell death was studied at 24 h p.i. It is evident from that inhibition of JNK significantly inhibited A. hydrophila-induced HKM apoptosis. We extended our observation in which we pre-incubated the HKM with SP600125 and studied the changes in total and phospho-Akt consequent to A. hydrophila infection. We observed that inhibiting JNK activity resulted in significant increase in phospho-Akt levels () and inhibited HKM apoptosis (). Pre-incubation with SP600125 had no effect on total Akt levels.
Collectively, these observations suggest (i) JNK activation is downstream event of ER-stress and plays a critical role in HKM apoptosis and (ii) JNK exerts its pro-apoptotic effect by inhibiting Akt activation.
ER-stress mtROS crosstalk is critical in A. hydrophila-induced HKM apoptosis
Mitochondrial ROS (mtROS) plays an important role in microbial pathogenesis and host defense [Citation47,Citation48]. However, the role of mtROS has not been studied in A. hydrophila-pathogenesis. To look into this, HKMs were infected with A. hydrophila and mtROS production monitored at indicated time intervals p.i. We observed maximum mtROS production at 4 h p.i. (Fig S2) and choose this time interval for further studies. Dead (Heat-killed) A. hydrophila failed to induce mtROS production (). Electron transport chain (ETC) inhibitor antimycin A induces mtROS [Citation49] and was used as positive control in the study (). Parallelly, HKM pre-incubated with mtROS inhibitor YCG063 were infected with A. hydrophila and apoptotic cell death studied at 24 h p.i. We observed pre-treatment with YCG063 alleviated HKM apoptosis () suggesting mtROS production is critical in A. hydrophila pathogenesis.
Figure 3. A. hydrophila induces pro-apoptotic mtROS production. HKM pre-incubated with Antimycin A, YCG063, 4-PBA, 2-APB, Dant, Ru360, Cyt D, heat-killed bacteria and Rapa were infected with or without A. hydrophila and (a) changes in mtROS levels were studied at 4 h p.i. and (b) HKM apoptosis was studied at 24 h p.i. Vertical bars represent mean ± SE (n = 3). Asterisk indicates significant difference between indicated groups (*p < 0.05). HKM, uninfected HKM; HKM+B, HKM infected with A. hydrophila; HKM+YCG063 + B, HKM pre-incubated with YCG063 infected with A. hydrophila; HKM+4-PBA+B, HKM pre-incubated with 4-PBA infected with A. hydrophila; HKM+2-APB +B, HKM pre-incubated with 2-APB infected with A. hydrophila; HKM+Dant+B, HKM pre-incubated with dantrolene infected with A. hydrophila; HKM+Ru360 + B, HKM pre-incubated with Ru360 infected with A. hydrophila; HKM+Cyt D + B, HKM pre-incubated with Cyt D infected with A. hydrophila; HKM+heat-killed B, HKM infected with heat-killed A. hydrophila; HKM+Rapa+B, HKM pre-incubated with rapamycin infected with A. hydrophila, HKM+Antimycin A, HKM pre-incubated with Antimycin A.
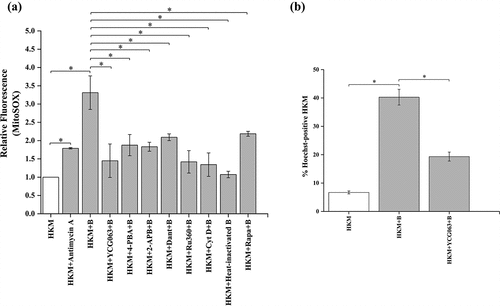
(Ca2+)ER is released through membrane resident IP3R and RyR channels, which gets mobilized into the mitochondria influencing mtROS generation [Citation15,Citation22]. To study this, HKMs pre-incubated with IP3R and RyR antagonist 2-APB and dantrolene (Dant), respectively, were infected with A. hydrophila and the changes in mtROS levels monitored at 4 h p.i. We observed significant reduction in mtROS levels in the presence of both 2-APB and Dant in A. hydrophila-infected HKM (). In parallel study, HKM was pre-incubated with 4-PBA, and the changes in A. hydrophila-induced mtROS production measured at 4 h p.i. We observed that alleviating ER-stress attenuated mtROS production in A. hydrophila-infected HKM (). These results established the importance of ER-stress and ensuing (Ca2+)ER dynamics on mtROS generation in A. hydrophila-infected HKM.
Our previous studies suggested the proximity between mitochondria and ER facilitate the uptake of (Ca2+)ER by mitochondria [Citation3]. We reasoned that if the proximity between the two organelles is prevented, (Ca2+)ER mobilization would be interrupted with a concomitant decline in mtROS levels. To study this, HKMs were pre-incubated with Cyt D which inhibits mitochondrial motility [Citation3,Citation50], then infected with A. hydrophila and the changes in mtROS production monitored at 4 h p.i. We observed significant reduction in mtROS production in the presence of Cyt D (), that clearly established co-localization of ER and mitochondria is important for the uptake of (Ca2+)ER and mtROS generation in A. hydrophila-infected HKM.
We followed this by identifying the molecular mechanism of (Ca2+)ER dynamics. Mitochondrial calcium uniporters (MCU) present on the mitochondrial membrane facilitates the uptake of (Ca2+)ER into the mitochondria [Citation51]. Additionally, HKMs were pre-incubated with Ru360, a selective inhibitor of MCU [Citation17], and measured mtROS levels at 4 h p.i. Pre-treatment with Ru360 led to significant decline in mtROS production in the infected HKM (), suggesting MCU to be responsible for regulating (Ca2+)ER influx in mitochondria.
We were interested to know whether mtROS per se has any role in the induction of ER-stress in A. hydrophila-infected HKM. Toward that end, HKMs were pre-incubated with mtROS inhibitor YCG063 [Citation52], and then infected with A. hydrophila and chop expression studied by RT-qPCR at 2 h p.i. We noted that pre-treatment with YCG063 significantly repressed chop expression in the infected HKM (). Additionally, antimycin A treatment also upregulated chop-mRNA expression in HKM (). These results collectively implicated that the interplay between (Ca2+)ER and mtROS signaling is critical for A. hydrophila pathogenesis.
A. hydrophila inhibits mitophagy triggering mtDNA release into cytosol
It has been observed that mtROS causes irreversible damage to mitochondria triggering the release of mitochondrial DNA (mtDNA) [Citation27]. First, we investigated the cytosolic translocation of mtDNA in A. hydrophila pathogenesis. HKMs were infected with A. hydrophila and the cytosolic migration of mtDNA studied. The mito-genome of C. gariepinus has been sequenced [Citation53] and using that information specific primers for the mitochondrial gene, cox2 of C. gariepinus was designed (). HKMs were lysed at indicated time intervals, mtDNA was isolated from the cytosol, PCR amplified using cox2 primers and the presence of cytosolic mtDNA studied. Our results suggested the presence of mtDNA in cytosol of infected HKM at 1 h p.i., (Fig S3); therefore, this time point was selected for further experiments. We hypothesized that A. hydrophila-induced mtROS triggers the release of mtDNA into cytosol, thereby impacting host immunity. For this, HKMs were pre-incubated with YCG063 following A. hydrophila infection. At 1 h p.i., mtDNA was quantitated using RT-PCR and the amount of cytosolic mtDNA (absolute quantification) was interpolated from the standard curve (see M&M section). Our results suggested inhibition of mtROS attenuated mtDNA release into cytosol (). Furthermore, treatment with antimycin A increased the amount of cytosolic mtDNA in A. hydrophila-infected HKM (). These results confirm previous findings suggesting the role of mtROS on releasing mtDNA in cytosol [Citation27].
