
ABSTRACT
Streptococcus suis epidemic strains were responsible for two outbreaks in China and possessed increased pathogenicity which was featured prominently by inducing an excessive inflammatory response at the early phase of infection. To discover the critical genes responsible for the pathogenicity increase of S. suis epidemic strains, the genome-wide transcriptional profiles of epidemic strain SC84 were investigated at the early phase of interaction with BV2 cells. The overall low expression levels of 89K pathogenicity island (PAI) and 129 known virulence genes in the SC84 interaction groups indicated that its pathogenicity increase should be attributed to novel mechanisms. Using highly pathogenic strain P1/7 and intermediately pathogenic strain 89–1591 as controls, 11 pathogenicity increase crucial genes (PICGs) and 38 pathogenicity increase-related genes (PIRGs) were identified in the SC84 incubation groups. The PICGs encoded proteins related to the methionine biosynthesis/uptake pathway and played critical roles in the pathogenicity increase of epidemic strains. A high proportion of PIRGs encoded surface proteins related to host cell adherence and immune escape, which may be conducive to the pathogenicity increase of epidemic strains by rapidly initiating infection. The fact that none of PICGs and PIRGs belonged to epidemic strain-specific gene indicated that the pathogenicity increase of epidemic strain may be determined by the expression level of genes, rather than the presence of them. Our results deepened the understanding on the mechanism of the pathogenicity increase of S. suis epidemic strains and provided novel approaches to control the life-threatening infections of S. suis epidemic strains.
Introduction
Streptococcus suis (S. suis) is an important zoonotic pathogen which can cause meningitis, septicemia, endocarditis, arthritis, pneumonia, and peritonitis in pigs and humans [Citation1]. Serotype 2 and sequence type (ST) 1 are the most frequently reported strains in both infected humans and pigs [Citation1,Citation2]. S. suis epidemic strains (serotype 2) were responsible for the outbreaks in 1998 and 2005 in China, characterized by the Streptococcal toxic-shock-like syndrome (STSLS) with high mortality [Citation3,Citation4]. In our previous study, the pathogenicity of three STs in serotype 2 strains was well-characterized and nominated as epidemic ST7, highly pathogenic ST1, and intermediately pathogenic ST25 strains, respectively [Citation5]. Compared with highly pathogenic ST1 and intermediately pathogenic ST25 strains, the pathogenicity of the epidemic strain increased and was featured prominently by inducing an excessive inflammatory response at the early phase of infection [Citation5]. Clarifying the mechanism of pathogenicity increase of epidemic strains is urgently needed to develop effective control and treatment strategies. To date, over 100 virulence genes have been reported to be involved in the pathogenesis of S. suis virulent strains [Citation6–10]. They were classified into four categories based on their properties and functions, including surface/secreted elements, enzymes/proteases, transcription factors/regulatory systems, and transporters/secretion systems [Citation7]. Moreover, novel virulence genes were continually identified in S. suis strains. However, the crucial genes responsible for the pathogenicity increase of the epidemic strains remain unclear.
The interaction with host cells is crucial in the pathogenesis of S. suis strains. Considering the interdependence of various virulence genes during the interaction, the analysis of the genome-wide transcriptional profiles rather than individual genes at specific time points post interaction with host cells could be a valuable strategy to link gene expression to the pathogenic phenotype of S. suis strains. In a previous study, microarray analysis was performed to identify the transcriptional profiles of a highly virulent S. suis strain at different infection sites in vivo infection [Citation11]. The regulated genes were mainly related to transcriptional regulation, metabolism, nutrient acquisition, stress defences, and virulence [Citation11]. However, the information on the regulated genes of S. suis strains post-interaction with host cells is still limited. Previous transcriptome studies mainly focused on the responses of host cells (such as THP-1 monocytes, porcine alveolar macrophages, and primary porcine choroid plexus epithelial cells et al) infected with S. suis strains. The genes of host cells involved in the inflammatory response, host defence, apoptosis, and signal transduction pathways were upregulated, while the genes concerned with cellular proliferation and metabolic process were downregulated [Citation12–15]. The innate immune response plays critical roles in the excessive production of pro-inflammatory cytokine and chemokine induced by S. suis epidemic strains [Citation16]. Microglia are the macrophage-like population responsible for the innate immune defence in the brain. The BV2 cells, as cell line that exhibits morphological and functional characteristics of microglia, play important roles in the CNS innate immune inflammation triggered by S. suis infection [Citation17]. Our previous study demonstrated that epidemic strains possessed a higher ability to elevate the phosphorylation of MAPK pathway proteins in immortal microglia cell BV2 than ST1 strains did at 4 h post-infection, leading to sequential active DNA binding activity of several transcription factors and then excessive pro-inflammatory cytokine release [Citation18]. Clarifying the transcriptome characteristics of epidemic strain before 4 h post-interaction with BV2 cells was important to elucidate its mechanisms to induce the excessive inflammatory response of BV2 cells. In the present study, we compared the genome-wide transcriptional profile of epidemic strain SC84 to those of highly pathogenic ST1 strain P1/7 and intermediately pathogenic ST25 strain 89–1591 at 2 h and 4 h-post interaction with BV2 cells using next-generation sequencing (RNA-seq). Among three interaction groups at the same time points, the upregulated genes of SC84 interaction groups with the highest transcription level were identified. The contributions of the identified genes to the pathogenesis of epidemic strains were further evaluated.
Materials and methods
Culture of BV2 cells and S. suis strains
BV2 cells were purchased from the Cell Resource Center, IBMS, CAMS/PUMC (Beijing, China). The cells were cultured in Dulbecco’s minimal essential medium (DMEM) with high glucose (Gibco, Carlsbad, USA) supplemented with 10% heat-inactivated foetal bovine serum (Gibco). For each experiment, 3 × 105 cells per well was plated into a 24-well flat-bottomed plates (Becton Dickinson and Company, Minneapolis, USA) and maintained at 5% CO2 37°C for 48 h to allow the cells to grow to confluency (1.5 × 106 cells per well) before the interaction with S. suis strains. The medium was changed every 24 h.
S. suis strain SC84 (the complete genome accession no. FM252031) as representative of epidemic strains, P1/7 (the complete genome accession no. AM946016) as representative of highly pathogenic strains, and 89–1591 as representative of intermediately pathogenic strains were used in the present study. The complete genome of 89–1591 was sequenced using PacBio Sequel platform and Illumina NovaSeq PE150 in the present study. The S. suis strains were grown overnight on Columbia blood base agar plates (Oxoid, London, UK) at 5% CO2 37°C, and isolated colonies were inoculated into 5 ml Todd – Hewitt broth (THB; Oxoid) without agitation at 5% CO2 37°C until OD600 nm value reaching 0.6 in the logarithmic growth phase, which corresponded to 1 × 109 CFU/ml, 1 × 109 CFU/ml, and 5 × 108 CFU/ml for SC84, P1/7, and 89–1591, respectively. The strains were washed twice in PBS (pH 7.4; Gibco). The pellets were then resuspended in DMEM with high glucose medium to obtain an expected concentration of 1 × 107 CFU/ml for each strain. Serial dilutions of the suspension were plated onto THB agar plates to determine the final number of CFU for each experiment.
Interaction with BV2 microglial cells
Bacterial suspensions (1 ml per well, 72 wells per strain per time point) were added to BV2 cells (MOI was about 7 for each strain). The plates were centrifuged at 500 g for 10 min and incubated at 5% CO2 37°C for 0, 2 and 4 h. The 0 h post-interaction groups were used as blank for each strain. Experiments were biologically repeated at least three times for each strain at each time point.
Enrichment of bacteria RNA and depletion of rRNA
After the interaction, the medium of each well was removed, and bacterial cells in each well were collected in 1 ml PBS by repeated washing 10 times. The strain suspension was filtered by 5 μm minisart syringe filter (Sartorius, Gottingen, German) twice to remove the BV2 cells, then washed twice in PBS. The total RNA of the pallet was extracted using the TRIzol (Thermo Fisher Scientific, Loughborough, U.K.) according to the manufacturer’s instructions. The MICROB Express Kit (Ambion, Austin, USA) and MICROB Enrich Kit (Ambion) were used to enrich bacterial mRNAs in total RNA samples by removing rRNAs and polyadenylated mRNAs according to the manufacturer’s instructions. Prior to sequencing, RNA samples were further quantified and examined for protein and reagent contamination using a Nanodrop ND‐1000 spectrophotometer. The integrity of RNA was determined by running the RNA sample on an Agilent Bioanalyzer RNA 6000 Nano/Pico Chip. The 100 ng purified total RNA samples with RIN (RNA Integrity Number) exceeded seven were used to construct RNA-Seq libraries. Totally, 34 samples (12 for the SC84 interaction and blank groups, 13 for the P1/7 interaction and blank groups, and nine for the 89–1591 interaction and blank groups) were obtained (Supplemental ).
Table 1. The information of PICGs identified in the SC84 interaction groups.
RNA-Seq sequencing and gene expression analysis
Illumina TruSeq RNA libraries were prepared using the NEB Next Ultra RNA Library Prep Kit (Illumina, San Diego, USA), following the manufacturer’s instructions. cDNA was then ligated to sequencing adapters. After purification and enrichment by PCR, the quality of the cDNA library was assessed on an Agilent Bioanalyzer DNA high-sensitivity chip. The constructed cDNA libraries with fragment lengths of approximately 300 bp were sequenced on an Illumina X-TEN platform (Illumina), sequencing 100 bp length for each read. Quality control of the resulting raw reads was performed using Trimmomatic software (v.0.35). The raw RNA-Seq reads were trimmed and filtered by fastQ software (v.0.21.0). For interaction samples, 1.58 to 8 Gb of >Q20 clean data (MAPQ > 20) were obtained (Supplemental ). The >Q20 clean RNA-Seq reads were uniquely mapped and aligned to ORFs of the corresponding S. suis genome by Burrows-Wheeler-Alignment Tool (BWA, v.0.7.17) to produce unigenes. The numbers of reads aligning to ORFs was calculated by cufflinks. The ratio of uniquely aligned reads to >Q20 clean RNA-Seq reads for interaction samples ranged from 12.43% to 92.13%, with average coverage to the corresponding S. suis genome from 152◊to 1207◊ (Supplemental ).
For differential expression gene (DEG) analysis, the Fragments Per Kilobase per Million (FPKM) value of each gene was calculated by cufflinks. The FPKM value of each gene in the SC84 interaction groups was compared with that of its homologous gene in the SC84 blank group using the edgeR program in BioConductor [Citation19]. DEGs of the SC84 interaction groups were identified when the p-value was<0.05 and the fold change was>2. In addition, the FPKM values of genes in the SC84 interaction groups were further compared with these of their homologous genes in the P1/7 and/or 89–1591 interaction groups at the same time point using the edgeR program in BioConductor. The differences in the FPKM values between homologous genes of different groups were significant when the p-value was<0.05 and the fold change was>2. The biological functions of DEGs were analysed by mapping the clusters of orthologous groups (COG) database by EggNOG V5.0.0 using default parameters (http://eggnogdb.embl.de/#/app/emapper).
The upregulated DEG in the SC84 interaction groups was nominated as pathogenicity increase crucial gene (PICG) if it met one of the following conditions: i. it belonged to epidemic strain-specific gene; ii. its FPKM value was simultaneously higher than that of its homologous gene in the P1/7 and 89–1591 interaction groups at the same time point; iii. its FPKM value was higher than that of its homologous gene in the P1/7 or 89–1591 interaction group at the same time point if its homologous gene was absent in one of genomes. Correspondingly, the non-DEGs in the SC84 interaction groups were nominated as pathogenicity increase-related genes (PIRGs) if they met one of the aforementioned second and third conditions.
To identify the homologous genes among three genomes, a matrix describing the gene content of three genomes was constructed using OrthoMCL. The cut-off value of BLAST E value, identity, and coverage was 1e5, 80%, and 80%, respectively. Mobile genes were excluded from three genomes based on the insertion sequence (IS) and transposon databases from EBI and GenBank. Multi-copy genes were also excluded from three genomes.
Construction of mutants deficient for PICGs of the SC84 2 h post-interaction group
Genomic DNA of SC84 was extracted and purified with the Wizard Genomic DNA Purification kit (Promega, Madison, USA). Restriction enzymes were purchased from New England Biolabs (NEB, Ipswich, USA) and used according to the manufacturer’s recommendations. Corresponding PICGs deletion fragments were constructed using splicing-by-overlap-extension PCR. Primers for the construction of mutants are shown in Supplemental . The ID5 and ID8 primers of each mutants were spliced with restriction enzyme PstI, BamHI/EcoRI restrictive site 6 bp and corresponding protective base 3 or 4 bp at 5’ side (Supplemental ). The PCR amplicons were digested with corresponding double restriction enzymes and subcloned into the thermosensitive E. coli-S. suis shuttle vector pSET4s [Citation20] digested with identical double restriction enzymes. The recombinant vectors were transformed into TOP10 competent cells (TIANGEN, Beijing, China) as recommended by the manufacturer. The PICG-deletion constructs were extracted and purified with Plasmid Mini Kit (Omega Bio-Tek, Guangzhou, China), then electroporated into competent S. suis strain SC84 using Gene Pulser X cell apparatus (Bio-Rad, Hercules, USA) under specific conditions (2.5 kV/cm, 200Ω, and 25 μF). After electroporation, the competent cells were plated on THA supplemented with Spectinomycin (100 μg/ml, THA + Sp) and incubated for 3 days at 28°C. Several Sp-resistant colonies were then subcultured again on THA + Sp for 2 days at 28°C. The colonies were next cultured on THA + Sp and incubated for 1 days at 37°C for two successive passages and then screened for the first crossing-over event. Loss of vector was induced by incubation of candidates at 28°C at least 10 successive passages. Temperature- and Sp-resistant clones were successively cultured on THA and THA + Sp at 37°C to obtain Sp-sensitive candidates. Deletion of the genes was confirmed by PCR and sequencing analysis. The draft genomes of constructed mutants were sequenced by Illumina sequencing to ensure that the sequences between the wild-type strain and mutants were identical, except for the PICGs deletion.
Table 2. The information of PIRGs identified in the SC84 interaction groups.
Experimental infection
Survival assay. C57BL/6 mice (6 weeks old, female) were injected intraperitoneally with 1 × 107 CFU of the wild-type strain SC84 and ∆PICGs mutants in 1 mL THB or 1 mL THB only as the control group. Each infected group contained ten mice and the control group contained five mice. The mortality was recorded per 6 h within 24 h post-infection and per 12 h from 24 h to 72 h post-infection. The experiment was performed independently twice. In the zebrafish infection model [Citation21], 15 adult zebrafish per group were intraperitoneally injected with 3 × 105 CFU and 3 × 106 CFU of wild-type strain SC84 and ∆PICGs mutants in 1 mL THB or 1 mL THB only as the control group. Mortality was monitored until 96 h post-infection. Survival rates of both infection models were calculated via the Kaplan–Meier method.
Cytokine production in vivo and bacterial loads in peripheral blood. C57BL/6 mice (6 weeks old, female) were injected intraperitoneally with 1 × 106 CFU of the wild-type strain SC84 and ∆PICGs mutants in 1 mL THB. Each group contained five mice. At 8 h post-infection, the mice were killed, and the peripheral blood of each infected mouse was collected aseptically. These experiments were performed independently twice. One hundred microlitres of serial tenfold dilutions of peripheral blood of each infected mouse were plated onto blood agar plates. Colonies were counted and expressed as CFU/ml. All serum samples were tested with concentrations of IL-6 and TNF-α using the ELISA kit (R&D Systems, Minneapolis, USA), as recommended by the manufacturer.
Statistical analysis
The survival rates of different infected groups were compared using the Log-rank test. Statistical analyses of the cytokine data and bacterial counts between wild-type strain SC84 and ∆PICGs mutants infected groups were performed by using the Wilcoxon’s two-sample test. The growth rates of different strains were compared using the Student unpaired t test. For these tests, a p-value <0.05 was considered to be significant. The statistical analysis was performed using GraphPad Prism software.
Nucleotide sequence accession number
The sequence of the 89–1591 genome sequenced in the present study was deposited in the GenBank under accession number PRJNA822771. The transcriptome data obtained in the study were deposited in the GenBank under accession numbers listed in Supplemental .
Ethical statement
This C57BL/6 mouse infection experiment was approved by the ethics committee of the National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention (Permit code: 2022–004). This zebrafish infection experiment was approved by the Laboratory Animal Monitoring Committee of Jiangsu Province (Permit code: SYXK (Su) 2017–0007).
Results
The homologous and specific genes identified among three genomes
Overall, 1576 genes shared by three genomes were nominated as core genes of three genomes. Among non-core genes, 272 genes only shared by SC84 and P1/7 genomes, 10 genes only shared by SC84 and 89–1591 genomes, and one gene only shared by P1/7 and 89–1591 genomes were identified. In addition, 58, 15, and 456 specific genes were identified in SC84, P1/7, and 89-1591 genomes, respectively (Supplemental ).
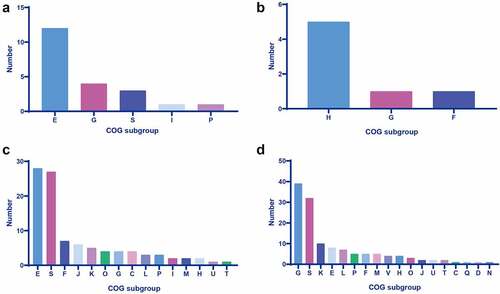
Figure 1. Cog analysis of DEGs in the sc84 interaction groups. A. Upregulated DEGs in the 2 h post-interaction group. B. Downregulated DEGs in the 2 h post-interaction group. C. Upregulated DEGs in the 4 h post-interaction group. D. Downregulated DEGs in the 4 h post-interaction group. The subgroups of cog are indicated in the bars with different colour.
The DEGs identified in the SC84 interaction groups
In the present study, 28 and 231 DEGs were identified in the SC84 2 h and 4 h post-interaction groups, respectively. Eighteen DEGs were identified in both 2 h and 4 h post-interaction groups, including 13 upregulated DEGs and 5 downregulated DEGs (Supplemental Table S3–1 and 3–2). Totally, 241 DEGs were identified in the SC84 interaction groups. Notably, none of the epidemic strain-specific genes were upregulated in the SC84 interaction groups, while nine epidemic strain-specific genes were downregulated in the SC84 4 h post-interaction group.
i. 2 h post-interaction group
Compared with the blank group, a total of 28 DEGs were identified, of which 21 displayed an upregulation and 7 displayed a downregulation. Except for homologous genes of three DEGs SSUSC84_0921, SSUSC84_1835, and SSUSC84_1836 absent in the 89–1591 genome, the remaining 25 DEGs belonged to core genes of three genomes. The homologous genes of SSUSC84_1406, SSUSC84_1605, and SSUSC84_1606 were also upregulated in both P1/7 and 89–1591 2 h post-interaction groups. The upregulation of the homologous genes of SSUSC84_0921 and SSUSC84_0739 was also found in the P1/7 2 h post-interaction group. The homologous genes of SSUSC84_0446, SSUSC84_0674, SSUSC84_1603, and SSUSC84_1842 also displayed the upregulated transcription level in the 89–1591 2 h post-interaction group. Remaining 12 DEGs were specifically upregulated in the SC84 2 h post-interaction group (Supplemental Table S3–1).
Seven downregulated DEGs were discovered in the SC84 2 h post-interaction group. All of them were the core genes of three genomes. Moreover, their homologous genes were also downregulated in the P1/7 2 h post-interaction group (Supplemental Table S3–2).
In the present study, the biological function of DEGs was investigated by the COG analysis. The COG of upregulated DEGs was predominantly categorized into subgroup of amino acid transport and metabolism, whereas downregulated DEGs mainly encoded coenzyme transport and metabolism-related protein (Supplemental Table S3–1, S3–2, and ).
ii. 4 h post-interaction group
Significantly more DEGs were identified in the SC84 4 h post-interaction group, consisting of 99 upregulated and 132 downregulated DEGs. The majority of the DEGs (196/231) were core genes of three genomes. No homologous genes of seven upregulated and 24 downregulated DEGs were found in the 89–1591 genome, whereas the homologous genes of 13 downregulated DEGs were absent in the P1/7 genome (Supplemental Table S4–1 and S4–2).
Among 99 upregulated DEGs, 28 of them were specifically upregulated in the SC84 4 h post-interaction group, whereas the homologous genes of 29 DEGs also displayed the upregulated transcription level in both P1/7 and 89–1591 4 h post-interaction groups (Supplemental Table S4–1). In addition, the homologous genes of 16 and 26 DEGs were upregulated in the P1/7 and 89–1591 interaction groups at the same time point, respectively. Forty-six DEGs were specifically downregulated in the SC84 4 h post-interaction group. The remaining 86 downregulated DEGs displayed a downregulation in the P1/7 and/or 89–1591 interaction groups at the same time point (Supplemental Table S4–2).
The predominant COG subgroup of 70 upregulated DEGs with known function was amino acid transport and metabolism (n = 28). In contrast, 99 downregulated DEGs with known function were mainly categorized into the COG subgroup of carbohydrate transport and metabolism (n = 39) (Supplemental Table S4–1, S4–2 and ).
The clustering distribution of DEGs
About 60% of DEGs (160/241) consisted of two or more contiguous genes in clusters ranging from 0.5 to 7.7 kbp in size. Totally, 53 clusters were identified, among which, 23 upregulated DEG cluster (UDCs) composed of 61 upregulated DEGs and 30 downregulated DEG cluster (DDCs) composed of 99 downregulated DEGs were found (Supplemental Table S3–1, S3–2, S4–1, and S4–2). Five UDCs and two DDCs were present in the SC84 2 h post-interaction group, whereas 20 UDCs and 29 DDCs were present in the SC84 4 h post-interaction group. Among them, UDC14, UDC20, and DDC21 were consecutively regulated in both 2 h and 4 h post-interaction groups (Supplemental Table S3–1 and S4–2).
Transcriptional activity of 89K PAI, known virulence genes and cps locus in the SC84 interaction groups
None of upregulated DEG was identified among transcriptionally active genes of 89K PAI in either 2 h or 4 h interaction groups, whereas only 13 downregulated 89K PAI genes were found in the 4 h post-interaction group (Supplemental Table S4–2).
In the present study, we also investigated the expression patterns of S. suis 129 known virulence genes in the SC84 interaction groups (Supplemental Table S5). Most of these virulence genes (103/129) genes were core genes of three genomes, whereas 25 and 7 genes were absent in the 89–1591 and P1/7 genome, respectively. Six upregulated and nine downregulated virulence genes were identified in the SC84 interaction groups (Supplemental Table S3–1, S4–1, S4–2, and S5). However, the genes encoding the virulence marker preferentially present in highly pathogenic S. suis serotype 2 strains, including mrp, sly, epf, sof, nisK, nisR, salK, salR, revS, SSU05_0473, neuB and neuC [Citation22,Citation23], were identified as non-DEGs in the SC84 interaction groups.
Only the glutamate dehydrogenase (gdh) gene was upregulated in the SC84 2 h post-interaction group (Supplemental Table S3–1 and S5). It was a core gene of three genomes. Interestingly, the gene was subsequently upregulated in the P1/7 and 89–1591 4 h post-interaction groups (data were not shown).
Fourteen virulence genes were identified as DEGs in the SC84 4 h post-interaction group. They were core genes of three genomes, except for hp197 and pnuc absent in the 89–1591 genome. The transcription levels of hp197, grpe, arcA, ciaH, ciaR, SSU05_1311, feoB, gtfA, and snt genes were downregulated, whereas 38 kDa, igA1, mutT, pnuc, and nadR genes were upregulated (Supplemental Table S4–1, S4–2, and S5).
Low expression levels of cps cluster genes were also discovered in the SC84 interaction groups. Four genes were downregulated in the 4 h post-interaction group, and none of the genes were upregulated during the whole interaction (Supplemental Table S4–2).
The PICGs identified in the SC84 interaction groups
Ten PICGs SSUSC84_1405, SSUSC84_1406, SSUSC84_1600, SSUSC84_1601, SSUSC84_1602, SSUSC84_1603, SSUSC84_1605, SSUSC84_1606, SSUSC84_1835, and SSUSC84_1836 were identified in the 2 h post-interaction group, while only the SSUSC84_0130 was identified as PICG in the 4 h post-interaction group ().
Notably, 10 PICGs of the 2 h interaction group also constituted UDC14, UDC15, UDC16, and UDC20 ranging from 2.3 to 4.1 kb in size, respectively. One PICG of the 4 h post-interaction group constituted UDC3 with an upregulated DEG SSUSC84_0131 ().
The PICGs SSUSC84_1405 and SSUSC84_1406 of UDC14 encoded cystathionine β- lyase and cystathionine γ-synthase, respectively. The function of proteins encoded by four PICGs SSUSC84_1600, SSUSC84_1601, SSUSC84_1602, and SSUSC84_1603 of UDC15 was methionine ABC transporter permease, methionine ABC transporter ATP-binding protein, peptidase M20, and MetQ, respectively. The UDC16 was composed of two PICGs SSUSC84_1605 and SSUSC84_1606 which encoded 5-methyltetrahydropteroyltriglutamate-homocysteine methyltransferase (MetE) and 5,10-methylenetetrahydrofolate reductase (MetF), respectively. UDC20 contained two PICGs SSUSC84_1835 and SSUSC84_1836 which encoded homocysteine S-methyltransferase and homocysteine S-methylpermease, respectively. Besides MetE and MetH, homocysteine S-methyltransferase and homocysteine S-methylpermease constituted a third methionine synthase in E. coli [Citation24]. The only PICG SSUSC84_0130 of the SC84 4 h post-interaction group encoded folate family ECF transporter S component. Notably, the COG of all PICGs was categorized into amino acid transport and metabolism-related protein. All of them were related to the pathway of methionine biosynthesis and uptake.
The PICGs of the 2 h post-interaction group played different roles in pathogenesis of SC84
The role of PICG SSUSC84_0130 of the 4 h post-interaction group in the pathogenesis of S. suis has been well studied [Citation25,Citation26]. Therefore, we focus on the PICGs of the 2 h post-interaction group in the present study. To assess the contribution of PICGs identified in the 2 h post-interaction group to the pathogenicity of SC84, the knock-out mutants deficient for UDC14, UDC15, UDC16, and UDC20 were constructed.
The wild-type strain SC84 and mutants reached the exponential phase at 3 h post incubation in THB medium. No significant difference in viable counts was observed between SC84 and mutants (Supplemental ). The comparative pathogenicity analysis was performed in C57BL/6 mouse and zebrafish infection models between mutants and wild-type strains. The survival rate of mice infected with wild-type strain SC84 was 60% at 12 h post-infection, then decreased to 30% at 18 h post-infection. Compared with that of mice infected with wild-type strain SC84, the significantly higher percentage of mice infected with ∆UDC14, ∆UDC15, and ∆UDC16 mutants survived throughout the experiment. The mice infected with ∆UDC14, ∆UDC15, and ∆UDC16 mutants still had a 100%, 90%, and 100% survival rate at 72 h post-infection, respectively. Moreover, the survival rates of mice infected with three mutants were similar to that of mock-infected mice. In contrast, no significant difference in survival rate between the mice infected with ∆UDC20 mutant and wild-type strain SC84 was observed. The survival rate of ∆UDC20 mutant infected mice was 50% at 12 h post-infection, then dramatically decreased to 20% at 24 h post-infection and 0% at 48 h post-infection ().
Figure 2. Survival curves of mice injected with 1 × 107 CFU of wild-type strain SC84, △UDC14 mutant, △UDC15 mutant, △UDC16 mutant, △UDC20 mutant, and PBS only as control group (A). Survival rates were calculated via the Kaplan–Meier method. The experiment was performed independently in duplicate. Mean of survival rates were present. The survival rates of different groups were compared using Log-rank test. # significant difference in survival level between the infected group and control group. * significant difference in survival level between the mutant and SC84 infected group. Production of proinflammatory cytokines IL-6 (B) and TNF-α (C) in serum and bacterial loads in peripheral blood (D) of C57BL/6 mice at 8 h post infected with 1 × 106 CFU of wild-type strain SC84 and △udcs mutants. The experiment was repeated twice with similar results. Median of cytokine levels in serum were presented. Peripheral blood bacterial loads were presented in median with interquartile range. Statistical analysis of the cytokine and bacterial counts was performed using the Wilcoxon two-sample test. p <0.05 was considered significant. * the cytokine levels were significantly higher than those of mice infected with wild-type strain SC84. § the cytokine levels were significantly lower than those of mice infected with wild-type strain SC84. # the bacterial loads in blood were significantly lower than those of mice infected with wild-type strain SC84.
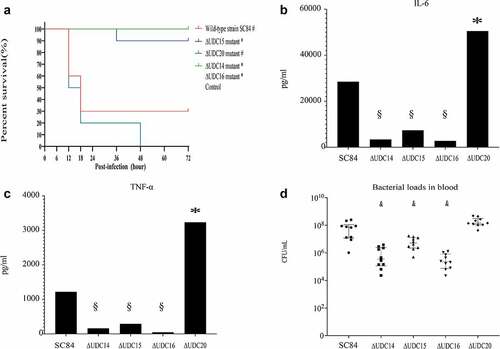
Excessive proinflammatory cytokines played critical roles in the pathogenesis of epidemic strains [Citation5,Citation16,Citation27]. The capacity of ∆UDC mutants to induce the production of pro-inflammatory cytokines in C57BL/6 mice was compared with that of wild-type strain SC84. Production of proinflammatory cytokines TNF-a and IL-6 in mice infected with ∆UDC20 mutant, was significantly higher than those of mice infected with wild-type strain SC84 at 8 h post-infection ( B and C). No significant difference was observed in bacterial loads of the peripheral blood between two infected groups. The bacterial counts in the peripheral blood of both wild-type strain SC84 and ∆UDC20 mutant infected groups reached 108 CFU/mL at 8 h post-infection (). In contrast, the pro-inflammatory cytokine levels in serum and bacterial loads in peripheral blood of the mice infected with ∆UDC14, ∆UDC15, and ∆UDC16 mutants were significantly lower than those of mice infected with wild-type strain SC84 at 8 h post-infection ( B and C).
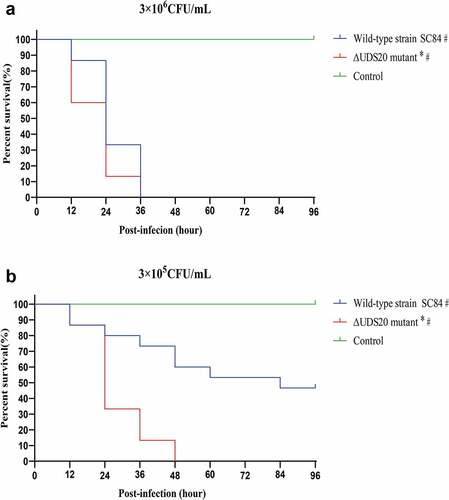
In order to further evaluate the pathogenicity of ∆UDC20 mutant, the zebrafish infection model was also used (). The zebrafish as a convenient, reliable, and standardized model was wildly applied to the assess the virulence level of S. suis [Citation21,Citation28–30]. In contrast to the result of mouse infection model, the survival rate of ∆UDC20 mutant infected group was significantly lower than that of the wild-type strain SC84 infected groups in the zebrafish infection model at both 105 and 106cfu infection doses. At 105cfu infection dose, ∆UDC20 mutant had a 100% mortality rate to zebrafish at 48 h post-infection, whereas the mortality rate to zebrafish infected with the wild-type strain SC84 was 40% at the same time point. At 106cfu infection dose, the difference in survival level between the zebrafish infected with ∆UDC20 mutant and wild-type strain SC84 was mainly observed at the early phase of infection. Zebrafish infected with ∆UDC20 mutant had a 60% survival rate at 12 h post-infection, while survival rate of zebrafish infected with wild-type strain SC84 was 100% at the same time point. The survival rates of both infected groups dramatically decreased to 0% at 36 h post-infection. These data indicated that ∆UDC20 mutant possessed a higher pathogenicity than wild-type strain SC84 did.
Figure 3. Survival curves of zebrafish infected with 3 × 106 CFU (A) and 3 × 105 CFU (B) of wild-type strain SC84 and △UDC20 mutant. PBS only was used in control group. Survival rates were calculated via the Kaplan–Meier method. The experiment was performed independently in duplicate. Mean of survival rate were presented. The survival rates of different groups were compared using Log-rank test. #, significant difference in survival level between infected group and control group. *, significant difference in survival level between △UDC20 mutant and wild-type strain SC84 infected groups. P < 0.05 was considered significant.
The PIRGs identified in SC84 interaction groups
Among 1576 core genes of three genomes, 12 and 11 PIRGs were identified in the SC84 2 h and 4 h post-interaction group, respectively. None of them were identified as PIRG in both 2 h and 4 h post-interaction groups. Among 272 genes only shared by the SC84 and P1/7 genomes, 13 and 3 PIRGs were identified in SC84 2 h and 4 h post-interaction group, respectively (). Amongst, only hp272 gene was identified as PIRG in both 2 h and 4 h post-interaction groups. Totally, 38 PIRGs were identified in the SC84 interaction groups. It is noteworthy that six of them belonged to known virulence genes, consisting of S. suis classical virulence gene epf, hp197, hp272, ciaR, sspep, and pili ( and Supplemental Table 5).
Interestingly, a high proportion of PIRGs encoded surface/membrane proteins. It indicated that surface/membrane proteins play important roles in the excessive inflammatory response induced by epidemic strains. In addition, PIRG SSUSC84_0348 and SSUSC84_0932 genes encoded methionine-regulated ECF transporter and O-acetylhomoserine (thiol)-lyase, respectively. Both of them may be involved in the pathway of methionine metabolism.
The feature of clustering distribution was also present in PIRGs. Twenty contiguous PIRGs constituted eight clusters, five (C1, C3, C4, C5, and C7) and three clusters (C2, C6, and C8) were identified in the 2 h and 4 h post-interaction group, respectively ().
Discussion
The pathogenicity increase of epidemic strain is characterized by inducing an excessive host inflammatory response at the early phase of infection [Citation5]. In our previous study, the inflammatory responses of BV2 cells infected with epidemic strain as well as highly pathogenic ST1 strain were compared [Citation18]. The phosphorylation level of the MAPK pathway in BV2 cells infected with epidemic strain was significantly higher than that of cells infected with ST1 strain at 4 h post-interaction. Subsequently, BV2 cells challenged epidemic strain produced significantly higher levels of the pro-inflammatory cytokines than BV2 cells infected with ST1 strain did from 8 h post-infection [Citation18]. Considering the fact that the differentiated response of infected BV2 cells was observed at 4 h post-interaction, the whole-genome transcriptome profiles of epidemic strain SC84 at 2 h and 4 h post-interaction with BV2 cells were analysed and compared with those of ST1 strain P1/7 in the present study. In order to eliminate potential false-positive targets and obtain a stringent list of genes related to the pathogenicity increase of epidemic strain, S. suis strain 89–1591 with intermediate virulence was also included in the present study.
The regulation of genes in the SC84 interaction groups was time-dependent, whereas 28 and 231 DEGs were identified in the 2 h and 4 h post-interaction group, respectively. Notably, the distribution of two or more contiguous DEG in clusters was wildly present in DEGs of the SC84 interaction groups. These DEG clusters may participate in the infection of epidemic strain as functional modules. Obvious differences were observed in functional categories between upregulated and downregulated DEGs. The predominant COG subgroup of upregulated DEGs was amino acid transport and metabolism. In contrast, downregulated DEGs mainly encoded proteins involved in carbohydrate transport and metabolism. Nutrient availability is closely linked to the adaptations and virulence of pathogens. Our data suggested that metabolites of amino acid metabolism may be indispensable at the early phase of epidemic strain interaction with host cells. Further metabonomic analysis may provide a novel approach to deepen the understanding of the pathogenesis of epidemic strains.
In the present study, the overall transcription level for genes of 89K PAI was low in the SC84 interaction groups. Similar results were also reported in two epidemic strains purely grown in THB medium [Citation31]. T4SS-like system and two-component signal transduction (TCST) systems located in 89K PAI contributed to the development of STSLS and the production of excessive pro-inflammatory cytokines [Citation32–34]. Our results showed that the expression levels of these systems was not regulated throughout the interaction. It indicated that 89K PAI of epidemic strains might not play critical roles at the early phase of infection. Likewise, low transcriptional activities of S. suis known virulence genes were also found in the SC84 interaction groups. These results indicated that the pathogenicity increase of epidemic strains might be attributed to novel mechanisms.
Compared with transcriptome profiles of highly pathogenic ST1 strain P1/7 and intermediately pathogenic ST25 strain 89–1591, 10 and one upregulated genes were identified as PICGs in the SC84 2 h and 4 h post-interaction group, respectively. The fact that none of them belonged to epidemic strain-specific genes suggested the pathogenicity increase of epidemic strain may be determined to a great extent by the expression level of genes, rather than their presence. It is noteworthy that 10 PICGs of 2 h post-interaction group constituted four clusters, consisting of UDC14, UDC15, UDC16, and UDC20.
Both PICGs SSUSC84_1405 and SSUSC84_1406 constituting UDC14 are pyridoxal phosphate-dependent enzymes and their homologous genes were involved in the transsulfuration pathway of methionine biosynthesis in Streptococcus anginosus [Citation35,Citation36]. The proteins encoded by four PICGs of UDC15 were predicted to be involved in the methionine uptake. The genes SSUSC84_1600, SSUSC84_1601, SSUSC84_1602, and SSUSC84_1603 encoded methionine ABC transporter permease, methionine ABC transporter ATP-binding protein, peptidase, and MetQ, respectively. The arrangement of UDC15 was similar to S. pneumoniae metQNP locus encoding a methionine ABC transporter [Citation37]. Moreover, MetQ and ABC transporter were constituents of the Methionine ABC (ATP binding cassette) Uptake Transporter (MUT) family in Escherichia coli and Bacillus subtilis [Citation38,Citation39]. MetQ was responsible for methionine delivery to the ABC transporter. The metQ gene was known to be relevant to the survival of S. suis in blood [Citation11], while a serotype 2 S. suis strain knocked out metQ gene showed decreased anti-phagocytosis ability in infected RAW264.7 cells [Citation40].
The UDC16 was composed of two PICGs SSUSC84_1605 and SSUSC84_1606 which encoded 5-methyltetrahydropteroyltriglutamate-homocysteine methyltransferase (MetE) and 5,10-methylenetetrahydrofolate reductase (MetF), respectively. MetE catalysed the synthesis and transfer of methyl group from 5-methyltetrahydrofolate directly to homocysteine [Citation41,Citation42]. The methyl group transferred by MetE to homocysteine is donated by 5-methyltetrahydrofolate which is synthesized from N5,10-methylene tetrahydrofolate by the catalyzation of MetF [Citation43]. Disruption of metF leads to methionine auxotrophy in Streptomyces lividans [Citation44].
UDC20 contained two PICGs SSUSC84_1835 and SSUSC84_1836 which encoded homocysteine S-methyltransferase and permease, respectively. The homologous gene of SSUSC84_1835 in E. coli was YagD gene (renamed mmuM gene) that catalyzes the last step in methionine biosynthesis and plays an important role in the metabolism process [Citation45]. The homologous gene of SSUSC84_1836 in E. coli was ykfD gene (renamed mmuP gene) that was responsible for the transporter of S-methylmethionine and co-transcribed with the mmuM gene [Citation24]. The physiological roles of the mmuM and mmuP products appear to uptake the external S-methylmethionine and convert homocysteine into methionine by transferring the methyl to homocysteine [Citation24], which is catalysed by either of two transmethylase enzymes MetE and MetH in enterobacteria [Citation41].
It is noteworthy that the proteins encoded by 10 PICGs of 2 h interaction group were all involved in the pathway of methionine biosynthesis and uptake. It indicates that the methionine and its metabolites are indispensable at the early phase of epidemic strain interaction with BV2 cells. Methionine, a vital amino acid in bacterial metabolism, acts as the initiator of protein synthesis and elongation. In addition, some methionine derivatives (e.g. S-adenosylmethionine) serve as methyl donors for the biosynthesis of phospholipids and nucleic acids [Citation46].
To further evaluate the combined contribution of PICGs in the pathogenicity increase of epidemic strains, the virulence levels of knock-out mutants deficient for UDC14, UDC15, UDC16, or UDC20 were compared with that of their parental strain SC84 using C57BL/6 mouse and zebrafish infection models. The ∆UDC14, ∆UDC15, and ∆UDC16 mutants showed significantly decreased lethality to C57BL/6 mice and pro-inflammatory cytokine levels in serum, which may mainly attribute to their lower bacterial loads in peripheral blood. The results indicated that the upregulation of three UDC14, UDC15, and UDC16 significantly bolstered the capacity of epidemic strains to resist phagocytosis by the host immunological system and rapidly establish infection.
On the contrary, the pathogenicity of ∆UDC20 mutant was higher than that of its parental strain since it showed a higher capacity to stimulate the production of pro-inflammatory cytokines in C57BL/6 mice than its parental strain did at the early phase of infection. In addition, the mortality rate of zebrafish infected with ∆UDC20 mutant was significantly higher than that of the group infected with the wild-type strain SC84 at the early phase of infection. These results indicated that the overexpression of UDC20 negatively regulated the virulence of the epidemic strain.
The differentiated pathogenic phenotypes of the four UDCs indicated they may play different roles in the biosynthesis and metabolism of methionine. The deletion of UDC14, UDC15, and UDC16 may result in methionine auxotrophy owing to the detriment of methionine biosynthesis and uptake, then the capacity of epidemic strains to survive and disseminate in the bloodstream significantly attenuated. On the contrary, significantly increased pathogenicity was observed in the ∆UDC20 mutant. It is possible that the deletion of UDC20 resulted in the accumulation of its substrates (such as S-methylmethionine and S-adenosylmethionine) or the enhanced production of selenomethionyl protein [Citation47], which may in turn lead to the pathogenicity increase of epidemic strains. The deletion of UDC20 may also result in excessive methionine production by upregulated other methionine synthase at the early phase of infection that significantly bolstered the pathogenicity of ∆UDC20 mutant. So far, the pathway of methionine biosynthesis and metabolism in the S. suis are still not well characterized. Further studies are urgently needed to elucidate the pivotal components of methionine cycling and their mechanisms in the pathogenicity increase of epidemic strains.
It is noteworthy that six of 10 PICGs were consistently upregulated in the SC84 4 h post-interaction group, consisting of SSUSC84_1405, SSUSC84_1406, SSUSC84_1600, SSUSC84_1602, SSUSC84_1835, and SSUSC84_1836. They were no longer identified as PICGs in the SC84 4 h post-interaction group in that their homologous genes were upregulated in the 89–1591 and/or P1/7 4 h post-interaction groups. It indicated that the extremely rapid initiation of methionine biosynthesis is crucial for the pathogenicity increase of the epidemic strains.
The only PICG identified in the SC84 4 h post-interaction group was the SSUSC84_0130 gene. The gene encoded folate family ECF transporter S component. Folate is essential for methionine biosynthesis in Streptomyces, Enterobacteria, and Mycobacterium tuberculosis [Citation44,Citation48]. It is possible that the protein encoded by SSUSC84_0130 gene was also involved in the methionine biosynthesis of S. suis strains. In a previous study, a hypervirulent S. suis strain (S735-pCOM1-V10) was generated by transforming a 3-kb fragment containing orf2 gene from virulent serotype 2 strain 10 into the weakly virulent serotype 2 strain S735 [Citation26]. The expression level and specific nucleotide polymorphisms of the fragment were responsible for the pathogenicity increase of S735-pCOM1-V10 [Citation25]. The sequence of SSUSC84_0130 was identical to that of orf2 gene. The homologous gene of SSUSC84_0130 was also upregulated in the 89–1591 2 h and 4 h post-interaction groups, although its FPKM value was lowest among three interaction groups at the same time point. Compared with the sequence of SSUSC84_0130 gene, 17 base-pair substitutions were identified in the homologous gene of the 89–1591 genome. In a previous study, the expression level of orf2 was strongly associated with sequences of the predicted −35 region upstream of its promoter [Citation25]. An SNP present in the corresponding region of the 89–1591 genome may be partially responsible for its low FPKM value in the 89–1591 interaction groups.
In addition to PICGs, 38 PIRGs were identified in the SC84 interaction groups, including two genes related to methionine metabolism and six known virulence genes. In addition, the expression level of hp272 in epidemic strain 05ZY033 5 h post-inoculate in THB medium were significantly higher than that of P1/7 [Citation31]. Although none of the “classical” S. suis virulence markers mrp, sly and epf were upregulated in the SC84 interaction groups, epf gene was identified as PIRG in the SC84 2 h post-interaction group. It is noteworthy that mrp and sly genes were upregulated in the P1/7 4 h post-interaction group (data were not shown), although no differences in FPKM values were observed between P1/7 and SC84 interaction groups at the same time point. Compared with mrp and sly genes, epf gene may play critical role in the pathogenicity increase of epidemic strains. In addition, surface proteins encoded by genes hP197 [Citation49] and hP272 [Citation50], secretary/immunogenic protein encoded by gene sspep [Citation51], and two-component regulatory system encoded by gene ciaR [Citation52] promoted the adherence of the S. suis to host cells. SSUSC84_1906 gene (separated into Sbp2’ and Sbp2’’ because of a nonsense mutation) coding a major pilin subunit Sbp2. Pili on the surface of Gram-positive strains played important roles in adherence to and invasion of host cells and biofilm formation. Deletion of Sbp2’ clearly attenuated the virulence of S. suis strain P1/7 in a zebrafish model [Citation53].
Interestingly, six upregulated known virulence genes in the SC84 interaction groups also encoded surface proteins. Among them, gdh and 38 kDa gene was mainly related to amino acid and carbohydrate metabolism, respectively [Citation8,Citation54,Citation55]. igA1 gene assisted S. suis strains to break the host mucosal immune barrier [Citation56], mutT, pnuc, and nadR genes involved in the biosynthesis and utilization pathway of NAD (Nicotinamide Adenine Dinucleotide) that were responsible for the resistance to oxidative stress and evasion of host immunity phagocytosis during the infection [Citation57–59]. In the present study, high proportion of PIRGs and upregulated known virulence genes encoded surface proteins that were involved in host cell adherence, in vivo survival, and immune escape, which were first step for epidemic strains to initiate the infection. The capacity to more rapidly initiate the infection may partially be responsible for the pathogenicity increase of epidemic strains. Consistent with our results, it was previously shown that most of the upregulated genes in epidemic strains grown in THB medium in vitro encoded surface-anchored proteins [Citation31]. The roles of many PIRGs in the pathogenesis of epidemic strains are still poorly characterized and worthy of further investigation. Surprisingly, the global expression level of capsular polysaccharide (CPS) conferring antiphagocytic properties to epidemic strains was low. Our data indicated that CPS of epidemic strains might not play critical roles in the stage of initiating infection.
In conclusion, the genome-wide transcriptional profiles of epidemic strain SC84 at the early phase of interaction with BV2 cells were investigated. The overall low expression levels of 89K PAI genes and known virulence genes in the SC84 interaction groups indicated that the pathogenicity increase of epidemic strains should be attributed to novel mechanisms. Using highly pathogenic strain P1/7 and intermediately pathogenic strain 89–1591 as controls, we revealed that methionine biosynthesis/uptake pathway played critical roles in the pathogenicity increase of epidemic strains. The proteins related to host cell adherence and immune escape may contribute to the extremely rapid initiation of the infection which is conducive greatly to the pathogenicity increase of epidemic strains. Our study deepened the understanding of the pathogenesis of epidemic strains, meanwhile provided novel approach and valuable information to control the infection of epidemic strains.
Contributions
Conceptualization, H.Z.; Methodology, J.W., P.L., H.S., Z.W., and K.Q.; Data Analysis, H.Z., J.W., P.L., and M.G.; Writing – Original Draft Preparation, H.Z., J.W. and P.L.; Writing – Review & Editing, M.G. and Z.W.; Funding Acquisition, H.Z. All authors have read and approved the final version of the manuscript.
Supplemental Material
Download Zip (782 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The sequence of the genome sequenced in the study was deposited in the GenBank under accession number PRJNA822771. The transcriptome data obtained in the study were deposited in the GenBank under accession numbers SRR18778325-SRR18778333, SRR18778712 -SRR18778724, and SRR18779406-SRR18779417. The data supporting the results presented in the study are available from the corresponding author upon reasonable request.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/21505594.2022.2116160
Additional information
Funding
References
- Goyette-Desjardins G, Auger JP, Xu J, et al. Streptococcus suis, an important pig pathogen and emerging zoonotic agent-an update on the worldwide distribution based on serotyping and sequence typing. Emerging Microbes & Infections. 2014 Jun;3(6):e45. DOI:10.1038/emi.2014.45
- Huong VT, Ha N, Huy NT, et al. Epidemiology, clinical manifestations, and outcomes of Streptococcus suis infection in humans. Emerg Infect Dis. 2014 Jul;20(7):1105–1114. DOI:10.3201/eid2007.131594
- Yu H, Jing H, Chen Z, et al. Human Streptococcus suis outbreak, Sichuan, China. Emerg Infect Dis. 2006 Jun;12(6):914–920. DOI:10.3201/eid1206.051194
- Ye C, Zhu X, Jing H, et al. Streptococcus suis sequence type 7 outbreak, Sichuan, China. Emerg Infect Dis. 2006 Aug;12(8):1203–1208. DOI:10.3201/eid1708.060232
- Ye C, Zheng H, Zhang J, et al. Clinical, experimental, and genomic differences between intermediately pathogenic, highly pathogenic, and epidemic Streptococcus suis. J Infect Dis. 2009 Jan 1 199;(1)97–107. 10.1086/594370
- Roodsant TJ, Van Der Putten BCL, Tamminga SM, et al. Identification of Streptococcus suis putative zoonotic virulence factors: a systematic review and genomic meta-analysis. Virulence. 2021 Dec;12(1):2787–2797. DOI:10.1080/21505594.2021.1985760
- Feng Y, Zhang H, Wu Z, et al. Streptococcus suis infection: an emerging/reemerging challenge of bacterial infectious diseases? Virulence. 2014 May 15 5;(4)477–497. 10.4161/viru.28595
- Fittipaldi N, Segura M, Grenier D, et al. Virulence factors involved in the pathogenesis of the infection caused by the swine pathogen and zoonotic agent Streptococcus suis. Future Microbiol. 2012 Feb;7(2):259–279. DOI:10.2217/fmb.11.149
- Dutkiewicz J, Zajac V, Sroka J, et al. Streptococcus suis: a re-emerging pathogen associated with occupational exposure to pigs or pork products. Part II - Pathogenesis. Ann Agric Environ Med. 2018 Mar 14 25;(1)186–203. 10.26444/aaem/85651
- Segura M, Calzas C, Grenier D, et al. Initial steps of the pathogenesis of the infection caused by Streptococcus suis: fighting against nonspecific defenses. Febs Lett. 2016 Nov;590(21):3772–3799. DOI:10.1002/1873-3468.12364
- Arenas J, Bossers-de Vries R, Harders-Westerveen J, et al. In vivo transcriptomes of Streptococcus suis reveal genes required for niche-specific adaptation and pathogenesis. Virulence. 2019 Dec;10(1):334–351. DOI:10.1080/21505594.2019.1599669
- Schwerk C, Adam R, Borkowski J, et al. In vitro transcriptome analysis of porcine choroid plexus epithelial cells in response to Streptococcus suis: release of pro-inflammatory cytokines and chemokines. Microbes Infect. 2011 Oct;13(11):953–962. DOI:10.1016/j.micinf.2011.05.012
- Liu M, Tan C, Fang L, et al. Microarray analyses of THP-1 cells infected with Streptococcus suis serotype 2. Vet Microbiol. 2011 May 12 150;(1–2)126–131. 10.1016/j.vetmic.2010.12.014
- de Greeff A, Benga L, Wichgers Schreur PJ, et al. Involvement of NF-kappaB and MAP-kinases in the transcriptional response of alveolar macrophages to Streptococcus suis. Vet Microbiol. 2010 Feb 24 141;(1–2)59–67. 10.1016/j.vetmic.2009.07.031
- Lauer AN, Scholtysik R, Beineke A, et al. A comparative transcriptome analysis of human and porcine choroid plexus cells in response to Streptococcus suis serotype 2 infection points to a role of hypoxia. Front Cell Infect Microbiol. 2021;11:639620.
- Zheng H, Luo X, Segura M, et al. The role of toll-like receptors in the pathogenesis of Streptococcus suis. Vet Microbiol. 2012 Apr 23 156;(1–2)147–156. 10.1016/j.vetmic.2011.10.014
- Dominguez-Punaro Mde L, Segura M, Contreras I, et al. In vitro characterization of the microglial inflammatory response to Streptococcus suis, an important emerging zoonotic agent of meningitis. Infect Immun. 2010 Dec;78(12):5074–5085. DOI:10.1128/IAI.00698-10
- Zheng H, Sun H, Dominguez-Punaro Mde L, et al. Evaluation of the pathogenesis of meningitis caused by Streptococcus suis sequence type 7 using the infection of BV2 microglial cells. J Med Microbiol. 2013 Mar;62(Pt 3):360–368. DOI:10.1099/jmm.0.046698-0
- Robinson MD, McCarthy DJ, Gk S. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010 Jan 1;26(1):139–140. DOI:10.1093/bioinformatics/btp616
- Takamatsu D, Osaki M, Sekizaki T. Thermosensitive suicide vectors for gene replacement in Streptococcus suis. Plasmid. 2001 Sep;46(2):140–148.
- Wu Z, Zhang W, Lu Y, et al. Transcriptome profiling of zebrafish infected with Streptococcus suis. Microb Pathog. 2010 May;48(5):178–187. DOI:10.1016/j.micpath.2010.02.007
- Huang W, Wang M, Hao H, et al. Genomic epidemiological investigation of a Streptococcus suis outbreak in Guangxi, China, 2016. Infect Genet Evol. 2019 Mar;68:249–252.
- Zheng X, Zheng H, Lan R, et al. Identification of genes and genomic islands correlated with high pathogenicity in Streptococcus suis using whole genome tiling microarrays. PLoS One. 2011 Mar 30 6;(3)e17987. 10.1371/journal.pone.0017987
- Thanbichler M, Neuhierl B, Bock A. S-Methylmethionine metabolism in Escherichia coli. J Bacteriol. 1999 Jan;181(2):662–665.
- de Greeff A, Buys H, Wells JM, et al. A naturally occurring nucleotide polymorphism in the orf2/folc promoter is associated with Streptococcus suis virulence. Bmc Microbiol. 2014 Nov 12 14;(1)264. 10.1186/s12866-014-0264-9
- Smith HE, Buijs H, Wisselink HJ, et al. Selection of virulence-associated determinants of Streptococcus suis serotype 2 by in vivo complementation. Infect Immun. 2001 Mar;69(3):1961–1966. DOI:10.1128/IAI.69.3.1961-1966.2001
- Zheng H, Punaro MC, Segura M, et al. Toll-Like receptor 2 is partially involved in the activation of murine astrocytes by Streptococcus suis, an important zoonotic agent of meningitis. J Neuroimmunol. 2011 May;234(1–2):71–83. DOI:10.1016/j.jneuroim.2011.02.005
- Wang X, Sun J, Bian C, et al. The population structure, antimicrobial resistance, and pathogenicity of Streptococcus suis cps31. Vet Microbiol. 2021 Jun 12;259:109149. DOI:10.1016/j.vetmic.2021.109149
- Zaccaria E, Cao R, Wells JM, et al. A zebrafish larval model to assess virulence of porcine Streptococcus suis Strains. PLoS One. 2016;11(3):e0151623. DOI:10.1371/journal.pone.0151623
- Neely MN, Pfeifer JD, Caparon M Streptococcus-Zebrafish model of bacterial pathogenesis. Infect Immun. 2002 Jul;70(7):3904–3914.
- Zhang D, Du N, Ma S, et al. In vitro transcriptome analysis of two Chinese isolates of Streptococcus suis serotype 2. Genomics Proteomics & Bioinformatics. 2014 Dec;12(6):266–275. DOI:10.1016/j.gpb.2014.11.001
- Xu J, Fu S, Liu M, et al. The two-component system NisK/nisr contributes to the virulence of Streptococcus suis serotype 2. Microbiol Res. 2014 Jul-Aug;169(7–8):541–546. DOI:10.1016/j.micres.2013.11.002
- Zhao Y, Liu G, Li S, et al. Role of a type IV-like secretion system of Streptococcus suis 2 in the development of streptococcal toxic shock syndrome. J Infect Dis. 2011 Jul 15 204;(2)274–281. 10.1093/infdis/jir261
- Li M, Wang C, Feng Y, et al. SalK/salr, a two-component signal transduction system, is essential for full virulence of highly invasive Streptococcus suis serotype 2. PLoS One. 2008;3(5):e2080. DOI:10.1371/journal.pone.0002080
- Yoshida Y, Negishi M, Nakano Y. Homocysteine biosynthesis pathways of Streptococcus anginosus. FEMS microbiology letters. 2003 Apr 25; 221(2):277–284. 10.1016/S0378-1097(03)00215-5
- Yoshida Y, Nakano Y, Amano A, et al. Lcd from Streptococcus anginosus encodes a C-S lyase with alpha,beta-elimination activity that degrades L-cysteine. Microbiology (Reading). 2002 Dec;148(Pt 12):3961–3970. DOI:10.1099/00221287-148-12-3961
- Basavanna S, Chimalapati S, Maqbool A, et al. The effects of methionine acquisition and synthesis on Streptococcus pneumoniae growth and virulence. PLoS One. 2013;8(1):e49638. DOI:10.1371/journal.pone.0049638
- Hullo MF, Auger S, Dassa E, et al. The metNPQ operon of Bacillus subtilis encodes an ABC permease transporting methionine sulfoxide, D- and L-methionine. Res Microbiol. 2004 Mar;155(2):80–86. DOI:10.1016/j.resmic.2003.11.008
- Merlin C, Gardiner G, Durand S, et al. The Escherichia coli metD locus encodes an ABC transporter which includes Abc (MetN,YaeE (MetI) and YaeC (MetQ). J Bacteriol. 2002 Oct;184(19):5513–5517.
- Pei X, Liu J, Liu M, et al. Quantitative proteomics revealed modulation of macrophages by MetQ gene of Streptococcus suis serotype 2. Amb Express. 2020 Oct 30 10;(1)195. 10.1186/s13568-020-01131-2
- Ferla MP, Patrick WM. Bacterial methionine biosynthesis. Microbiology (Reading). 2014 Aug;160(Pt 8):1571–1584.
- Weissbach H, Brot N. Regulation of methionine synthesis in Escherichia coli. Mol Microbiol. 1991 Jul;5(7):1593–1597.
- Saint-Girons I, Parsot C, Zakin MM, et al. Methionine biosynthesis in Enterobacteriaceae: biochemical, regulatory, and evolutionary aspects. Crc Crit Rev Biochem. 1988;23(1):S1–42. DOI:10.3109/10409238809083374
- Blanco J, Coque JJ, Martin JF. The folate branch of the methionine biosynthesis pathway in Streptomyces lividans: disruption of the 5,10-methylenetetrahydrofolate reductase gene leads to methionine auxotrophy. J Bacteriol. 1998 Mar;180(6):1586–1591.
- Ying J, Wang H, Bao B, et al. Molecular variation and horizontal gene transfer of the homocysteine methyltransferase gene mmuM and its distribution in clinical pathogens. Int J Biol Sci. 2015;11(1):11–21. DOI:10.7150/ijbs.10320
- Kozak M. Comparison of initiation of protein synthesis in procaryotes, eucaryotes, and organelles. Microbiol Rev. 1983 Mar;47(1):1–45.
- Kitajima T, Chiba Y, Jigami Y. Mutation of high-affinity methionine permease contributes to selenomethionyl protein production in Saccharomyces cerevisiae. Appl Environ Microbiol. 2010 Oct;76(19):6351–6359.
- Nixon MR, Saionz KW, Koo MS, et al. Folate pathway disruption leads to critical disruption of methionine derivatives in Mycobacterium tuberculosis. Chem Biol. 2014 Jul 17 21;(7)819–830. 10.1016/j.chembiol.2014.04.009
- Yuan ZZ, Yan XJ, Zhang AD, et al. Molecular mechanism by which surface antigen HP0197 mediates host cell attachment in the pathogenic bacteria Streptococcus suis. J Biol Chem. 2013 Jan 11 288;(2)956–963. 10.1074/jbc.M112.388686
- Chen B, Zhang A, Li R, et al. Evaluation of the protective efficacy of a newly identified immunogenic protein, HP0272, of Streptococcus suis. Fems Microbiol Lett. 2010 Jun;307(1):12–18. DOI:10.1111/j.1574-6968.2010.01944.x
- Tan C, Liu M, Li J, et al. SsPep contributes to the virulence of Streptococcus suis. Microb Pathog. 2011 Nov;51(5):319–324. DOI:10.1016/j.micpath.2011.07.008
- Li J, Tan C, Zhou Y, et al. The two-component regulatory system CiaRH contributes to the virulence of Streptococcus suis 2. Vet Microbiol. 2011 Feb 24 148;(1)99–104. 10.1016/j.vetmic.2010.08.005
- Shao J, Zhang W, Wu Z, et al. The truncated major pilin subunit Sbp2 of the srtBCD pilus cluster still contributes to Streptococcus suis pathogenesis in the absence of pilus shaft. Curr Microbiol. 2014 Nov;69(5):703–707. DOI:10.1007/s00284-014-0642-4
- Okwumabua O, Chinnapapakkagari S. Identification of the gene encoding a 38-kilodalton immunogenic and protective antigen of Streptococcus suis. Clin Diagn Lab Immunol. 2005 Apr;12(4):484–490.
- