
ABSTRACT
Candida species are the most commonly isolated opportunistic fungal pathogens in humans. Candida albicans causes most of the diagnosed infections, closely followed by Candida glabrata. C. albicans is well studied, and many genes have been shown to be important for infection and colonization of the host. It is however less clear how C. glabrata infects the host. With the help of fungal RNA enrichment, we here investigated for the first time the transcriptomic profile of C. glabrata during urinary tract infection (UTI) in mice. In the UTI model, bladders and kidneys are major target organs and therefore fungal transcriptomes were addressed in these organs. Our results showed that, next to adhesins and proteases, nitrogen metabolism and regulation play a vital role during C. glabrata UTI. Genes involved in nitrogen metabolism were upregulated and among them we show that DUR1,2 (urea amidolyase) and GAP1 (amino acid permease) were important for virulence. Furthermore, we confirmed the importance of the glyoxylate cycle in the host and identified MLS1 (malate synthase) as an important gene necessary for C. glabrata virulence. In conclusion, our study shows with the support of in vivo transcriptomics how C. glabrata adapts to host conditions.
Introduction
Opportunistic fungal infections, either superficial or invasive, contribute significantly to morbidity and mortality of hospitalized patients [Citation1]. The majority of these infections are caused by Candida species, including, principally Candida albicans and Candida glabrata [Citation2–5]. Interestingly, C. glabrata is more closely related to the baker’s yeast Saccharomyces cerevisiae compared to C. albicans [Citation6–8]. Both Candida species developed the ability to infect their host independently, leading to very different infection strategies [Citation5]. For C. albicans, several virulence factors have been described. Virulence is considered to be mainly dependent on the ability to switch from yeast to hyphae. This enables the fungus to disseminate through the bloodstream, as yeast, while penetrating epithelial layers, as hyphae. This morphogenic switch is also important for the formation of biofilms, which protect against host defences and antifungal treatment [Citation9]. On top of that, specific gene families responsible for adhesion, proteolytic activity and defence against reactive oxygen species contribute significantly to virulence depending on the infected niche [Citation10,Citation11]. More recently, the metabolic flexibility of C. albicans was added to its list of virulence attributes, as it allows the fungus to grow in diverse environments, contributes to lower susceptibility to antifungals and helps in evading host defences [Citation12]. The pathogenesis of C. albicans is well studied and more than 300 genes have been associated with reduced virulence in a mammalian host, as well as about 40 genes to avirulence according to the curated data in the candida genome database (Candidagenome.org). For C. glabrata on the other hand, much fewer genes have been reported critical for virulence. Some gene families are linked to virulence in C. glabrata and among them several cell wall-associated genes. They include proteases called yapsins (YPS-genes) and several adhesins with EPA1 as an important mediator of adhesion [Citation13,Citation14]. C. glabrata has also gained the ability to survive in the environment of macrophages [Citation15] and possesses intrinsic high resistance to oxidative stress [Citation16], both of which can contribute to C. glabrata persistence in the host.
S.cerevisiae and C. glabrata underwent a whole genome duplication, which did not occur in C. albicans. Furthermore, fewer genes are found in C. glabrata compared to S. cerevisiae. Gene loss occurred in central carbon metabolism and nicotinic acid biosynthesis [Citation7]. C. glabrata is unable to grow on galactose, maltose, sucrose, raffinose or lactose as the sole carbon source, while assimilation of ethanol, glycerol and acetate is strain-dependent [Citation17]. This fungal pathogen can, however, use specific amino acids as carbon source as well as a nitrogen source [Citation18]. Contrary to gene loss, C. glabrata gained some genes, specifically in its telomeric regions. These genes encode cell wall proteins with an important function in adhesion to different surfaces [Citation7].
Only three classes of antifungals are regularly used in the clinic, including the fungicidal polyenes and echinocandins and the fungistatic azoles. Polyenes bind directly to ergosterol in the fungal cell membrane, while azoles target the biosynthesis of this sterol. Echinocandins, on the other hand, target the biosynthesis of glucans in the fungal cell wall [Citation19]. C. glabrata exhibits intrinsically higher epidemiological cut-off values for azoles compared to other Candida spp and rapidly acquires resistance to azoles [Citation20]. Resistance to echinocandins also occurs but is less frequent, while resistance to polyenes is rare [Citation19–21]. Interestingly, C. glabrata isolates acquiring rapidly resistance often contain mutations in MSH2, a gene of the DNA mismatch repair pathway [Citation22]. The few antifungal targets and the occurrence of drug resistance motivates the need to investigate C. glabrata in more detail with regard to its virulence and possible novel targets for therapeutic strategies.
In vivo studies with C. glabrata are rare and mainly quantified using fungal burden, due to the low morbidity and mortality in mice. This requires euthanasia of the animals followed by organ extraction and homogenization [Citation23–25]. Following developments in C. albicans, bioluminescence was recently used to study C. glabrata urinary tract infections (UTIs) [Citation26]. This facilitates the tracing of infection in living animals, allowing the use of organs for other applications including transcriptional studies.
Transcriptomics analysis can unveil the pathways that are needed for fungal adaptation inside the host. Host-mimicking conditions have been used for that purpose with C. glabrata cells engulfed by macrophages, or in contact with human blood or epithelial cells [Citation27–29]. Transcriptional studies from host tissues face the issue of limited fungal RNA recovery, since only a small percentage of the RNA extracted from infected organs belongs to the fungus. In C. albicans, a study was performed using RNA enrichment during the preparation of the sequencing library to increase the recovery of C. albicans RNA from total RNA [Citation30]. This transcriptomics study yielded data consistent with other published studies that investigated genes important for virulence [Citation30]. Such an RNA enrichment approach for C. glabrata is challenging, since fungal burdens are usually low in infected hosts. Among available animal models for C. glabrata infections, urinary tract infections (UTIs) are attractive in this respect, since high fungal burdens can be maintained in infected bladders [Citation26].
UTIs caused by Candida species are usually related to indwelling urinary catheters, with fungi causing 2–15% of the infections [Citation31–34]. C. glabrata is an important causative agent with 19–48% of UTIs linked to this pathogen, especially when azole antifungals are used [Citation31–38]. In mice, these infections develop in the bladder but rapidly progress into the kidneys and the spleen [Citation26] congruent with kidney infection causing severe symptoms in humans, leading to high mortality [Citation32,Citation35]. Treatment is challenging, especially when the catheter cannot be removed. As such, it is necessary to study the mechanism of pathogenesis of C. glabrata and to identify genes important for virulence that could be used as targets for new antifungals in the future.
In this work, we carried out an in vivo transcriptomics study, including extensive RNA enrichment for C. glabrata RNA. This led to the identification of multiple pathways and four specific genes involved in C. glabrata virulence during UTI. We also show that the C. glabrata transcriptome diverges from C. albicans during infection.
Materials and methods
Strains and growth media
The C. glabrata luminescent strain SSY21 was pre-grown for UTI infection as described [Citation26]. Deletion and insertion mutants were pre-grown in yeast peptone dextrose (YPD) (2% Bacto Peptone, 1% yeast extract and 2% glucose) at 30°C under continuous shaking. Spot assays were performed on nitrogen starvation (NS) medium (0,17% YNB without amino acids and without ammonium sulphate, 2% glucose, 2% Bacto agar) supplemented with either 0.5% Urea (PlusOne, GE healthcare), 5 mM L-Citrulline (Sigma) or 5 mM L-Tryptophan (Sigma) for DUR1,2 and GAP1 mutants and on YNB (0.67% YNB without amino acids, 2% Bacto agar) supplemented with either 2% potassium acetate or 2% ethanol for MLS1 and VMA22 mutants. All strains are summarized in Table S1.
Urinary tract infection in mice using bioluminescence
All animal experiments were carried out according to the approval of the Institutional Animal Use Committee, Affaires Veterinaires du Canton de Vaud, Switzerland (authorization VD1734.5a) at the University Hospital Center of Lausanne. Animals were housed in ventilated cages with ad libidum access to food and water.
The infections were carried out as described in [Citation26]. Briefly, one hour before the infection, 6-week-old female balb/C mice (Charles River, France) were injected with 0.1 mg/kg buprenorphine. Upon anaesthesia with isoflurane (3%), the bladders were emptied and 2.5 x 108C. glabrata cells were injected through a catheter (Intramedick,28 mm-61 mm, Clayton medick, Becton Dickinson). On day 3 post infection, 100 microlitre D-Luciferin (18 mg/ml) was injected and bioluminescence was measured in the Bruker in vivo Xtreme II (Bruker BioSpin MRI GmbH, Ettlingen, Germany) while under anaesthesia. Mice were then sacrificed using CO2 and the bladder and kidneys were extracted and stored in RNAlater until RNA extraction.
RNA extraction
For in vitro RNA extraction, exponential phase C. glabrata cells were centrifuged and the pellet was snap-frozen in liquid nitrogen and further processed similarly to organs. RNA from organs was extracted similarly according to Amorim-Vaz et al. [Citation30]. Briefly, organs were removed from RNAlater, snap-frozen in liquid nitrogen and subsequently mashed using a mortar. The resulting powder was resuspended in RNA buffer (0.1 M TrisHCl pH 7.5, 0.1 M LiCl, 10 mM EDTA, 0.5% SDS), TrizolReagent (Ambion) and phenol-chlorophorm-isoamylalcohol (PCI) (Sigma). Acid-washed glass beads (Sigma) were added and mechanical lysis was carried out in the Precellys Evolution instrument (Bertin Technologies). Upon centrifugation, the supernatant was mixed with PCI, vortexed and centrifuged. The resulting supernatant was then mixed with 100% ethanol and further processed with the Directzol RNA MiniPrep kit (Zymo Research) according to instructions. RNA quality was verified using a 5200 Fragment Analyser (Agilent) upon preparation with the high sensitivity RNA kit (Agilent).
Probe design for RNA enrichment
Non-overlapping head-to-tail 120-nucleotide probes were designed using the eArray software (Agilent Technologies, Santa Clara, CA). Two probe groups were designed. The first one (“set 1”) included a total of 49,789 bait probes to cover 4995 C. glabrata ORFs (assembly GCA_002219205.121) lacking cell wall adhesins. The second design (“set 2”) included a total of 49,964 bait probes to cover 5134 C. glabrata ORFs that included cell wall adhesins. Gene lists included in the two designs are given in File S1. To avoid cross-hybridization of probes to multiple cell wall adhesin genes, only gene-specific probes were selected. The first 250 nucleotides of each gene were not covered in the bait designs. It was verified that none of the probes could be mapped to the mouse and human cDNA sequences from Ensembl.
Library preparation, enrichment, and sequencing
RNA libraries for RNA-seq were prepared using the SureSelectXT multiplexed sequencing kit with RNA target enrichment for Illumina (Agilent Technologies) according to the manufacturer’s instructions. The RNA library preparation was essentially similar to a previously published study that established the RNA enrichment but for C. albicans RNA [Citation30]. For non-enriched samples of infected organs and in vitro grown C. glabrata, RNA libraries for RNA-seq were prepared with a TruSeq stranded total RNA library prep kit (Illumina). Before sequencing, libraries were analysed with a fragment analyser automated CE system (Advanced Analytical) to assess quality and fragment size and with a Qubit fluorometer (Invitrogen) to determine cDNA concentration. Libraries were kept at -20°C until sequencing. The resulting libraries were sequenced on an Illumina HiSeq 4000 system at the LGTF (Lausanne Genomic Technologies Facility) from the University of Lausanne.
Data analysis
Read processing was performed using a homemade bioinformatics pipeline called ANAPURNAseq. A version of the software is available on GitHUB (https://github.com/Navrique/ANAPURNAseq). ANAPURNAseq (Automated Nextflow Alignment Pipeline for Unprocessed RNAseq) performs various read correction steps, aligns corrected reads to one or two reference genomes and finally counts the number of genes that can be associated with a chosen feature (often gene or CDS). The reference genome used in the alignment procedure (the other possible reference genome was not included) was from isolate DSY562 (Bioproject PRJNA374542, assembly GCA_002219205.1). After running ANAPURNAseq, read counts were normalized using the EdgeR package that filtered for i) low read counts and ii) composition bias with TMM (Trimmed means of M-value) [Citation39]. Reads were next transformed with the Voom function of the limma package in R [Citation40] and also corrected for batch effect using the SVA package [Citation41]. Differential gene expression (DGE) was performed with a linear model in the limma package [Citation40]. A principal component analysis (PCA) plot of individual samples (Figure S1) showed little variations within replicate samples. Lists of differentially expressed genes can be found in File S2. Raw sequencing reads can be found under Bioproject PRJNA766605.
Gene set enrichment analysis (GSEA) was carried out with a list of genes with P-values ≤0.05 and log2 fold-change ≥2 and ≤ -2 (bladder in vivo conditions compared to in vitro). A file consisting of 167 differential expression datasets (File S4) from published data in C. glabrata was used in the GSEA software 4.10, with following analysis parameters: norm, meandiv; scoring_scheme, weighted; set_min, 15; nperm, 1000; set_max, 500. GSEA results were uploaded into Cytoscape 3.0 with the following parameters: P value cut-off, 0.01; FDR q value, 0.05. KEGG pathway enrichment was performed with the online tool ShinyGO (http://bioinformatics.sdstate.edu/go/) [Citation42] with a filtered C. glabrata gene list of up- and down-regulated genes (log2 fold-change ≥2 and ≤ -2; bladder in vivo conditions compared to in vitro conditions).
The GO term enrichment was obtained with the over-representation analysis on the four gene sets that are commonly down- and up-regulated in C. albicans and C. glabrata (File S5), using the clusterProfiler package (v4.0.5) [Citation43]. The GO annotation for C. albicans SC5314 (genome A22-s07-m01-r126) and C. glabrata CBS138 (genome s03-m01-r12) were used. P-values were BH-adjusted [Citation44]. The hierarchical clustering was performed on the Wang-measured semantic similarity of GO terms, using the GOSemSim package (v2.18.1) [Citation45]. The plots of GO terms were created with the enrichplot package (v1.12.3) in R v4.1.2.
qPCR
To validate the RNA-seq data, expression levels of seven genes (ATH1, DUR1,2, GAP1, MLS1, VMA22, B1J91_K00825 g and B1J91_K02035 g) were measured with qPCR. One µg of RNA extracted from infected bladders or in vitro grown cells was converted into cDNA using the PrimeScript™ RT Reagent Kit (Takara) according to manufacturer’s instructions. qPCRs were carried out using 0.2 µM of each primer and 0.2 µM of the TaqMan probe, using the iTaq Universal Probe kit (Biorad) according to manufacturer’s instructions. Sequences of primers and probes are listed in Table S2. Fold change expression was calculated using the ΔΔCt method, amplifying RDN58 as the housekeeping gene. qPCRs were carried out with a Quantstudio 3 device.
Construction of Candida glabrata deletion and reintegration mutants
Deletion mutants were constructed with CRISPR/Cas9 approaches, using a fusion PCR product as a repair template. Each repair template was constructed by fusing three different fragments: fragment 1, a homologous region to the promoter of the gene; fragment 2, the nourseothricin resistance marker (NAT1) and fragment 3, a homologous region to the terminator of the gene, following a PCR reaction using nested primers. All used primers are listed in Table S3. The repair fragment and the in vitro reconstituted RNA-protein complexes (RNPs) were transformed into strain ATCC2001 using electroporation [Citation46]. Gene-specific RNA guides (Table S4) were designed in silico using Geneious Prime (Biomatters, Ltd., Auckland, New Zealand). Gene-specific RNA guides were obtained from IDT (Integrated DNA Technologies, Inc., Coralville, IO, USA). Transformants were selected on YPD supplemented with 200 µg/ml nourseothricin. Correct insertions were verified by PCR with primers listed in Table S3.
Reintegrants were constructed using specific plasmid constructs. Each gene was amplified by PCR, including its promoter and terminator, using primers containing restriction sites (SacI and SpeI) (Table S5). PCR products were next cloned into vector pVS29 using SacI and SpeI restriction sites. pVS29 is originating from pVS20 [Citation47] into which the URA3 gene was replaced by NaeI-SbfI subcloning with the Hygromycin B resistance marker (HygB) from pAP599 [Citation48] after PCR amplification with primers HPH-NaeI 5’-CTTAGCCGGCAAGAAATTACCGTCGCTCG-3’ and HPH-SbfI 5’-AATTCCTGCAGGCCCTTTACCTCTATATCG-3’. Plasmids were transformed into each deletion mutant by electroporation and transformants were selected on YPD supplemented with 300 µg/ml hygromycin B.
Serial dilution assays
Strains were pre-grown overnight in YPD at 30°C under continuous agitation before being 10-fold serially diluted starting from a density with OD600 value of 1. Five microlitres of cell suspension were spotted on each plate and plates were incubated at 30°C ranging from 4 to 6 days. Pictures were taken in Alpha Imager 3400 (Alpha Innotech).
Results
RNA sequencing of infected bladders and kidneys
Mice were inoculated with luminescent C. glabrata cells (isolate SSY21) directly into the bladder using a catheter. At 3 days post infection, luminescence was measured as described previously [Citation26] to determine which bladders were strongly infected. These bladders, as well as the kidneys from the same animals, were stored in RNAlater immediately after sacrificing the mice until RNA was extracted. RNA was also extracted from non-infected kidneys and from in vitro grown luminescent C. glabrata cells. Upon RNA quality control, sequencing libraries were prepared and these libraries were subsequently enriched for C. glabrata RNA using specific probes, a strategy that we previously applied successfully with mice tissues infected with C. albicans [Citation30]. After quality control of the sequencing libraries and Illumina sequencing, reads were processed and aligned to the C. glabrata genome (isolate DSY562). These different steps are summarized in .
Figure 1. Overview of the RNA sequencing experiment. Stationary phase, luminescent C. glabrata cells were injected directly into the bladder of immunocompetent mice via a catheter. At day 3 post infection, bioluminescence was measured and highly infected mice were selected for RNA extraction and processed up to RNA sequencing.
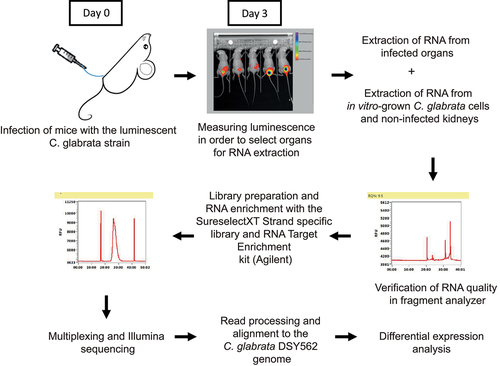
File S3 lists all obtained RNA-seq samples, and, if enriched, with which probe set and the amount of total aligned reads for each sample. Enrichment efficiency was calculated based on the percentage of total reads that aligned to the C. glabrata genome. shows that enrichment of the in vitro spiked C. glabrata RNA mixed with non-infected kidney RNA and the RNA of infected bladders was very effective. From less than 1% C. glabrata RNA in starting material, enrichment yielded more than 60% fungal RNA. The enrichment factor reached values ranging between 750- to 970-fold as compared to non-enriched samples (). Differences were observed between probe set 2 and probe set 1, the later did not include specific probes for adhesins. Probe set 1 resulted in a slightly more efficient enrichment compared to probe set 2. RNA enrichment from infected kidneys was also effective (about 2000-fold), yet it did not result in high percentages of reads aligning to the C. glabrata genome (). The low percentage of C. glabrata reads in the starting material (0.003%) could have been a limiting factor in the enrichment efficiency. Even if our enrichment procedure encountered some limitations, the correlation between normalized read counts of enriched in vitro samples (probe set 2) and the non-enriched in vitro samples was satisfactory (R^2 = 0.89, ). While some genes were not enriched due to absence of corresponding probes, a few other genes (3.6% of all probed genes) were not proportionally enriched, which was close to enrichment biases observed with C. albicans [Citation30]. More than 1000 genes were significantly upregulated (≥2-fold) in the bladder compared to in vitro grown cells and a similar amount was downregulated (≤2-fold). Similar numbers were found for the comparison between infected kidneys and in vitro grown cells (File S2).
Figure 2. RNA enrichment validity and differential expression between in vivo and in vitro conditions. (a) Quality of enrichment was assessed by correlating the normalized read counts of the enriched in vitro samples (enriched from a mix with RNA from a non-infected kidney) and the non-enriched in vitro samples. Red-labelled data points indicate enrichment bias. (b) Differential expression diagram: more than 1000 genes are up- and downregulated (Log2(fc) ≥1 and ≤1) compared to in vitro conditions. FC: fold-change.
Table 1. Enrichment efficiency for infected organs and spiked samples.
RNA-seq data were validated by measuring the expression of several selected genes (see below) using qPCR with RNA samples extracted from infected bladders or in vitro grown cells. The five genes (DUR1,2, ATH1, GAP1, MLS1, and VMA22) that were strongly upregulated in the bladder compared to in vitro grown cells yielded qPCR results concordant with RNA-seq data (Figure S2). Two genes (B1J91_K00825 g, similar to YGR208w, a S. cerevisiae phosphoserine phosphatase; B1J91_K02035 g, similar to YER134c, a putative a S. cerevisiae magnesium-dependent phosphatase) significantly downregulated in the bladder compared to in vitro conditions by RNA-seq results were also validated by qPCR (Figure S2).
Kidneys and bladder transcriptomes compared to in vitro conditions were highly overlapping given that about 84% of genes upregulated in the bladder were also upregulated in the kidneys. Conversely, 77% of genes downregulated in the bladder were also downregulated in the kidneys (Figure S3; see below for additional comments).
Gene set enrichment analysis shows expected links to other C. glabrata transcriptional studies
The differential expression in the bladder compared to in vitro grown cells was analysed with 167 gene lists of C. glabrata regulated genes from several published data sets (File S4). As shown in (green circled nodes), our data have several genes in common with those up- and down-regulated in contact with epithelial cells at different time points, especially at 24 h. The convergence of transcriptional profile between these two different conditions suggests that C. glabrata cells are in contact with the epithelial cell layers of the bladder. Furthermore, a significant overlap was observed between our data and genes upregulated while grown in urine (black circled node), a significant nutrient source in the bladder, as well as weak acid stress (sorbic acid: black circled node), which probably reflects the low pH that prevails in the bladder [Citation49]. In addition, niacin starvation (black circled node), a condition often linked to the environment of the bladder [Citation50] includes regulated genes with overlap to our data. Data shown in also indicate a significant overlap of bladder-dependent transcription with C. glabrata profiles obtained with immune cells and blood (orange circled nodes). These results highlight that host-like in vitro conditions mirrored the in vivo transcriptional landscape of C. glabrata. Surprisingly, PDR1-dependent expression (violet circled nodes) overlapped with in vivo data. Gain-of-function PDR1 mutations are associated with drug resistance and increased virulence of C. glabrata [Citation51], therefore the in vivo profile suggests that C. glabrata has kept a PDR1-like signature to support its lifestyle in the host. We also noticed that a substantial number of regulated genes in the bladder behaved in a similar manner compared to those regulated by DNA damage (green hatched circled node). This suggests a possible link between the response to DNA damaging agents and C. glabrata adaptation to in vivo conditions.
Figure 3. (a) GSEA (Gene set enrichment analysis) of C. glabrata genes regulated in vivo. The gene list was produced from data in file S4 (“Cg14.Gmt”), in which genes with P values of ≤0.05 and log fold-change ≥ 2 (in vivo compared to in vitro) were chosen. The Cg14.Gmt file contains 167 differential expression data sets from published transcriptional data performed with C. glabrata. The list was then imported into the GSEA software 4.10. Analysis parameters were as follows: norm, meandiv; scoring_scheme, weighted; set_min, 15; nperm, 1000; set_max, 500. GSEA results were uploaded into cytoscape 3.0 with the following parameters: P value cut-off, 0.01; FDR q value, 0.05. Red nodes represent enriched gene lists in upregulated genes from the GSEA. Blue nodes represent enriched gene lists in downregulated genes from the GSEA. Nodes are connected by edges when overlaps exist between nodes. The size of nodes reflects the total number of genes that are connected by edges to neighbouring nodes. Edge thickness reflects the level of confidence between nodes. Labels of nodes indicate specific classes of genes listed in the Cg14.Gmt file. (b) and (c) KEGG pathway enrichment of C. glabrata genes up- and down regulated regulated in vivo, respectively. The enrichment analysis was performed with the online tool ShinyGO (http://bioinformatics.Sdstate.edu/go/) [Citation42] with a filtered C. glabrata gene list of up- and down-regulated genes (log2(fc) ≥ 2 compared to in vitro growth)
![Figure 3. (a) GSEA (Gene set enrichment analysis) of C. glabrata genes regulated in vivo. The gene list was produced from data in file S4 (“Cg14.Gmt”), in which genes with P values of ≤0.05 and log fold-change ≥ 2 (in vivo compared to in vitro) were chosen. The Cg14.Gmt file contains 167 differential expression data sets from published transcriptional data performed with C. glabrata. The list was then imported into the GSEA software 4.10. Analysis parameters were as follows: norm, meandiv; scoring_scheme, weighted; set_min, 15; nperm, 1000; set_max, 500. GSEA results were uploaded into cytoscape 3.0 with the following parameters: P value cut-off, 0.01; FDR q value, 0.05. Red nodes represent enriched gene lists in upregulated genes from the GSEA. Blue nodes represent enriched gene lists in downregulated genes from the GSEA. Nodes are connected by edges when overlaps exist between nodes. The size of nodes reflects the total number of genes that are connected by edges to neighbouring nodes. Edge thickness reflects the level of confidence between nodes. Labels of nodes indicate specific classes of genes listed in the Cg14.Gmt file. (b) and (c) KEGG pathway enrichment of C. glabrata genes up- and down regulated regulated in vivo, respectively. The enrichment analysis was performed with the online tool ShinyGO (http://bioinformatics.Sdstate.edu/go/) [Citation42] with a filtered C. glabrata gene list of up- and down-regulated genes (log2(fc) ≥ 2 compared to in vitro growth)](/cms/asset/77d57430-be0e-46fa-9944-d8169a9e8ecd/kvir_a_2095716_f0003_oc.jpg)
Taken together, the GSEA revealed that the in vivo obtained transcriptional profiles overlapped with other transcriptional studies that included conditions mimicking the host environment.
KEGG pathway analysis points to the importance of a metabolic shift during in vivo growth
A KEGG pathway analysis was carried out for the differential gene expression in the bladder compared to in vitro grown cells. As shown in , the largest difference between the two conditions lies in “metabolic pathways” in both up- and down-regulated genes. Biosynthesis of secondary metabolites, as well as the glyoxylate cycle and fatty acid metabolism seem to be specifically upregulated in vivo (). In line with the GSEA, where several genes are upregulated in common with the response to DNA damaging agents, pathways for both homologous recombination and non-homologous end joining are upregulated in the bladder. In addition, the signature of MAPK signalling is among the in vivo upregulated genes which highlights the need of C. glabrata to respond to the unfavourable host conditions. Furthermore, metabolism of specific amino acids (alanine, aspartate and glutamate) seems to be upregulated, while metabolism of other amino acids (arginine and proline) is downregulated. The process of ribosome biogenesis is among down-regulated genes thus probably reflecting that the bladder environment has negative impact on growth of C. glabrata compared to in vitro conditions.
In vivo expression of known virulence genes
Proteolytic activity has been linked to virulence both in C. albicans and C. glabrata [Citation13,Citation52]. C. glabrata contains one family of eleven proteases (YPS genes), which are glycosylphosphatidylinositol (GPI) anchored cell wall proteins. These proteases are involved in cell wall integrity, adherence to mammalian cells, survival in macrophages and virulence [Citation13]. In our study, we observed that their expression profile differs between the niches of the bladder and the kidneys. While YPS4, YPS6 and YPS11 were upregulated in both organs, YPS1, YPS8 and YPS9 showed bladder-specific upregulation. YPS2, YPS5, and YPS10 were only upregulated in the kidneys ().
Table 2. In vivo expression of the YPS genes.
Another gene family that is important for virulence includes genes encoding adhesins. The most well-known family is the EPA-family [Citation14]. However, many other adhesins are present in the C. glabrata genome and this varies greatly between strains. The wild-type strain used in our study, DSY562, contains about 101 adhesins or adhesion-like proteins, compared to 63 identified in the reference strain, ATCC2001 [Citation53,Citation54]. However, recent data suggest ATCC2001 may contain up to 83 adhesin-like genes [Citation54]. DSY562 does not contain copies of EPA3, EPA7, EPA13, and EPA21 [Citation53]. We found that five of the EPA-genes were upregulated in the bladder, including EPA6, EPA23, EPA9, EPA10, and EPA20 in addition to four other adhesins (CAGL0I11011g, CAGL0C00253g, CAGL0C01133g, and CAGL0C00896g). Expression patterns of these adhesins are listed in .
Table 3. In vivo expression of adhesin genes.
Several C. glabrata genes have been described as resulting in decreased virulence upon deletion according to the Candida Genome Database including ATG1, AUS1, HOG1, SKN7, VPH2, VPS15, VPS34, CNA1, and CNB1 [Citation55–61]. However, only three of these genes (AUS1, SKN7, and CNA1) were significantly upregulated in our study compared to in vitro conditions (File S2).
Comparison between the bladder and the kidneys transcriptomes
Our data revealed high overlap between the blabber and the kidney transcriptomes (Figure S3). Given that these host environments are quite different, this was unexpected. Besides the niche-specific gene expression as mentioned above, several other genes exhibited inversed regulation patterns that were dependent on the infection site. For example, WAR1, a transcription factor involved in the regulation of genes involved in weak acid stress [Citation62] is upregulated in the bladder (log2 FC:1.4), while downregulated in the kidneys (log2 FC:-3.6). These data are consistent with the differences in environmental pH between the bladder and the kidneys [Citation49]. ATO3, a putative ammonium transporter, is downregulated in the bladder (log2 FC: -1.6), however strongly upregulated in the kidneys (log2 FC: 4.6). These data reflect that C. glabrata grows in the two organs in which ammonium availability is different. It could also link to the necessity of ammonium export to counteract acidification of the phagolysosome upon macrophage phagocytosis, as was shown for C. albicans [Citation63].
Deletion of specific highly differentially expressed genes decreases virulence
In order to address the relevance of highly regulated genes in the process of C. glabrata infection in the UTI model, we selected four genes (DUR1,2, GAP1, MLS1, and VMA22) that were highly upregulated in the bladder compared to in vitro conditions. In vivo upregulated genes only were chosen, since we hypothetized that they were important for the lifestyle of C. glabrata in host tissues. In addition, these genes were selected since they have a known homolog in S. cerevisiae, which facilitates in vitro phenotypic verifications. C. glabrata deletion mutants of each of these genes were tested with serial dilution assays and then used to infect mice. In general, deletion of each of the four selected genes did not affect growth compared to the wild-type parent (Figure S4).
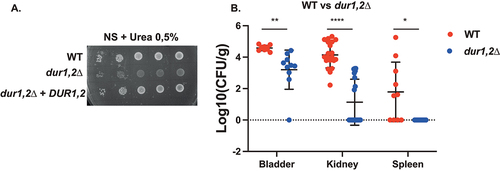
Deletion of the urea amidolyase, DUR1,2, resulted in strongly impaired growth on nitrogen starvation (NS) medium containing urea as the sole nitrogen source () [Citation64]. Given that urea is a dominant nitrogen source in urine, DUR1,2 could be important for colonization of C. glabrata of the bladder. Consistent with this hypothesis, DUR1,2 deletion resulted in significantly reduced virulence in the UTI model. The deletion strain showed reduced colonization of the bladder and the kidneys and seemed unable to colonize the spleen ().
Figure 4. Deletion of DUR1,2 strongly impairs growth on urea as the sole nitrogen source, as well as virulence in the urinary tract. (a) Serial dilutions of each strain were spotted on nitrogen starvation (NS) medium, containing 0.5% urea as the sole nitrogen source and grown for 4 days at 30°C. (b) Ten mice per group were infected with either the wild type or the mutant strain, by injection (through a catheter) into the bladder. On day 3 post infection, animals were euthanized and bladders, kidneys and spleens were collected for quantification of the fungal burden. (Significance symbols: **** p < 0.0001, *** 0.0001 < p < 0.0001, ** 0.001 < p < 0.01, * 0.01 < p < 0.05).
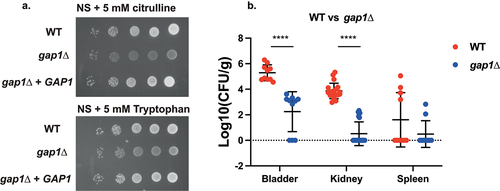
The general amino acid permease and receptor, GAP1, is well studied in S. cerevisiae as a major regulator of nitrogen metabolism in yeast [Citation65–67]. Since several nitrogen-metabolism-related pathways were shown to be differentially regulated in vivo according to the KEGG pathway analysis, we hypothesized that amino acid uptake and signalling could be important for virulence. Deletion of GAP1 in C. glabrata resulted in impaired growth on NS medium supplemented with either L-citrulline or L-tryptophan as the sole nitrogen source (), which is consistent with S. cerevisiae phenotypes [Citation65]. Furthermore, GAP1 deletion resulted in a strong decrease in colonization of the bladder and kidneys in the UTI model, without abolishing spleen colonization ().
Figure 5. Deletion of GAP1 strongly impairs growth on citrulline or tryptophan as the sole nitrogen source, as well as virulence in the urinary tract. (a) Serial dilutions of each strain were spotted on nitrogen starvation (NS) medium, containing 5 mM of either L-citrulline or L-tryptophan as the sole nitrogen source and grown for 4 days at 30°C. (b) Ten mice per group were infected with either the wild type or the mutant strain, by injection (through a catheter) into the bladder. On day 3 post infection, animals were euthanized and bladders, kidneys and spleens were collected for quantification of the fungal burden. (Significance symbols: **** p < 0.0001, *** 0.0001 < p < 0.0001, ** 0.001 < p < 0.01, * 0.01 < p < 0.05).
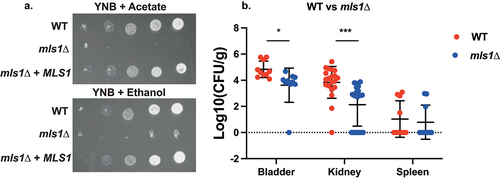
The malate synthase gene, MLS1, is a part of the glyoxylate cycle that bypasses part of the Krebs cycle in order to retain carbon in limiting environments [Citation68]. Given that the glyoxylate cycle was upregulated in the UTI model according to the KEGG pathway analysis, MLS1 could be important for virulence in this model. Deletion of MLS1 in C. glabrata resulted in abolishment of growth on minimal medium (YNB) with either acetate or ethanol as a carbon source (). This is consistent with S. cerevisiae phenotypes; however, in baker’s yeast the defect is also displayed on rich medium (YP) supplemented with acetate or ethanol [Citation68,Citation69], which was not the case for C. glabrata (data not shown). The deletion strain was significantly impaired in colonization of the bladder and the kidney, while spleen colonization was not affected ().
Figure 6. Deletion of MLS1 abolishes growth on acetate or ethanol as the sole carbon source, as well as virulence in the urinary tract. (a) Serial dilutions of each strain were spotted on minimal medium (YNB), containing 2% of either acetate or ethanol as the sole carbon source and grown for 6 days at 30°C. (b) Ten mice per group were infected with either the wild type or the mutant strain, by injection (through a catheter) into the bladder. On day 3 post infection, animals were euthanized and bladders, kidneys and spleens were collected for quantification of the fungal burden. (Significance symbols: **** p < 0.0001, *** 0.0001 < p < 0.0001, ** 0.001 < p < 0.01, * 0.01 < p < 0.05).
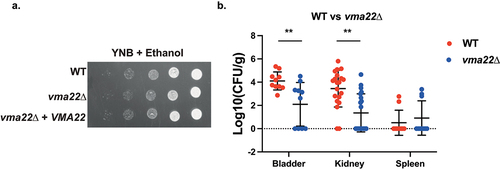
Vacuolar acidification is important for cellular pH homoeostasis, as well as ion balance and nutrient trafficking throughout the cell and critical for cellular function [Citation70]. VMA22 is an adaptor that helps assemble the Vacuolar-ATPase, the main actor in vacuolar acidification. Deletion of this gene in S. cerevisiae results in strong phenotypes, such as reduced growth in the presence of hydrogen peroxide or inability to metabolize glycerol [Citation71,Citation72]. VMA22 deletion in C. glabrata, however, did not lead to clearly different in vitro phenotypes compared to wild type (). It did, however, cause a significant decrease in colonization of the bladder ().
Figure 7. Deletion of VMA22 results in decreased virulence in the urinary tract, even though no strong growth defects were found in vitro. (a) Serial dilutions of each strain were spotted on minimal medium (YNB), containing 2% ethanol as the sole carbon source and grown for 6 days at 30°C. (b) Ten mice per group were infected with either the wild type or the mutant strain, by injection (through a catheter) into the bladder. On day 3 post infection, animals were euthanized and bladders, kidneys and spleens were collected for quantification of the fungal burden. (Significance symbols: **** p < 0.0001, *** 0.0001 < p < 0.0001, ** 0.001 < p < 0.01, * 0.01 < p < 0.05).
Comparison between gene expression during C. albicans and C. glabrata infection
In the work performed in our group by Amorim-Vaz et al. [Citation30], in vivo transcriptomics was carried out for C. albicans during invasive infection in mice by RNA enrichment from infected kidneys. Even though both the model (invasive infection vs UTI) and the timepoint (16 h vs 3 days) are different, it is interesting to look at the most highly significantly expressed genes in their work compared to the in vivo expression profiles of C. glabrata. lists the top 20 most upregulated genes in C. albicans during invasive infection and, if existing, their C. glabrata counterpart and associated differential expression levels. Twelve of the C. albicans genes did not have orthologs in C. glabrata and only three orthologs were upregulated during C. glabrata UTI. These upregulated genes in common between both studies include a sugar transporter, HGT12, a secreted aspartyl protease, SAP5, and a transcription factor, UME6. Only UME6 was linked to virulence in C. albicans up to now [Citation73]. Transcription factors do not always regulate the same processes in different fungal species (so-called “transcriptional re-wiring”) and as such the UME6 ortholog may not be necessarily involved in virulence in C. glabrata.
Table 4. Comparison between most highly significantly upregulated genes in C. albicans invasive infection vs C. glabrata UTI.
When comparing the most highly differentially expressed genes in C. glabrata in the kidneys to C. albicans genes upregulated in invasive infection (from kidneys), only nine genes have orthologs in C. albicans (). Four of these orthologs were not differentially expressed in C. albicans during infection. Only HST6 and orf19.7588 are upregulated in vivo in C. albicans as well, although to a much lower extent than in C. glabrata. Interestingly, UPC2, GAT1, and ATC1 are downregulated during infection in C. albicans while being strongly upregulated in C. glabrata. UPC2 and GAT1 are transcriptional regulators involved in sterol synthesis and nitrogen catabolite repression, respectively. UPC2 has two orthologs in C. glabrata, UPC2A (CAGL0C01199g; downregulated in vivo) and UPC2B (CAGL0F07865g: upregulated in vivo), but only UPC2A is involved in sterol homoeostasis in this species [Citation74]. Therefore, it is possible that the true C. glabrata UPC2 ortholog to C. albicans is UPC2A (CAGL0C01199g) (see also below). We showed here that nitrogen uptake and metabolism are important for C. glabrata virulence. It is possible that the GAT1 homolog is important for this function. Downregulation of this gene in C. albicans could mean that nitrogen catabolite repression is less important in vivo for this species, or that it is regulated differently. Atc1 is a cell wall-linked trehalase enzyme. C. glabrata trehalase enzymes were recently shown to be important for stress resistance [Citation75], which is one of the factors important for the maintenance of fungal virulence.
Table 5. Comparison between strongest upregulated genes in C. glabrata UTI (kidneys) vs C. albicans invasive infection.
The divergence between the C. glabrata and C. albicans in vivo transcriptomes can be best observed when comparing all regulated ortholog genes (593) between the two species (). When examining regulated orthologs between the two species, 49% were coregulated, while the remaining were inversely regulated.
Figure 8. In vivo regulation of C. albicans and C. glabrata orthologs. The orthologs between both species were obtained from the public Candida genome database. The C. albicans RNAseq data were obtained from the work of Amorim-Vaz et al. [Citation30] taking the 16 h time point of mice infection. The C. glabrata RNAseq data were from infected kidneys (File S5). Data were filtered by p-value (<0.05).
![Figure 8. In vivo regulation of C. albicans and C. glabrata orthologs. The orthologs between both species were obtained from the public Candida genome database. The C. albicans RNAseq data were obtained from the work of Amorim-Vaz et al. [Citation30] taking the 16 h time point of mice infection. The C. glabrata RNAseq data were from infected kidneys (File S5). Data were filtered by p-value (<0.05).](/cms/asset/23c4c1c8-162e-4eee-8859-57c948d30b22/kvir_a_2095716_f0008_b.gif)
The set of commonly regulated genes compared to in vitro conditions comprises 120 and 172 up- and downregulated genes between both species recovered from kidneys, respectively (File S5). A GO term enrichment analysis based on gene ontology specific for each species revealed commonly regulated processes. Commonly upregulated genes included biological processes related to response to stress and starvation as well as positive regulation of transcription (Figure S5). This signature reflects that the two fungal species have to cope with an unfavourable host environment. Among the commonly downregulated genes (Figure S6), enriched biological processes participated to translation and ergosterol biosynthesis. Decreased translation revealed by downregulation of several ribosomal genes suggests slow growth of both fungal species in the host. Decreased expression of genes involved in sterol biosynthesis during infection is still difficult to interpret, however is consistent with downregulation of UPC2A (a regulator of sterol biosynthesis) in C. glabrata, as mentioned above.
Discussion
In this work, we successfully carried out in vivo transcriptomics upon RNA enrichment with the Sureselect RNA enrichment method. Similar to the work of Amorim-Vaz et al. performed in our group [Citation30], we managed to quantitatively recover fungal RNA from most fungal genes in total RNA extracted from infected animal organs. Amorim-Vaz et al. [Citation30] discovered that only few genes were not proportionally enriched, which was explained by insufficient bait coverage and low GC content of the probes. In addition, we noticed that a limiting factor in the enrichment procedure to high read mapping percentages was the amount of fungal RNA in the starting material. Samples containing 0.0066–0,3% of fungal RNA could be enriched to high percentages, while samples originating from infected kidneys (only 0,003%) did lead to efficient enrichment, but not to high read mapping percentages (). This is consistent with enrichment efficiencies reported in the previous study [Citation30]. Another alternative to measure in vivo gene expression for pathogens is the nanoString nCounter platform. It can, however, only test a limited amount of genes at the same time, making it unable to get a full transcriptome [Citation76,Citation77]. So far, this approach has been used to investigate specific genes during infections of C. albicans, Aspergillus fumigatus and Cryptococcus neoformans [Citation77–81].
With a probe set including known adhesin genes from isolate DSY562, we showed that several adhesins were strongly upregulated in the bladder compared to in vitro growth conditions. Interestingly, the strain DSY562 used in the present study contains significantly more adhesin genes compared to the reference strain ATCC2001 [Citation53]. DSY562 was originally recovered from an AIDS patient with an oral Candida infection [Citation82]. Its adhesin repertoire might have been adapted to this host niche. It would be interesting to assess expression of adhesins in this strain during oral infection to determine if a specific adhesion profile exists for each niche.
The Gene Set Enrichment Analysis (GSEA) used in the present study showed that our data overlapped significantly with several other in vitro conditions mimicking the host environment, which was also the case for the C. albicans study [Citation30]. However, overlap was not only detected with conditions specifically mimicking the in vivo environment. Interestingly, many of in vivo upregulated genes overlapped with genes upregulated in response to DNA damaging agents [Citation83]. Part of these overlapping genes are homologs to the S. cerevisiae mating genes, which were identified by Shor and Perlin [Citation84]. The significance of the DNA damage response in the context of in vivo conditions still remains puzzling. The factor that is shared between both this in vitro (DNA damage) and the in vivo (UTI) situation is stress. Therefore, it is possible that the overlap between these two conditions rather reflects the signature of stress.
In the UTI model, we infected mice directly into the bladder, from where infection spreads towards the kidneys and the spleen [Citation26]. As of yet, it is not clear how the fungus migrates to the kidney and the spleen; it is, however, likely that colonization of the kidneys happened through the ureter. In the invasive infection model, fungal cells are directly injected into the bloodstream, which carries them towards the kidneys and many other organs, where they are either killed or can persist depending on the species [Citation5]. The question remains how the transcriptomics data presented here are relevant in the context of a different infection model, such as invasive infection. Interestingly, DUR1,2 was shown to be important during invasive infection of C. albicans in mice. In the absence of this gene, a more balanced immune response was measured compared to wild type, suggesting that DUR1,2 contributes to renal failure by unbalancing the host response [Citation85]. In our UTI model experiments, deletion of DUR1,2 led to a strong reduction in colonization of both the bladder and the kidneys and the inability to colonize the spleen. It remains to be determined whether deletion of DUR1,2 has an effect on the host immune response or whether its effect on virulence mainly stems from the inability to assimilate urea as a nitrogen source. In C. albicans DUR1,2 is coregulated with the urea transporter DUR3 [Citation25], which is not the case in C. glabrata taken from our data. It is possible that this transporter is post-translationally regulated in C. glabrata. Additionally, Brunke et al. [Citation86] showed that DUR1,2 deletion resulted in reduced virulence in the fruit fly as well as during invasive infection in mice. It is as such possible that the other virulence genes identified in our work are important during invasive infection.
The general amino acid permease, GAP1, has been studied extensively in S. cerevisiae. It is required for transport of L-citrulline, while it also transports a wide variety of other amino acids. Furthermore, GAP1 is necessary for the activation of the PKA pathway upon addition of amino acids in the presence of a fermentable carbon source [Citation65,Citation66]. Interestingly, PKA activation and transport are independent [Citation65,Citation66]. Whether the C. glabrata GAP1 is also a transceptor is not known. Deletion of the gene did strongly affect virulence in the UTI model. Interestingly, C. albicans does not have a clear GAP1 homolog; it has six genes that are more or less homologous to the S. cerevisiae GAP1. Each of these homologs has their own amino acid transport specificity, while GAP4 even transports S-adenosylmethionine (SAM) [Citation87,Citation88]. Signalling activity was not shown for any of the C. albicans GAP genes. We identified two genes involved in nitrogen metabolism, uptake and possibly regulation to be involved in virulence, namely DUR1,2 and GAP1. However, other genes involved in nitrogen sensing and response were strongly upregulated in the bladder. One of them is the C. glabrata homolog of S. cerevisiae SSY1 and C. albicans CSY1, CAGL0E01089g. SSY1 is a part of the SPS amino acid sensing system, which senses amino acids extracellularly and then activates amino acid catabolism [Citation89]. C. albicans CSY1 was shown to be required for amino-acid-induced morphogenesis [Citation90], a process that has been linked to virulence in this organism [Citation91]. Furthermore, the transcription factor GLN3, recently described in C. glabrata as the main regulator of nitrogen catabolite repression (NCR) [Citation92], was strongly upregulated in mice. Interestingly, GLN3 also seems to negatively regulate the ABC transporters CDR1 and CDR2, both involved in fluconazole resistance [Citation93].
Next to nitrogen, carbon is also essential for survival in the host. Since glucose, the preferred carbon source for most yeasts, is rarely present extracellularly, other carbon sources must be used. When carbon sources are not readily available, yeasts use an adapted version of the Krebs cycle for its metabolism, the glyoxylate cycle, which bypasses the CO2 expelling steps of the pathway [Citation68]. In our work, we observed a strong upregulation of one enzyme in this cycle, MLS1, while the other predicted enzyme, ICL1 was downregulated in the host. Deletion of MLS1 led to a significant decrease in virulence in the UTI model. Interestingly, the in vitro defect of MLS1 deletion in C. glabrata was not as strong as in S. cerevisiae. In baker’s yeast, the deletion mutant cannot grow on acetate as the sole carbon source, independent of the richness of the medium [Citation68]. The growth defect in C. glabrata only occurs on minimal medium, not on rich medium. Possibly C. glabrata can more efficiently use specific amino acids as a carbon source, as well as a nitrogen source [Citation18]. As mentioned earlier, ICL1 is downregulated in our study in vivo as compared to in vitro conditions. In C. albicans, this gene was shown to be important for survival inside macrophages [Citation94]. It is either possible that the ICL1 gene is incorrectly predicted from the genome or that its activity is regulated post-translationally in the host in C. glabrata. It would be interesting to further address how ICL1 deletion affects virulence and how the glyoxylate cycle is regulated in C. glabrata.
A fourth gene, VMA22, is involved in assembly of the vacuolar ATPase (V-ATPase), which is essential for acidification of this organelle. Acidification of cell organelles creates an electrochemical gradient, which is used, in protein sorting, endocytosis and storage of nutrients [Citation95,Citation96]. V-ATPases are conserved in fungi, plants, and animals, yet some assembly factors are kingdom specific, such as VMA22 [Citation95]. Deletion of VMA22 in S. cerevisiae results in several growth defects on, among others, glycerol, hydrogen peroxide, alkaline pH, and boric acid [Citation71,Citation97–99]. The C. albicans ortholog was unviable in a large-scale survey [Citation100]. Surprisingly, VMA22 deletion in the present study did not result in any growth defects on the tested media, even though organelle acidification has been linked to oxidative stress resistance and virulence in C. glabrata [Citation60,Citation101]. VMA22 function may differ in C. glabrata compared to S. cerevisiae and C. albicans. Interestingly, in the transposon mutagenesis of Gale et al. [Citation102], VMA22 was scored as a putative essential gene in C. glabrata. Further studies are needed on the function of VMA22, given that its deletion resulted in reduced virulence in the UTI model.
Since only a few antifungal targets are known in C. glabrata, this work may help the development of potential novel antifungals. Here, we identified four new virulence genes in this species. All four proteins (Dur1,2, Gap1, Mls1, and Vma22) show low identity to human proteins, and as such have potential as antifungal targets. As mentioned earlier, DUR1,2 was shown to be involved in virulence in both C. albicans and C. glabrata [Citation85]. Its enzymatic function makes it possible to use existing strategies in development of potential inhibitors. Furthermore, the glyoxylate cycle, of which MLS1 is a part, does not exist in animals, making it another potentially interesting target.
Our work does confirm that C. albicans and C. glabrata use divergent different infection strategies, which is reflected by different transcriptional profiles in the host. Recent transcriptional studies performed with different Candida spp. incubated with whole blood confirm this observation [Citation103]. C. albicans relies heavily on morphogenetic switching, candidalysin secretion and defence against reactive oxygen species (ROS) [Citation30], two of which are absent in C. glabrata. The defence against ROS is mainly carried out by superoxide dismutase (SOD) and glutathione biosynthesis and recovery genes in C. albicans. Interestingly, C. albicans expresses multiple orthologs of both genes, while only one of each is identified in C. glabrata. C. glabrata does show anti-ROS activity [Citation104], yet neither of the genes involved is upregulated in vivo in our work. C. glabrata seems to depend mainly on metabolic adaptation to the host environment and expression of adhesins. Both adhesion and metabolic flexibility are also considered as virulence attributes in C. albicans [Citation11].
Conclusions
In conclusion, fungal RNA enrichment and subsequent sequencing is a powerful tool to obtain a full view of the genes and pathways that are differentially regulated during growth inside a host. We show that two classes of virulence genes, the yapsins and the adhesins, probably have a niche-specific expression profile. Furthermore, we confirmed the importance of metabolic flexibility and the ability to use different nitrogen and carbon sources for the virulence capacity of C. glabrata. Three out of the four virulence genes identified in this work belong to important, fungal-specific metabolic pathways and therefore may represent useful antifungal targets. Additionally, we illustrate differences between the infection strategy of C. glabrata compared to C. albicans, and highlight specific virulence genes in either species not having direct homologs in the other.
Author’s contributions
SS designed and performed experiments and contributed to writing, ED and VDT contributed to analysis, DS designed experiments and contributed to writing.
Supplemental Material
Download Zip (11.4 MB)Acknowledgement
Authors are thankful to Danielle Brandalise for excellent technical assistance. Authors acknowledge the laboratory of Bernhard Hube for providing strains used in this study. Authors thanks collaborators of the Lausanne Genomic Technology Facility (LGTF) for technical help. This work was supported by a grant of the Swiss National Research Foundation grant 31003A_172958 to DS.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/21505594.2022.2095716
Additional information
Funding
Related Research Data
References
- Magill SS, Edwards JR, Bamberg W, et al. Multistate point-prevalence survey of health care-associated infections. N Engl J Med. 2014 Mar 27;370(13):1198–1208.
- Lamoth F, Lockhart SR, Berkow EL, et al. Changes in the epidemiological landscape of invasive candidiasis. J Antimicrob Chemother. 2018 Jan 1;73(suppl_1):i4–i13.
- Pfaller MA, Diekema DJ, Turnidge JD, et al. Twenty years of the SENTRY antifungal surveillance program: results for Candida species from 1997-2016. Open Forum Infect Dis. 2019;6(Suppl 1):S79–S94.
- Bolotin-Fukuhara M, Fairhead C. Candida glabrata: a deadly companion? Yeast. 2014 Aug;31(8):279–288.
- Maccallum DM. Hosting infection: experimental models to assay Candida virulence. Int J Microbiol. 2012;2012:363764.
- Kurtzman CP, Robnett CJ. Three new insect-associated species of the yeast genus Candida. Can J Microbiol. 1998 Oct;44(10):965–973.
- Gabaldon T, Martin T, Marcet-Houben M, et al. Comparative genomics of emerging pathogens in the Candida glabrata clade. BMC Genomics. 2013 Sep 14;14:623.
- Roetzer A, Gabaldon T, Schuller C. From Saccharomyces cerevisiae to Candida glabratain a few easy steps: important adaptations for an opportunistic pathogen. FEMS Microbiol Lett. 2011 Jan;314(1):1–9.
- Kim J, Sudbery P. Candida albicans, a major human fungal pathogen. J Microbiol. 2011 Apr;49(2):171–177.
- Calderone RA, Fonzi WA. Virulence factors of Candida albicans. Trends Microbiol. 2001 Jul;9(7):327–335.
- Poulain D. Candida albicans, plasticity and pathogenesis. Crit Rev Microbiol. 2015 Jun;41(2):208–217.
- Padder SA, Ramzan A, and Tahir I, et al. Metabolic flexibility and extensive adaptability governing multiple drug resistance and enhanced virulence in Candida albicans. Crit Rev Microbiol. 2021;48(2): 1–20.
- Kaur R, Ma B, Cormack BP. A family of glycosylphosphatidylinositol-linked aspartyl proteases is required for virulence of Candida glabrata. Proc Natl Acad Sci U S a. 2007 May 1;104(18):7628–7633.
- Lopez-Fuentes E, Gutierrez-Escobedo G, and Timmermans B, et al. Candida glabrata’s genome plasticity confers a unique pattern of expressed cell wall proteins. J Fungi (Basel). 2018 Jun 5;4:(2). DOI:10.3390/jof4020067
- Kasper L, Seider K, Hube B. Intracellular survival of Candida glabrata in macrophages: immune evasion and persistence. FEMS Yeast Res. 2015 Aug;15(5):fov042.
- Cuellar-Cruz M, Briones-Martin-Del-Campo M, Canas-Villamar I, et al. High resistance to oxidative stress in the fungal pathogen Candida glabrata is mediated by a single catalase, Cta1p, and is controlled by the transcription factors Yap1p, Skn7p, Msn2p, and Msn4p. Eukaryot Cell. 2008;7(5):814–825.
- Chew SY, Ho KL, and Cheah YK, et al. Physiologically relevant alternative carbon sources modulate biofilm formation, cell wall architecture, and the stress and antifungal resistance of Candida glabrata. Int J Mol Sci. 2019 Jun 28;20:(13). DOI:10.3390/ijms20133172
- Freese S, Vogts T, Speer F, et al. C- and N-catabolic utilization of tricarboxylic acid cycle-related amino acids by Scheffersomyces stipitis and other yeasts. Yeast. 2011;28(5):375–390. DOI:10.1002/yea.1845
- Rasheed M, Battu A, Kaur R. Host-Pathogen interaction in Candida glabrata infection: current knowledge and implications for antifungal therapy. Expert Rev Anti Infect Ther. 2020 Nov;18(11):1093–1103.
- Vale-Silva LA, Sanglard D. Tipping the balance both ways: drug resistance and virulence in Candida glabrata. FEMS Yeast Res. 2015 Jun;15(4):fov025.
- Whaley SG, Rogers PD. Azole resistance in Candida glabrata. Curr Infect Dis Rep. 2016 Dec;18(12):41.
- Healey KR, Zhao Y, Perez WB, et al. Prevalent mutator genotype identified in fungal pathogen Candida glabrata promotes multi-drug resistance. Nat Commun. 2016 Mar 29;7:11128.
- Ferrari S, Sanguinetti M, De Bernardis F, et al. Loss of mitochondrial functions associated with azole resistance in Candida glabrata results in enhanced virulence in mice. Antimicrob Agents Chemother. 2011;55(5):1852–1860.
- Ferrari S, Sanguinetti M, Torelli R, et al. Contribution of CgPDR1-regulated genes in enhanced virulence of azole-resistant Candida glabrata. PLoS One. 2011 Mar 9;6(3):e17589. DOI:10.1371/journal.pone.0017589
- Nakayama H, Ueno K, Uno J, et al. Growth defects resulting from inhibiting ERG20 and RAM2 in Candida glabrata. FEMS Microbiol Lett. 2011;317(1):27–33.
- Schrevens S, and Sanglard D. Investigating Candida glabrata urinary tract infections (UTIs) in mice using bioluminescence imaging. J Fungi (Basel). 2021 Oct 9;7(10). DOI:10.3390/jof7100844
- Rasheed M, Battu A, Kaur R. Aspartyl proteases in Candida glabrata are required for suppression of the host innate immune response. J Biol Chem. 2018 Apr 27;293(17):6410–6433.
- Kammer P, McNamara S, and Wolf T, et al. Survival strategies of pathogenic Candida species in human blood show independent and specific adaptations. Mbio. 2020 Oct 6;11(5):e02435–20.
- Pekmezovic M, Hovhannisyan H, Gresnigt MS, et al. Candida pathogens induce protective mitochondria-associated type I interferon signalling and a damage-driven response in vaginal epithelial cells. Nat Microbiol. 2021;6(5):643–657.
- Amorim-Vaz S, Tran Vdu T, and Pradervand S, et al. RNA enrichment method for quantitative transcriptional analysis of pathogens in vivo applied to the fungus Candida albicans. Mbio. 2015 Sep 22;6(5): e00942–15.
- Silva S, Negri M, Henriques M, et al. Silicone colonization by non-Candida albicans Candida species in the presence of urine. J Med Microbiol. 2010;59(Pt 7):747–754.
- Gharaghani M, Taghipour S, Halvaeezadeh M, et al. Candiduria; a review article with specific data from Iran. Turk J Urol. 2018;44(6):445–452.
- Sierra-Diaz E, Hernandez-Rios CJ, Bravo-Cuellar A. Antibiotic resistance: microbiological profile of urinary tract infections in Mexico. Cir Cir. 2019;87(2):176–182.
- Peng D, Li X, Liu P, et al. Epidemiology of pathogens and antimicrobial resistance of catheter-associated urinary tract infections in intensive care units: a systematic review and meta-analysis. Am J Infect Control. 2018;46(12):e81–e90.
- Santana MMP, Hoffmann-Santos HD, Dias LB, et al. Epidemiological profile of patients hospitalized with candiduria in the Central-Western region of Brazil. Rev Iberoam Micol. 2019;36(4):175–180.
- Jain AK, Misra V, Ranjan N, et al. Speciation, Biofilm formation and antifungal susceptibility of Candida isolates from clinically diagnosed patient of UTI in a tertiary care hospital. J Assoc Physicians India. 2019;67(9):42–45.
- Gajdacs M, Doczi I, Abrok M, et al. Epidemiology of candiduria and Candida urinary tract infections in inpatients and outpatients: results from a 10-year retrospective survey. Cent European J Urol. 2019;72(2):209–214.
- Denis B, Chopin D, Piron P, et al. Candiduria in kidney transplant recipients: is antifungal therapy useful? Mycoses. 2018;61(5):298–304. DOI:10.1111/myc.12740
- Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11(3):R25.
- Law CW, Chen Y, Shi W, et al. Voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. [2014 Feb 3];15(2):R29.
- Leek JT, Johnson WE, Parker HS, et al. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012 Mar 15 28;(6):882–883. DOI:10.1093/bioinformatics/bts034
- Ge SX, Jung D, Yao R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics. 2020 Apr 15; 36(8):2628–2629. DOI:10.1093/bioinformatics/btz931
- Wu T, Hu E, Xu S, et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. The Innovation. 2021;2(3):100141. 2021/08/28/. DOI:10.1016/j.xinn.2021.100141
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Statistical Soc Ser B Methodol. 1995;57(1):289–300.
- Yu G. Gene ontology semantic similarity analysis using GOSemSim. In: Kidder B, editor. Stem cell transcriptional networks: Methods and protocols. New York, NY: Springer US; 2020. pp. 207–215.
- Kannan A, Asner SA, Trachsel E, et al. Comparative genomics for the elucidation of multidrug resistance in Candida lusitaniae. mBio. 2019 Dec 24;10(6). DOI:10.1128/mBio.02512-19
- Vale-Silva L, Ischer F, Leibundgut-Landmann S, et al. Gain of function mutations in CgPDR1, a regulator of antifungal drug resistance in Candida glabrata, control adherence to host cells. Infect Immun. 2013 Mar 4;81(5):1709–1720.
- Domergue R, Castaño I, De Las Peñas A, et al. Nicotinic acid limitation regulates silencing of Candida adhesins during UTI. Sci. 2005 May 6;308(5723):866–870.
- Landry DW, Bazari H. 116 - Approach to the patient with renal disease. In: Goldman L, Schafer AI, editors. Goldman’s cecil medicine (Twenty Fourth Edition). Philadelphia: W.B. Saunders; 2012. pp. 708–716.
- Domergue R, Castano I, De Las Penas A, et al. Nicotinic acid limitation regulates silencing of Candida adhesins during UTI. Sci. 2005 May 6;308(5723):866–870.
- Ferrari S, Ischer F, Calabrese D, et al. Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog. 2009 Jan 16;5(1):e1000268.
- Naglik J, Albrecht A, Bader O, et al. Candida albicans proteinases and host/pathogen interactions. Cell Microbiol. 2004;6(10):915–926.
- Vale-Silva L, Beaudoing E, and Tran VDT, et al. Comparative genomics of two sequential Candida glabrata clinical isolates. G3 (Bethesda). 2017 Aug 7; 7(8):2413–2426.
- Xu Z, Green B, Benoit N, et al. De Novo genome assembly of Candida glabrata reveals cell wall protein complement and structure of dispersed tandem repeat arrays. Mol Microbiol. 2020;113(6):1209–1224.
- Shimamura S, Miyazaki T, Tashiro M, et al. Autophagy-Inducing factor Atg1 is required for virulence in the pathogenic fungus Candida glabrata. Front Microbiol. 2019;10:27.
- Nagi M, Tanabe K, Ueno K, et al. The Candida glabrata sterol scavenging mechanism, mediated by the ATP-binding cassette transporter Aus1p, is regulated by iron limitation. Mol Microbiol. 2013;88(2):371–381.
- Srivastava VK, Suneetha KJ, Kaur R. The mitogen-activated protein kinase CgHog1 is required for iron homeostasis, adherence and virulence in Candida glabrata. Febs J. 2015 Jun;282(11):2142–2166.
- Saijo T, Miyazaki T, Izumikawa K, et al. Skn7p is involved in oxidative stress response and virulence of Candida glabrata. Mycopathologia. 2010;169(2):81–90. DOI:10.1007/s11046-009-9233-5
- Minematsu A, Miyazaki T, Shimamura S, et al. Vacuolar proton-translocating ATPase is required for antifungal resistance and virulence of Candida glabrata. PLoS One. 2019;14(1):e0210883. DOI:10.1371/journal.pone.0210883
- Rai MN, Sharma V, Balusu S, et al. An essential role for phosphatidylinositol 3-kinase in the inhibition of phagosomal maturation, intracellular survival and virulence in Candida glabrata. Cell Microbiol. 2015;17(2):269–287.
- Chen YL, Konieczka JH, and Springer DJ, et al. Convergent Evolution of calcineurin pathway roles in thermotolerance and virulence in Candida glabrata. G3 (Bethesda). 2012;2(6):675–691.
- Lebel K, MacPherson S, Turcotte B. New tools for phenotypic analysis in Candida albicans: the WAR1 gene confers resistance to sorbate. Yeast. 2006 Mar;23(4):249–259.
- Vylkova S, Lorenz MC. Modulation of phagosomal pH by Candida albicans promotes hyphal morphogenesis and requires Stp2p, a regulator of amino acid transport. PLoS Pathog. 2014 Mar;10(3):e1003995.
- Sprenger M, Brunke S, and Hube B, et al. A TRP1-marker-based system for gene complementation, overexpression, reporter gene expression and gene modification in Candida glabrata. FEMS Yeast Res. 2021 Jan 6;20(8). DOI:10.1093/femsyr/foaa066
- Van Zeebroeck G, Rubio-Texeira M, Schothorst J, et al. Specific analogues uncouple transport, signalling, oligo-ubiquitination and endocytosis in the yeast Gap1 amino acid transceptor. Mol Microbiol. 2014;93(2):213–233.
- Donaton MC, Holsbeeks I, Lagatie O, et al. The Gap1 general amino acid permease acts as an amino acid sensor for activation of protein kinase a targets in the yeast Saccharomyces cerevisiae. Mol Microbiol. 2003;50(3):911–929.
- Van Zeebroeck G, Bonini BM, Versele M, et al. Transport and signaling via the amino acid binding site of the yeast Gap1 amino acid transceptor. Nat Chem Biol. 2009;5(1):45–52.
- Lee YJ, Jang JW, Kim KJ, et al. TCA cycle-independent acetate metabolism via the glyoxylate cycle in Saccharomyces cerevisiae. Yeast. 2011;28(2):153–166. DOI:10.1002/yea.1828.
- Chen Y, Siewers V, Nielsen J. Profiling of cytosolic and peroxisomal acetyl-CoA metabolism in Saccharomyces cerevisiae. PLoS One. 2012;7(8):e42475.
- Nguyen TD, Walker ME, Gardner JM, et al. Appropriate vacuolar acidification in Saccharomyces cerevisiae is associated with efficient high sugar fermentation. Food Microbiol. 2018;70:262–268.
- Outten CE, Falk RL, Culotta VC. Cellular factors required for protection from hyperoxia toxicity in Saccharomyces cerevisiae. Biochem J. 2005 May 15;388(Pt 1):93–101.
- Merz S, Westermann B. Genome-Wide deletion mutant analysis reveals genes required for respiratory growth, mitochondrial genome maintenance and mitochondrial protein synthesis in Saccharomyces cerevisiae. Genome Biol. 2009;10(9):R95.
- Banerjee M, Thompson DS, Lazzell A, et al. UME6, a novel filament-specific regulator of Candida albicans hyphal extension and virulence. Mol Biol Cell. 2008;19(4):1354–1365.
- Nagi M, Nakayama H, Tanabe K, et al. Transcription factors CgUPC2A and CgUPC2B regulate ergosterol biosynthetic genes in Candida glabrata. Genes Cells. 2011;16(1):80–89.
- Van Ende M, Timmermans B, Vanreppelen G, et al. The involvement of the Candida glabrata trehalase enzymes in stress resistance and gut colonization. Virulence. 2021;12(1):329–345.
- Geiss GK, Bumgarner RE, Birditt B, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26(3):317–325.
- Cheng S, Clancy CJ, Xu W, et al. Profiling of Candida albicans gene expression during intra-abdominal candidiasis identifies biologic processes involved in pathogenesis. J Infect Dis. 2013 Nov 1;208(9):1529–1537.
- Xu W, Solis NV, Ehrlich RL, et al. Activation and alliance of regulatory pathways in C. albicans during mammalian infection. PLoS Biol. 2015;13(2):e1002076.
- O’-Meara TR, Xu W, Selvig KM, et al. The Cryptococcus neoformans Rim101 transcription factor directly regulates genes required for adaptation to the host. Mol Cell Biol. 2014;34(4):673–684.
- Fanning S, Xu W, Solis N, et al. Divergent targets of Candida albicans biofilm regulator Bcr1 in vitro and in vivo. Eukaryot Cell. 2012;11(7):896–904.
- Chung M, Teigen L, Liu H, et al. Targeted enrichment outperforms other enrichment techniques and enables more multi-species RNA-Seq analyses. Sci Rep. 2018 Sep 6;8(1):13377.
- Sanglard D, Ischer F, Calabrese D, et al. The ATP binding cassette transporter gene CgCDR1 from Candida glabrata is involved in the resistance of clinical isolates to azole antifungal agents. Antimicrob Agents Chemother. 1999;43(11):2753–2765.
- Shor E, Garcia-Rubio R, DeGregorio L, et al. A noncanonical DNA damage checkpoint response in a major fungal pathogen. mBio. 2020 Dec 15;11(6). DOI:10.1128/mBio.03044-20
- Shor E, Perlin DS. DNA damage response of major fungal pathogen Candida glabrata offers clues to explain its genetic diversity. Curr Genet. 2021 Jun;67(3):439–445.
- Navarathna DH, Lionakis MS, Lizak MJ, et al. Urea amidolyase (DUR1,2) contributes to virulence and kidney pathogenesis of Candida albicans. PLoS One. 2012;7(10):e48475.
- Brunke S, Quintin J, Kasper L, et al. Of mice, flies–and men? Comparing fungal infection models for large-scale screening efforts. Dis Model Mech. 2015;8(5):473–486.
- Kraidlova L, Van Zeebroeck G, Van Dijck P, et al. The Candida albicans GAP gene family encodes permeases involved in general and specific amino acid uptake and sensing. Eukaryot Cell. 2011;10(9):1219–1229.
- Kraidlova L, Schrevens S, and Tournu H, et al. Characterization of the Candida albicans amino acid permease family: Gap2 is the only general amino acid permease and Gap4 is an S-Adenosylmethionine (SAM) transporter required for SAM-Induced Morphogenesis. mSphere. 2016;1(6). DOI:10.1128/mSphere.00284-1616
- Zhang W, Du G, and Zhou J, et al. Regulation of Sensing, Transportation, and Catabolism of Nitrogen Sources in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2018;82(1). DOI:10.1128/MMBR.00040-17
- Brega E, Zufferey R, Mamoun CB. Candida albicans Csy1p is a nutrient sensor important for activation of amino acid uptake and hyphal morphogenesis. Eukaryot Cell. 2004 Feb;3(1):135–143.
- Schrevens S, Van Zeebroeck G, Riedelberger M, et al. Methionine is required for cAMP-PKA mediated morphogenesis and virulence of Candida albicans. Mol Microbiol. 2018;109(3):415–416.
- Perez-Delos Santos FJ, Riego-Ruiz L. Gln3 is a main regulator of nitrogen assimilation in Candida glabrata. Microbiol (Reading). 2016 Aug;162(8):1490–1499.
- Perez-de Los Santos FJ, Garcia-Ortega LF, Robledo-Marquez K, et al. Transcriptome Analysis Unveils Gln3 Role in Amino Acids Assimilation and Fluconazole Resistance in Candida glabrata. J Microbiol Biotechnol. 2021 May 28;31(5):659–666.
- Lorenz MC, Fink GR. The glyoxylate cycle is required for fungal virulence. Nature. 2001 Jul 5; 412(6842):83–86. DOI:10.1038/35083594
- Hill KJ, Stevens TH. Vma22p is a novel endoplasmic reticulum-associated protein required for assembly of the yeast vacuolar H(+)-ATPase complex. J Biol Chem. 1995 Sep 22;270(38):22329–22336.
- Kane PM. The where, when, and how of organelle acidification by the yeast vacuolar H±ATPase. Microbiol Mol Biol Rev. 2006 Mar;70(1):177–191.
- Dimmer KS, Fritz S, Fuchs F, et al. Genetic basis of mitochondrial function and morphology in Saccharomyces cerevisiae. Mol Biol Cell. 2002;13(3):847–853.
- Serrano R, Bernal D, Simon E, et al. Copper and iron are the limiting factors for growth of the yeast Saccharomyces cerevisiae in an alkaline environment. J Biol Chem. 2004 May 7;279(19):19698–19704.
- Schmidt M, Akasaka K, Messerly JT, et al. Role of Hog1, Tps1 and Sod1 in boric acid tolerance of Saccharomyces cerevisiae. Microbiol (Reading). 2012;158(Pt 10):2667–2678.
- Epp E, Vanier G, Harcus D, et al. Reverse genetics in Candida albicans predicts ARF cycling is essential for drug resistance and virulence. PLoS Pathog. 2010 Feb 5;6(2):e1000753.
- Nishikawa H, Miyazaki T, and Nakayama H, et al. Roles of vacuolar H±ATPase in the oxidative stress response of Candida glabrata. FEMS Yeast Res. 2016;16(5). DOI:10.1093/femsyr/fow054
- Gale AN, Sakhawala RM, and Levitan A, et al. Identification of essential genes and fluconazole susceptibility genes in Candida glabrata by profiling hermes transposon insertions. G3 (Bethesda). 2020 Oct 5; 10(10):3859–3870.
- Allert S, Schulz D, Kammer P, et al. From environmental adaptation to host survival: Attributes that mediate pathogenicity of Candida auris. Virulence. 2022;13(1):191–214.
- Mahl CD, Behling CS, Hackenhaar FS, et al. Induction of ROS generation by fluconazole in Candida glabrata: activation of antioxidant enzymes and oxidative DNA damage. Diagn Microbiol Infect Dis. 2015;82(3):203–208.