Figure 4. A. hydrophila triggers translocation of mtDNA via suppression of autophagy. (a) HKM were pre-incubated with Antimycin A, YCG063, CsA, Rapa, or transfected with DNase I and heat-inactivated DNase I were infected with or without A. hydrophila and presence of mtDNA in cytosol was studied at 1 h p.i. (b) HKMs were infected with A. hydrophila and expression of autophagic proteins (beclin-1, atg 5, and LC3B) were studied at by immunoblotting. (c) HKM incubated with rapamycin infected with A. hydrophila and percentage HKM viability was studied at 24 h p.i. using trypan blue dye exclusion method. Vertical bars represent mean ± SE (n = 3). Asterisk indicates significant difference between indicated groups (*p < 0.05). ns indicates not significant between indicated groups. HKM, uninfected HKM; HKM+B, HKM infected with A. hydrophila; HKM+YCG063 + B, HKM pre-incubated with YCG063 infected with A. hydrophila; HKM+CsA+B, HKM pre-incubated with Cyclosporin A infected with A. hydrophila; HKM+Rapa+B, HKM pre-incubated with rapamycin infected with A. hydrophila; HKM+DNase I + B, HKM transfected with DNase I infected with A. hydrophila; HKM+heat-inactivated DNase I + B, HKM transfected with heat-inactivated DNase I infected with A. hydrophila; HKM+Antimycin A, HKM pre-incubated with Antimycin A.
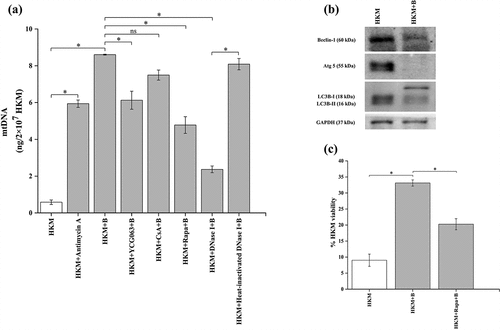
Table 2. List of RT-qPCR primer sequences
The next step was exploring the mechanisms underlying release of mtDNA into cytosol and studying its impact on A. hydrophila-infected HKM. It has been suggested that mitochondrial pore transition (MPT) facilitates the release of mtDNA into the cytosol [Citation27]. We observed that pre-incubation with MPT inhibitor CsA failed to inhibit the cytosolic migration of mtDNA in A. hydrophila-infected HKM (). Autophagy proteins have also been implicated in regulating the cytosolic migration of mtDNA [Citation27]. To investigate this, HKMs were infected with A. hydrophila and the expression of autophagy proteins Beclin-1, LC3B and Atg 5 studied by immunoblot. We observed down-regulation of Beclin-1, LC3B and Atg 5 expression in A. hydrophila-infected HKM (). Additionally, HKMs were pre-incubated with autophagy inducer, rapamycin and then infected with A. hydrophila and cytosolic translocation of mtDNA studied at 1 h p.i. and HKM viability monitored at 24 h p.i. respectively. We observed rapamycin inhibited the cytosolic translocation of mtDNA () with concomitant reduction in HKM death (). These findings clearly suggested that A. hydrophila inhibits mitophagy facilitating the release of mtDNA in cytosol, thereby triggering HKM death.
mtDNA triggers pro-inflammatory caspase-1/IL-1β cascade in A. hydrophila-infected HKM
We next explored how mtDNA impacts A. hydrophila pathogenesis. The role of mtDNA in activating pro-inflammatory caspase-1/IL-1β cascade is well established [Citation27]. This encouraged us to evaluate the role of mtDNA in activating the caspase-1/IL-1β axis in A. hydrophila-infected HKM. At the outset, HKMs were infected with A. hydrophila and the changes in caspase-1 activity and IL-1β levels measured. We observed maximum caspase-1 activity at 12 h p.i. and peak IL-1β levels at 24 h p.i. respectively and choose these time points for further studies [Citation54]. To correlate caspase-1 activation with A. hydrophila-pathogenesis, HKMs were pre-incubated with caspase-1 inhibitor, Z-YVAD-FMK and then infected with A. hydrophila and caspase-1 activity and HKM apoptosis studied. Pre-treatment with Z-YVAD-FMK inhibited caspase-1 activity () and HKM apoptosis () implicating the role of caspase-1 in A. hydrophila-pathogenesis. Subsequently, HKM pre-incubated with Z-YVAD-FMK were infected with A. hydrophila and IL-1β levels measured. We observed that inhibiting caspase-1 activity interfered with IL-1β production in the infected HKM ().
Figure 5. Cytosolic mtDNA triggers activation of pro-apoptotic caspase-1 in A. hydrophila-infected HKM. (a) HKMs pre-incubated with Z-YVAD-FMK, 4-PBA, YCG063 or transfected with mtDNA, DNase I, heat-inactivated DNase I were infected with or without A. hydrophila and caspase-1 activity studied at 12 h p.i. (b) HKMs incubated with Z-YVAD-FMK were infected with A. hydrophila and HKM apoptosis studied at 24 h p.i. Vertical bars represent mean ± SE (n = 3). Asterisk indicates significant difference between indicated groups (*p < 0.05). HKM, uninfected HKM; HKM+B, HKM infected with A. hydrophila; HKM+Z-YVAD-FMK+B, HKM pre-incubated with Z-YVAD-FMK infected with A. hydrophila; HKM+4-PBA+B, HKM pre-incubated with 4-PBA infected with A. hydrophila; HKM+YCG063 + B, HKM pre-incubated with YCG063 infected with A. hydrophila; HKM+mtDNA, HKM transfected with mtDNA; HKM+DNase I + B, HKM transfected with DNase I infected with A. hydrophila; HKM+heat-inactivated DNase I + B, HKM transfected with heat-inactivated DNase I infected with A. hydrophila.
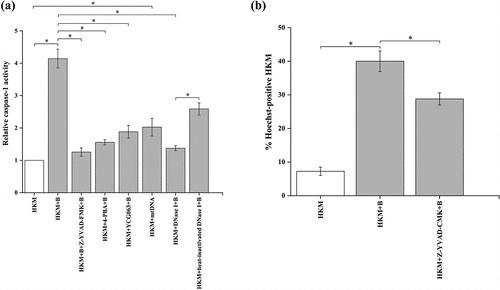
Figure 6. A. hydrophila-induced mtDNA/caspase-1 axis triggers IL-1β production. HKM pre-incubated with Z-YVAD-FMK, 4-PBA, YCG063 or transfected with sc-siRNA, il1b-siRNA, DNase I, heat-inactivated DNase I, mtDNA were infected with or without A. hydrophila and production of IL-1β protein was measured at 24 h p.i. Vertical bars represent mean ± SE (n = 3). Asterisk indicates significant difference between indicated groups (*p < 0.05). ns indicates not significant between indicated groups. HKM, uninfected HKM; HKM+B, HKM infected with A. hydrophila; HKM+Z-YVAD-FMK+B, HKM pre-incubated with Z-YVAD-FMK infected with A. hydrophila; HKM+4-PBA+B, HKM pre-incubated with 4-PBA infected with A. hydrophila; HKM+YCG063 + B, HKM pre-incubated with YCG063 infected with A. hydrophila; HKM+sc-siRNA, HKM transfected with sc-siRNA; HKM+sc-siRNA+B, HKM transfected with sc-siRNA infected with A. hydrophila, HKM+il1b-siRNA+B, HKM transfected with il1b-siRNA infected with A. hydrophila; HKM+DNase I + B, HKM transfected with DNase I infected with A. hydrophila; HKM+heat-inactivated DNase I + B, HKM transfected with heat-inactivated DNase I infected with A. hydrophila; HKM+mtDNA, HKM transfected with mtDNA.
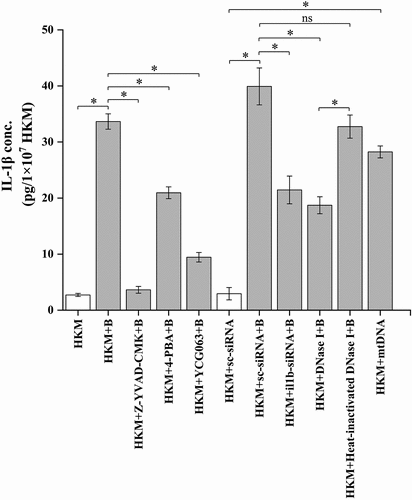
Our next step was elucidating the connection between ER-stress/mtROS axis and caspase-1/IL-1β cascade. HKMs were pre-incubated separately with 4-PBA and YCG063, respectively, then infected with A. hydrophila and caspase-1 activity and IL-1β levels studied. We observed that pre-incubation with 4-PBA and YCG063 inhibited caspase-1 () and suppressed IL-1β levels () in A. hydrophila-infected HKM. Collectively, these findings signify the role of ER-stress-induced mtROS in triggering caspase-1/IL-1β cascade activation in A. hydrophila-infected HKM.
We followed this by transfecting mtDNA and studied the effect on caspase-1 and IL-1β. It was observed that transfection of mtDNA augmented caspase-1 activation () and IL-1β production () in A. hydrophila infected HKM. To confirm the role of mtDNA in activating the caspase-1/IL-1β axis, HKMs were transfected with DNase I and then infected with A. hydrophila. We observed that DNase I transfection interfered with the cytosolic accumulation of mtDNA and inhibited caspase-1 activity and IL-1β production in the infected HKM. The transfection of heat-inactivated DNase I failed to reverse the translocation of mtDNA and inhibit caspase-1 activity in A. hydrophila-infected HKM (). To this, we concluded that cytosolic mtDNA triggers the caspase-1/IL-1β cascade in A. hydrophila-infected HKM, thereby influencing the pathogenesis induced by the bacterium in fish.
IL-1β triggers pro-apoptotic NO in A. hydrophila-infected HKM
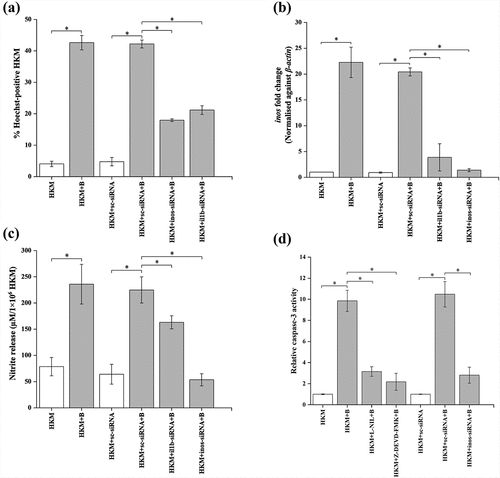
Previous studies suggested that IL-1β triggers necrosis [Citation55] and apoptosis [Citation56] under various conditions of stress. To investigate this, HKMs were transfected with il1b-siRNA and apoptosis studied at 24 h p.i. We noted significant decline in the number of Hoechst positive HKM () suggesting pro-apoptotic role of IL-1β in A. hydrophila pathogenesis. We wondered how IL-1β contributed toward A. hydrophila-induced HKM apoptosis. Several reports have implicated the contribution of NO in IL-1β mediated apoptosis [Citation57,Citation58]. We observed time dependent increase in inos-mRNA expression (Fig S4) and NO levels recorded at 24 h p.i [Citation3]. In view of this, HKM transfected with il1b-siRNA were infected with A. hydrophila and the changes in inos expression and NO levels monitored at 24 h p.i. It is clear from RNAi studies that there was marked reduction in inos expression () and NO production () with parallel decrease in HKM apoptosis () and caspase-3 activity () which suggested positive correlation between IL-1β production and NO-mediated apoptosis of A. hydrophila-infected HKM.
Figure 7. IL-1β induces activation of pro-apoptotic inos/NO axis in A. hydrophila-infected HKM. HKM transfected with sc-siRNA, il1b-siRNA, inos-siRNA or pre-incubated with L-NIL, Z-DEVD-FMK and infected with A. hydrophila and at 24 h p.i., (a) HKM apoptosis, (b) the expression of inos mRNA was studied, (c) nitrite levels were measured, and (d) caspase-3 activity was studied at 24 h p.i. Vertical bars represent mean ± SE (n = 3). Asterisk indicates significant difference between indicated groups (*p < 0.05). HKM, uninfected HKM; HKM+B, HKM infected with A. hydrophila; HKM+L-NIL+B; HKM pre-incubated with L-NIL and infected with A. hydrophila; HKM+Z-DEVD-FMK+B, HKM pre-incubated with Z-DEVD-FMK and infected with A. hydrophila; HKM+sc-siRNA, HKM transfected with sc-siRNA; HKM+sc-siRNA+B, HKM transfected with sc-siRNA infected with A. hydrophila; HKM+il1b-siRNA+B, HKM transfected with il1b-siRNA infected with A. hydrophila; HKM+inos-siRNA+B, HKM transfected with inos-siRNA infected with A. hydrophila.
Discussion
A. hydrophila displays wide host tropism that includes both poikilothermic and homoeothermic organisms, including mammals. How the bacterium infects such a wide range of hosts is still not known. Therefore, to have an insight of the molecular underpinning of A. hydrophila-pathogenesis we selected HKM as a model. Fish can be considered a good model for the present study because A. hydrophila is a natural fish pathogen [Citation19] and significant similarity between fish and mammalian immune systems [Citation59]. Studying A. hydrophila-HKM interactive pathways in fish will shed important light on the universal aspects of these regulations, beyond the fish syndrome which otherwise may not possible in other systems such as mammals.
Our previous studies suggested that A. hydrophila-induced alterations in (Ca2+)c levels is critical toward inducing ER-stress [Citation3]. We also observed that the PI3K/PLC axis plays an important role in inducing (Ca2+)c alterations in A. hydrophila-infected HKM [Citation14]. The two independent findings encouraged us to hypothesize a positive correlation between PI3K/PLC axis and ER-stress in A. hydrophila-pathogenesis. Toward that end, we studied the expression of well-known apoptotic ER-stress marker, CHOP in the absence of PI3K/PLC signaling. The marked decline in CHOP expression implicates PI3K/PLC mediated release of (Ca2+)c is indispensable for ER-stress in A. hydrophila-infected HKM. PI3K is well known to favor cell survival via activation of downstream kinase, Akt/protein kinase B. The involvement of PI3K in ER-stress induced apoptotic cascade is interesting and prompted us to study the underlying molecular mechanisms. Previous studies suggested that A. hydrophila suppresses Akt activation [Citation60], and Akt inhibition triggers apoptosis [Citation61]. The role of Akt in ER-stress is not well understood and we reasoned that prolonged ER-stress suppresses the activation of Akt, in A. hydrophila-infected HKM. We observed suppressed Akt activity in the infected HKM, which got reversed on ameliorating ER-stress leading to increased HKM survival. Our findings are in concordance with previous findings [Citation62] and suggest the central role of CHOP-Akt crosstalk in A. hydrophila pathogenesis.
We wondered how ER-stress inhibits Akt phosphorylation to promote HKM apoptosis. MAPK family members have been implicated in apoptosis and it has been suggested that CHOP inactivates Akt phosphorylation by activating MAPK [Citation63]. We observed a proportionate increase in phospho-JNK expression with apoptotic implication in A. hydrophila-infected HKM which was reduced in chop-knockdown HKM suggesting a positive relation between ER-stress and JNK activation. Concordantly, repressing JNK activation also resulted in increased Akt activity and HKM viability. Our results for the first time suggested that CHOP promotes apoptosis of A. hydrophila-infected HKM by inhibiting AKT activation and enhancing MAPK signaling.
ER-stress-induced mitochondrial dysfunction has a prominent role in triggering apoptosis of A. hydrophila-infected HKM [Citation3], but the molecular intermediates responsible for the process has not been addressed. Under stress, ER releases (Ca2+)ER, which is taken up by the mitochondria triggering the production of mtROS [Citation22,Citation64]. mtROS has not been implicated in A. hydrophila pathogenesis. Therefore, we evaluated mtROS production in A. hydrophila-infected HKM and observed significant mtROS generation which was inhibited on ameliorating ER-stress. Additionally, pre-treatment with (Ca2+)ER release modulators and MCU inhibitors also repressed mtROS levels altogether confirming the positive correlation between (Ca2+)ER and mtROS production in A. hydrophila-infected cells. Mitochondrial-Ca2+ uptake is expedited by the proximity between the mitochondria and ER [Citation3,Citation65]. The reduction in mtROS levels on pre-treatment with cyt D, which inhibits mitochondrial movement toward ER lends support to the hypothesis. We are presently trying to ascertain the molecular intermediates involved in ER-mitochondria cross-talk.
mtROS generated under varied conditions of stress is known for its ability to induce apoptosis [Citation66,Citation67], but the same has not been reported in fish. Our next step was correlating mtROS with A. hydrophila-induced HKM apoptosis and for that, we used mtROS augmenter (antimycin A) and inhibitor (YCG063). Interestingly, our results revealed pro-apoptotic implication of mtROS in A. hydrophila-infected HKM. This is the first report on the role of mtROS impacting microbial-pathogenesis in fish to the best of our knowledge. With the consensus of previous reports [Citation66,Citation67] and our findings, we posit that the potential of mtROS to induce apoptosis is conserved across species. Collectively, our findings lend support to earlier studies suggesting that the close connection between ER and mitochondria, and ensuing Ca2+ dynamics between them determine cell-fate, leading to different pathological conditions [Citation3,Citation68,Citation69].
We further aimed to study how mtROS influence HKM apoptosis. Increased mtROS levels affect several cellular targets including the organelle itself with pathological consequences [Citation70,Citation71]. mtROS induced structural-functional alterations in mitochondria release mtDNA into the cytosol [Citation27]. mtDNA is a DAMP which stimulates innate immune responses that influence antimicrobial responses, inflammatory pathology, and apoptosis [Citation25,Citation26]. We had previously observed A. hydrophila-induced structural-functional alterations in mitochondria of HKM [Citation3]. We hypothesized that elevated mtROS triggers the cytoplasmic migration of mtDNA thereby contributing in A. hydrophila pathogenesis. Indeed, our results confirmed that mtROS facilitates the cytosolic accumulation of mtDNA in infected HKM. Additionally, inhibiting the cytosolic accumulation of mtDNA attenuated HKM death which clearly established the pro-apoptotic implication of cytosolic mtDNA in A. hydrophila pathogenesis.
The mechanism underlying mtDNA escape to the cytosol is still not well understood [Citation24,Citation72]. MPT is observed to play a vital role in mitochondrial-pathology and previous studies have implicated its role in release of mtDNA in the cytosol [Citation27]. We could not observe significant role of MPT on the translocation of mtDNA in infected HKM. Several mechanisms have been suggested to be involved in the release of mtDNA [Citation71–73] and we are trying to ascertain the role of other factors in triggering mtDNA release in A. hydrophila-infected cells. Nonetheless, this is the first study linking mtDNA in A. hydrophila pathogenesis. Our results are of great significance as mtDNA is a critical innate immune agonist that impacts host responses.
Autophagy helps in the removal of damaged and malfunctioned cellular components [Citation74,Citation75]. It is also increasingly evident that autophagy is intimately associated with conferring innate protection against microbial pathogens [Citation76]. Earlier studies suggest, suppressed autophagy promotes mtROS generation and the cytosolic translocation of mtDNA [Citation27,Citation77,Citation78]. We observed suppressed expression of autophagic proteins in A. hydrophila-infected HKM. Furthermore, treatment of HKM with autophagy inducer, rapamycin resulted in the decreased accumulation of cytosolic mtDNA and inhibited HKM death. To this, we concluded that impaired mitophagy has a major role in A. hydrophila pathogenesis. The mechanism by which autophagy is suppressed in A. hydrophila-infected HKM is important to understand the pathogenesis of UDS.
The involvement of mtROS/mtDNA axis has been well elucidated as a platform for inflammasome activation in mammals [Citation76,Citation77]; however, the same has not been explained in fishes. The structure and function of Nod-like receptors (NLRs), the main component of inflammasome formation, are also poorly understood in fish. One important outcome of inflammasome is caspase-1/IL-1β activation [Citation77,Citation79]. The involvement of caspase-1/ IL-1β axis has been described earlier in several fish species [Citation32,Citation34]. Moreover, A. hydrophila related toxins induce caspase-1/IL-1β activation in bone marrow-derived macrophages of mice [Citation80], but their functional consequences are not completely known in fish. We noted enhanced caspase-1 activity in A. hydrophila-infected HKM was repressed on ameliorating ER-stress and mtROS production. This evidently indicated the ER-stress induced mtROS production is inevitable for caspase-1 activation in A. hydrophila-infected HKM.
An interesting outcome of this study was the pro-active role of cytosolic mtDNA in triggering caspase-1/IL-1β activity. Transfection of DNase I prevented the cytosolic aggregation of mtDNA and inhibited the activation of caspase-1/IL-1β axis in A. hydrophila-infected HKM. The role of cytosolic DNA (exogenous) has been reported in caspase-1 activation in several model systems [Citation81,Citation82]. Additionally, the role of mtDNA in caspase-1/IL-1β cascade has also been observed in mammalian cells under different conditions of stress [Citation27,Citation83]. This is the first study implicating mtDNA in the activation of caspase-1/IL-1β cascade and regulating microbial pathogenesis in fish. Based on these findings and earlier reports, we speculate this to be an evolutionary conserved host response against microbial pathogens.
We further monitored how IL-1β induce apoptosis of the infected HKM? IL-1β is mainly responsible for generating a pro-inflammatory response during host-pathogen interaction [Citation84], and there are also reports suggesting its involvement in apoptosis [Citation56]. However, there are reports documenting, IL-1β induces pro-apoptotic effects by affecting NO production [Citation57]. We had previously reported that NO primarily contributed in the activation of extrinsic pathway of apoptosis in A. hydrophila-infected HKM [Citation3]. Our RNAi studies indeed demonstrated marked inhibition in inos-mRNA and NO levels with concomitant decline in apoptosis of A. hydrophila-infected HKM, in absence of IL-1β. To this, we concluded that the caspase-1/IL-1β cascade culminates in NO-mediated activation of extrinsic caspases, triggering the apoptosis of A. hydrophila-infected HKM. Together, our results reveal an alternate pathway of caspase activation in A. hydrophila-pathogenesis.
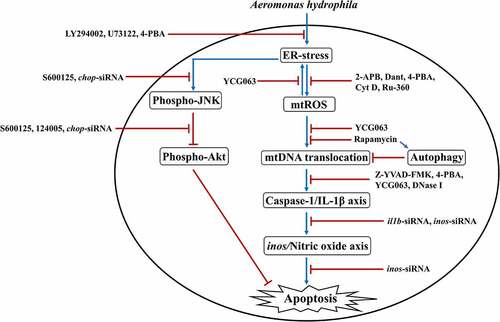
In this study, we have shown that PI3K-PLC signaling induced (Ca2+)C efflux results in the activation of pro-apoptotic ER-stress via modulation of Akt-JNK signaling pathway in A. hydrophila-infected HKM. Additionally, our observations demonstrated that interplay between (Ca2+)ER dynamics and mtROS signaling contributes to the cytosolic translocation of mtDNA. Based on our findings, we propose that impairment in autophagic machinery favors accretion of impaired mitochondria which release mtDNA in cytosol, amplifying the activation of inflammatory caspase-1/IL-1β signalosome and consequently NO-mediated HKM apoptosis (). Our findings would be helpful toward understanding A. hydrophila pathogenesis and controlling UDS.
Figure 8. Overview of the study. A. hydrophila-induced activation of PI3K/PLC axis triggers ER-stress leading to downstream phosphorylation of JNK and consequent inhibition of Akt phosphorylation. ER-stress instigates mtROS production leading to cytosolic translocation of mtDNA via suppression of autophagy. The cytosolic mtDNA activates inflammatory caspase-1/IL-1β axis leading to NO-mediated apoptosis of A. hydrophila-infected HKM.
Materials and methods
Animal maintenance
All the catfish (Clarias gariepinus) studies were carried out listed in the procedures issued by Committee for the purpose of Control and Supervision of Experiments on Animals (CPCSEA), Govt. of India and permitted by Animal Ethics Committee (DU/ZOOL/IAEC-R/2013/33), University of Delhi. Prior to experiments, C. gariepinus were acclimatized for 15 days and fed with chicken liver ad libitum [Citation19].
HKM isolation, infection with A. hydrophila and inhibitor studies
C. gariepinus and A. hydrophila (Strain 500,297) were used in the study. The protocols for HKM isolation, and infection of HKM with A. hydrophila (MOI 1:50) have been described earlier [Citation14].
zHKMs were incubated separately with ER stress inhibitor (4-PBA, 10 µM, Sigma), mtROS inhibitor (YCG063, 10 µM, Calbiochem), ETC inhibitor (antimycin A, 50 µM, Sigma), MPTP inhibitor (Cyclosporin A, 5 µM, Sigma), RyR inhibitor (Dantrolene, 10 µM, Sigma), Cytochalasin D (Cyt D, 5 µg/mL, Sigma), Autophagy inducer (Rapamycin, 20 µM, Sigma), Thapsigargin (1 µM, Sigma), Akt inhibitor (124,005, 10 µM, Calbiochem) for 1 h and mitochondrial Ca2+ uptake inhibitor (Ru360, 10 µM, Calbiochem), Caspase-1 inhibitor (Z-YVAD-FMK, 7.5 µM, Biovision), Caspase-3 inhibitor (Z-DEVD-FMK, 10 µM, Biovision), IP3R inhibitor (2-APB, 100 μM, Sigma), PLC inhibitor (U73122, 2 μM, Enzo Life Science), PI3-Kinase inhibitor (LY-294002 hydrochloride, 14.5 μM, Sigma), intracellular Ca2+chelator, (BAPTA-AM, 5 mM, Sigma), and JNK inhibitor (SP600125, 10 µM, Calbiochem) for 2 h followed by A. hydrophila infection as mentioned earlier [Citation3]. The inhibitor concentrations had no adverse effects on HKM viability and bacterial growth (data not shown).
siRNA transfection
The protocol for transfecting HKM with specific or scrambled siRNA (sc-siRNA) () has been described earlier [Citation14]. Silencing of genes was checked by RT-qPCR and HKM viability monitored throughout the experiment.
Table 1. List of siRNA sequences
mtROS production
mtROS was monitored using the MitoSOX™ Red mitochondrial superoxide indicator (Molecular Probes) [Citation85]. HKMs (1 × 106/mL) were incubated with inhibitors and infected with A. hydrophila. HKMs were washed, incubated with MitoSOX (5 μM) for 20 min and the fluorescence levels measured at A510nm excitation and A580nm emission (Molecular Devices, Spectramax).
Akt assay
HKMs (1 × 107 /mL) were incubated with inhibitors or transfected with chop-siRNA and infected with A. hydrophila. The total Akt and phospho-Akt levels in the cell lysates were measured with Duoset IC ELISA kit (R&D Systems) following the instructions specified by the manufacturer.
mtDNA isolation, detection, and transfection
mtDNA from cytosol was isolated as described [Citation27]. HKMs (1 × 107/mL) incubated with inhibitors were infected with A. hydrophila. HKMs were harvested at different time points, lysed, and homogenized in 100 mM Tricine-NaOH (pH 7.4) supplemented with sucrose (0.25 M), EDTA (1 mM), proteinase K (5 mg/mL) and centrifuged at 700 × g. Next, the supernatant was centrifuged at 10,000 × g at 4°C for 30 min. mtDNA was obtained from the supernatant (cytosolic fraction) using DNeasy Blood & Tissue Kit (Qiagen) according to the manufacturer’s instructions. The supernatant was mixed and incubated with Buffer AL (200 µL) at 56°C for 10 min. Ethanol (200 µL) was added and centrifuged at 6000 × g for 1 min. Then, Buffer AW1 (500 µL) was added and centrifuged at 6000 × g for 1 min. Flow-through was discarded and Buffer AW2 (500 µL) was added and centrifuged at 20,000 × g for 3 min. mtDNA was eluted using Buffer AE (200 µL), incubated at RT for 1 min, and then centrifuge for 1 min at 6000 × g. The presence of mtDNA in cytosolic fraction was confirmed by PCR using mitochondrial DNA encoded gene cox2 specific primers.
mtDNA (50 ng) was transfected using HiPerFect Transfection reagent (Qiagen). The HiPerFect-mtDNA complex was incubated and the complex gently introduced to the HKM. The transfected HKMs were incubated for 6 h at 30°C, washed and proceeded for subsequent studies. Transfection of mtDNA was confirmed as described above.
Immunoblot
HKMs (1 × 107/mL) incubated with inhibitors or transfected with sc-siRNA or chop-siRNA were infected with A. hydrophila. The HKMs were lysed for CHOP at 2 h p.i., LC3B, Beclin-1, Atg 5 at 4 h p.i. and for total and phospho-JNK at 24 h p.i., resolved on SDS-PAGE and transferred to PVDF membrane. Membrane was blocked with 1% BSA in TBST for 1 h and subsequently incubated with anti-CHOP (1: 100, Cell Signaling Technology), anti-LC3B (1: 1000, Abcam), anti-Beclin-1 (1: 1000, Abcam), anti-Atg 5 (1: 1000, Abcam) and anti-total JNK and anti-phosphorylated JNK (Cell Signaling Technology, 1: 500 dilution), respectively, overnight at 4°C. Anti-GAPDH antibody (Santacruz, 1: 5000) was used for the normalization of Beclin-1, Atg 5, LC3B, and anti-β-actin antibody (Cell Signaling Technology, 1:10,000 dilution) was used for CHOP and JNK.
Quantitative real time PCR (RT-qPCR)
HKMs (1 × 107/mL) incubated with inhibitors or transfected with sc-siRNA or chop-siRNA, il1b-siRNA, inos-siRNA were infected with A. hydrophila. The HKMs were washed, total RNA extracted using TRI reagent (Sigma) from which cDNA was prepared as described earlier [Citation14]. RT-qPCR (ABI ViiA) was done using SYBR green PCR Master Mix (ABI) with specific primers (). Comparative ∆∆CT method was used to study the expression of target genes and normalized against β-actin used as a housekeeping gene as described earlier [Citation14].
The absolute quantification of cytosolic mtDNA was done by RT-PCR using primers specific for mitochondrial gene cox2. Different concentrations of mtDNA (0.1 ng, 0.5 ng, 1 ng, 5 ng, and 10 ng) was used to generate a standard curve. The concentration of cytosolic mtDNA in the samples was interpolated from the standard curve using ct values.
Protein transfection
DNase 1 (3 μg) or heat-inactivated DNase I (3 μg) was transfected into HKM (1 × 107/mL) using PULSinTM Reagent (Polyplus transfectionTM) for 4 h at 30°C [Citation27]. HKM viability was checked, infected with A. hydrophila and proceeded for subsequent studies.
Caspase-1 and caspase-3 assay
Caspase-1 activity (YVADase) and caspase-3 activity (DEVDase) were studied using caspase-1 and caspase-3 assay kit (Biovision). HKMs (1 × 107/mL) pre-incubated with indicated inhibitors or transfected with specific siRNA, mtDNA, DNase I, heat-inactivated DNase I, were infected with A. hydrophila. HKMs were washed, lysed (at 12 h p.i. for caspase-1 and at 24 h p.i. for caspase-3) and 50 μL of cell lysate was mixed with 2 × reaction buffer containing 5 μL of YVAD-pNA substrate (Caspase-1) and DEVD-pNA substrate (Caspase-3). The absorbance was recorded at A405nm.
IL-1β assay
HKMs (1 × 107/mL) incubated with inhibitors, or transfected with il1b-siRNA or sc-siRNA, mtDNA, DNase I, heat-inactivated DNase I, respectively, were infected with A. hydrophila. The changes in IL-1β levels in the culture supernatant were determined using IL-1β assay kit (SunLong Biotech) following the instructions specified by the manufacturer. The IL-1β concentration in the samples was quantified from the standard curve.
NO production
HKMs (1 × 106/mL) transfected separately with sc-siRNA, il1b-siRNA, inos-siRNA were infected with A. hydrophila and at 24 h p.i., NO production was quantitated using Griess’ reagent [Citation3]. The quantity of nitrite produced was interpolated from sodium nitrite standard curve.
Apoptosis studies
HKMs (1 × 106/mL) incubated with inhibitors or transfected with specific or sc-siRNA were infected with A. hydrophila. HKMs were washed at 24 h p.i. and stained with Hoechst 33,342. The number of apoptotic HKM was calculated under fluorescence microscope (× 40, Zeiss Imager Z2) [Citation14].
Statistical analysis
The statistical analyses were done using one-way ANOVA followed by Bonferroni post-hoc test (IBM SPSS Software, Version 22). The data are presented as mean ± SE. The p value < 0.05 were considered statistically significant.
Author contributions
Conceived and designed the experiments: MK, AS, PD, SM, AR. Performed the experiments: MK, AS, PD, AR. Analyzed the data: MK, AS, PD, SM. Contributed reagents/materials/analysis tools: SM. Wrote the paper: MK, AS, SM.
Data availability:
Data sharing not applicable – no new data generated.
Supplemental Material
Download Zip (6.9 MB)Acknowledgments
The authors are grateful to S. Barik, Cleveland State University for helpful discussions and critically analyzing the manuscript. We thank Chaitali Banerjee for help in immunoblot studies and Tushar K. Maiti and Manjula Kalia, Regional Centre for Biotechnology (RCB), Faridabad and Dhiraj Kumar, ICGEB, New Delhi for help in protein transfection and apoptosis studies.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Janda JM, Abbott SL. The genus Aeromonas: taxonomy, pathogenicity, and infection. Clin Microbiol Rev. 2010 Jan 1;23:35–73. Pmid:20065325.
- Galindo CL, Fadl AA, Sha J, et al. Microarray analysis of Aeromonas hydrophila cytotoxic enterotoxin-treated murine primary macrophages. Infect Immun. 2004 Sep 1;72:5439–5445. Pmid:15322042.
- Banerjee C, Singh A, Das TK, et al. Ameliorating ER-stress attenuates Aeromonas hydrophila-induced mitochondrial dysfunctioning and caspase mediated HKM apoptosis in Clarias batrachus. Sci Rep. 2014 Jul 25;4(1):1–3. Pmid: 25059203.
- Van Nhieu GT, Clair C, Grompone G, et al. Calcium signalling during cell interactions with bacterial pathogens. Biol Cell. 2004 Feb;96(1):93–101. Pmid:15093131.
- Malhotra JD, Kaufman RJ. ER stress and its functional link to mitochondria: role in cell survival and death. Cold Spring Harb Perspect Biol. 2011 Sep 1;3:a004424. Pmid:21813400.
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011 Mar;13(3):184–190. Pmid:21364565.
- Afrazi A, Branca MF, Sodhi CP, et al. Toll-like receptor 4-mediated endoplasmic reticulum stress in intestinal crypts induces necrotizing enterocolitis. J Biol Chem. 2014Apr4;289(14):9584–9599. Pmid:24519940.
- Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium–apoptosis link. Nat Rev Mol Cell Biol. 2003 Jul;4(7):552–565. Pmid:12838338.
- Rutkowski DT, Arnold SM, Miller CN, et al. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006 Nov 7;4(11):e374. Pmid:17090218.
- Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Pmid:22251901. Nat Rev Mol Cell Biol. 2012 Feb;13(2):89–102.
- Szegezdi E, Logue SE, Gorman AM, et al. Mediators of endoplasmic reticulum stress‐induced apoptosis. EMBO Rep. 2006 Sep;7(9):880–885. Pmid:16953201.
- Ibarra C, Estrada M, Carrasco L, et al. Insulin-like growth factor-1 induces an inositol 1, 4, 5-trisphosphate-dependent increase in nuclear and cytosolic calcium in cultured rat cardiac myocytes. J Biol Chem. 2004 Feb 27;279(9):7554–7565. Pmid:14660553.
- Chun J, Prince A. Activation of Ca2+-dependent signaling by TLR2. J Immunol. 2006 Jul 15;177:1330–1337. Pmid:16818794.
- Shelly A, Banerjee C, Saurav GK, et al. Aeromonas hydrophila-induced alterations in cytosolic calcium activate pro-apoptotic cPKC-MEK1/2-tnfα axis in infected head kidney macrophages of Clarias gariepinus. Dev Comp Immunol. 2017 Nov 1;76:392–402. Pmid: 28713009.
- Clapham DE. Calcium signaling. Cell. 2007 Dec 14;131:1047–1058. Pmid:18083096.
- Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003 Jul;4(7):517–529. Pmid:12838335.
- Hajnóczky G, Csordás G, Das S, et al. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium. 2006 Nov 1;40(5–6)553–560. Pmid:17074387.
- Pinton P, Giorgi C, Siviero R, et al. Calcium and apoptosis: ER-mitochondria Ca 2+ transfer in the control of apoptosis. Oncogene. 2008 Oct;27(50):6407–6418. Pmid:18955969.
- Banerjee C, Khatri P, Raman R, et al. Role of calmodulin-calmodulin kinase II, camp/protein kinase A and ERK 1/2 on Aeromonas hydrophila-induced apoptosis of head kidney macrophages. PLoS Pathog. 2014 Apr 24;10:e1004018. Pmid:24763432.
- Rousset S, Emre Y, Join-Lambert O, et al. The uncoupling protein 2 modulates the cytokine balance in innate immunity. Cytokine. 2006 Aug 1;35:135–142. Pmid:16971137.
- West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011 Jun;11(6):389–402. Pmid:21597473.
- Brookes PS, Yoon Y, Robotham JL, et al. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004 Oct;287(4):C817–33. Pmid:15355853.
- Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008 Jul;454(7203):455–462. Pmid:18650916.
- Tiku V, Tan MW, Dikic I. Mitochondrial functions in infection and immunity. Trends Cell Biol. 2020 Apr 1;30:263–275. Pmid:32200805.
- Kausar S, Yang L, Abbas MN, et al. Mitochondrial DNA: a key regulator of anti-microbial innate immunity. Genes (Basel). 2020 Jan;111:86. Pmid:31940818.
- West AP, Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol. 2017 Jun;17(6):363. Pmid:28393922.
- Nakahira K, Haspel JA, Rathinam VA, et al. Autophagy proteins regulate innate immune response by inhibiting NALP3 inflammasome-mediated mitochondrial DNA release. Nat Immunol. 2011 Mar;123:222–230. Pmid:21151103.
- Bronner DN, O’Riordan MX. A near death experience: shigella manipulates host death machinery to silence innate immunity. EMBO J. 2014 Oct 1;33:2137–2139. Pmid:25180233.
- Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta Bioenerg. 2008 Sep 1;1777:1092–1097. Pmid:18519024.
- Schroder K, Tschopp J. The inflammasomes. Cell. 2010 Mar 19;140:821–832. Pmid:20303873.
- López-Castejón G, Sepulcre MP, Mulero I, et al. Molecular and functional characterization of gilthead seabream Sparus aurata caspase-1: the first identification of an inflammatory caspase in fish.Mol Immunol. 2008 Jan 1;45:49–57. Pmid:17610954.
- Vojtech LN, Scharping N, Woodson JC, et al.Roles of inflammatory caspases during processing of zebrafish interleukin-1β in Francisella noatunensis infection. Infect Immun. 2012 Aug 1;80:2878–2885. Pmid:22689811.
- Reis MI, Do Vale A, Pereira PJ, et al. Caspase-1 and IL-1β processing in a teleost fish. PLoS One. 2012 Nov 30;7:e50450. Pmid:23336286.
- Rojas V, Camus‐Guerra H, Guzmán F, et al. Pro‐inflammatory caspase‐1 activation during the immune response in cells from rainbow trout Oncorhynchus mykiss (Walbaum 1792) challenged with pathogen‐associated molecular patterns. J Fish Dis. 2015 Nov;38(11):993–1003. Pmid:25477241.
- Pancer Z, Cooper MD. The evolution of adaptive immunity. Annu Rev Immunol. 2006 Apr 23;24:497–518. Pmid:16551257.
- Pan P, Zhang Q, Liu W, et al. Dengue virus infection activates interleukin-1β to induce tissue injury and vascular leakage. Front Microbiol. 2019 Nov 22;10:2637. Pmid:31824450.
- Krishnan N, Robertson BD, Thwaites G. Pathways of IL-1β secretion by macrophages infected with clinical Mycobacterium tuberculosis strains. Tuberculosis. 2013 Sep 1;93:538–547. Pmid:23849220.
- Gov L, Karimzadeh A, Ueno N, et al. Human innate immunity to Toxoplasma gondii is mediated by host caspase-1 and ASC and parasite GRA15. mBio. 2013 Aug 30;4(4). Pmid:23839215.
- Scapigliati G, Buonocore F, Bird S, et al. Phylogeny of cytokines: molecular cloning and expression analysis of sea bass Dicentrarchus labrax interleukin-1β. Fish Shellfish Immunol. 2001 Nov 1;11(8)711–726. Pmid:11759041.
- Pelegrın P, Chaves-Pozo E, Mulero V, et al. Production and mechanism of secretion of interleukin-1β from the marine fish gilthead seabream. Dev Comp Immunol. 2004 Mar 1;28:229–237. Pmid:14642889.
- Wang Y, Wang Q, Baoprasertkul P, et al. Genomic organization, gene duplication, and expression analysis of interleukin-1β in channel catfish (Ictalurus punctatus). Mol Immunol. 2006 Apr 1;43:1653–1664. Pmid:16280165.
- Seppola M, Larsen AN, Steiro K, et al. Characterisation and expression analysis of the interleukin genes, IL-1β, IL-8 and IL-10, in Atlantic cod (Gadus morhua L.). Mol Immunol. 2008Feb1;45:887–897. Pmid:17875325.
- Bird S, Wang T, Zou J, et al. The first cytokine sequence within cartilaginous fish: IL-1β in the small spotted catshark (Scyliorhinus canicula). J Immunol. 2002 Apr 1;168:3329–3340. Pmid:11907090.
- Ogryzko NV, Renshaw SA, Wilson HL. The IL-1 family in fish: swimming through the muddy waters of inflammasome evolution.Dev Comp Immunol. 2014 Sep 1;46:53–62. Pmid:24690566.
- Gräb J, Rybniker J. The expanding role of p38 mitogen-activated protein kinase in programmed host cell death. Microbiol Insights. 2019 Jul;12:1178636119864594. Pmid:31384128.
- Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008 Oct 20;27(48):6245–6251. Pmid: 18931691.
- Dunn JD, Alvarez LA, Zhang X, et al. Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol. 2015 Dec 1;6:472–485. Pmid: 26432659.
- Stocks CJ, Schembri MA, Sweet MJ, et al.For when bacterial infections persist: toll‐like receptor‐inducible direct antimicrobial pathways in macrophages. J Leukoc Biol. 2018 Jan 2;103:35–51. Pmid: 29345056.
- Ahmad T, Aggarwal K, Pattnaik B, et al. Computational classification of mitochondrial shapes reflects stress and redox state. Cell Death Dis. 2013 Jan 17;4(1)e461–e. Pmid: 23328668.
- Ligon LA, Steward O. Role of microtubules and actin filaments in the movement of mitochondria in the axons and dendrites of cultured hippocampal neurons.J Comp Neurol.2000 Nov 20;427:351–361. Pmid:11054698.
- Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. 2006 Jan;86(1):369–408. Pmid: 16371601.
- Kim KH, Park JY, Jung HJ, et al.Identification and biological activities of a new antiangiogenic small molecule that suppresses mitochondrial reactive oxygen species. Biochem Biophys Res Commun. 2011 Jan 7;404:541–545. Pmid: 21144820.
- Han C, Li Q, Xu J, et al. Characterization of Clarias gariepinus mitochondrial genome sequence and a comparative analysis with other catfishes. Biologia. 2015 Sep;70(9):1245–1253.
- Kumar M, Kumar J, Sharma S, et al. TLR22-mediated activation of TNF-α-caspase-1/IL-1β inflammatory axis leads to apoptosis of Aeromonas hydrophila-infected macrophages. Mol Immunol. 2021 Sep 1;137:114–123. Pmid: 34242920.
- Medel-Matus JS, Álvarez-Croda DM, Martínez-Quiroz J, et al. IL-1β increases necrotic neuronal cell death in the developing rat hippocampus after status epilepticus by activating type I IL-1 receptor (IL-1RI). Int J Dev Neurosci. 2014 Nov 1;38:232–240. Pmid:25449684.
- Shen J, Xu S, Zhou H, et al. IL-1β induces apoptosis and autophagy via mitochondria pathway in human degenerative nucleus pulposus cells. Sci Rep. 2017 Jan 25;7(1):1–2. Pmid: 28120948.
- Zaitsev SV, Appelskog IB, Kapelioukh IL, et al. Imidazoline compounds protect against interleukin 1beta-induced beta-cell apoptosis. Diabetes. 2001 Feb 1;50(suppl 1):S70. Pmid:11272206.
- Størling J, Binzer J, Andersson AK, et al. Nitric oxide contributes to cytokine-induced apoptosis in pancreatic beta cells via potentiation of JNK activity and inhibition of Akt. Diabetologia. 2005 Oct;4810:2039–2050. Pmid: 16132952.
- Magnadóttir B. Innate immunity of fish (overview). Fish Shellfish Immunol. 2006 Feb 1;20:137–151. Pmid:15950491.
- Wiles TJ, Dhakal BK, Eto DS, et al. Inactivation of host Akt/protein kinase B signaling by bacterial pore-forming toxins. Pmid:18234841. Mol Biol Cell. 2008 Apr;19(4):1427–1438.
- de Frias M, Iglesias-Serret D, Cosialls AM, et al. Akt inhibitors induce apoptosis in chronic lymphocytic leukemia cells. Haematologica. 2009 Dec;9412:1698–1707. Pmid:19815839.
- Hu H, Tian M, Ding C, et al. The C/EBP homologous protein (CHOP) transcription factor functions in endoplasmic reticulum stress-induced apoptosis and microbial infection. Front Immunol. 2019 Jan 4;9:3083. Pmid:30662442.
- Li Y, Jiang W, Niu Q, et al. eIF2α-CHOP-BCl-2/JNK and IRE1α-XBP1/JNK signaling promote apoptosis and inflammation and support the proliferation of Newcastle disease virus. Cell Death Dis. 2019 Nov 26;10(12)1–5. Pmid:31767828.
- Delierneux C, Kouba S, Shanmughapriya S, et al. Mitochondrial calcium regulation of redox signaling in cancer.Cells. 2020 Feb 12;9:432. Pmid:32059571.
- Hayashi-Nishino M, Fujita N, Noda T, et al. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009 Dec;11(12):1433–1437. Pmid:19898463.
- Pinegin B, Vorobjeva N, Pashenkov M, et al. The role of mitochondrial ROS in antibacterial immunity. J Cell Physiol. 2018 May;233(5):3745–3754. Pmid:28771715.
- Wang J, Toan S, Zhou H. New insights into the role of mitochondria in cardiac microvascular ischemia/reperfusion injury. Pmid:32246225. Angiogenesis. 2020 Aug;23(3):299–314.
- Marchi S, Pinton P. Alterations of calcium homeostasis in cancer cells. Curr Opin Pharmacol. 2016 Aug 1;29:1–6. Pmid:27043073.
- Missiroli S, Patergnani S, Caroccia N, et al. Mitochondria-associated membranes (MAMs) and inflammation. Cell Death Dis. 2018 Feb 28;9(3)1–4. Pmid:29491386.
- Scheibye-Knudsen M, Fang EF, Croteau DL, et al.Protecting the mitochondrial powerhouse.Trends Cell Biol.2015 Mar 1;25:158–170. Pmid:25499735.
- West AP, Khoury-Hanold W, Staron M, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015 Apr 23;520(7548):553–557. Pmid:25642965.
- Kanneganti TD, Kundu M, Green DR. Innate immune recognition of mtDNA—an undercover signal?Cell Metab. 2015 Jun 2;21:793–794. Pmid:26039443.
- Boyapati RK, Tamborska A, Dorward DA, et al. Advances in the understanding of mitochondrial DNA as a pathogenic factor in inflammatory diseases. F1000Res. 2017 Feb 20;6:169. Pmid:28299196.
- Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010 May;221(1):3–12. Pmid:20225336.
- Zhu H, Toan S, Mui D, et al. Mitochondrial quality surveillance as a therapeutic target in myocardial infarction. Acta Physiol. 2021 Mar;231(3):e13590. Pmid:33270362.
- Deretic V. Autophagy: an emerging immunological paradigm.J Immunol. 2012 Jul 1;189:15–20. Pmid:22723639.
- Sorbara MT, Girardin SE. Mitochondrial ROS fuel the inflammasome. Cell Res. 2011 Apr;21(4):558–560. Pmid:21283134.
- Zhou H, Zhu P, Wang J, et al. DNA-PKcs promotes alcohol-related liver disease by activating Drp1-related mitochondrial fission and repressing FUNDC1-required mitophagy. Signal Transduct Target Ther. 2019 Dec 6;4:56. Pmid: 31839999.
- Naik E, Dixit VM. Mitochondrial reactive oxygen species drive proinflammatory cytokine production. J Exp Med. 2011 Mar 14;208:417–420. Pmid:21357740.
- McCoy AJ, Koizumi Y, Higa N, et al.Differential regulation of caspase-1 activation via NLRP3/NLRC4 inflammasomes mediated by aerolysin and type III secretion system during Aeromonas veronii infection.J Immunol. 2010 Dec 1;185:7077–7084. Pmid:21037094.
- Fernandes-Alnemri T, Yu JW, Juliana C, et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol. 2010 May;115:385–393. Pmid:20351693.
- Rathinam VA, Jiang Z, Waggoner SN, et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat Immunol. 2010 May;115:395–402. Pmid:20351692.
- Pereira CA, Carlos D, Ferreira NS, et al. Mitochondrial DNA promotes NLRP3 inflammasome activation and contributes to endothelial dysfunction and inflammation in type 1 diabetes. Front Physiol. 2020 Jan 17;10:1557. Pmid:32009974.
- Franchi L, Eigenbrod T, Muñoz-Planillo R, et al. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009 Mar;10(3):241–247. Pmid:19221555.
- Wang J, Zhu P, Li R, et al. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol. 2020 Feb;30:101415. Pmid: 31901590.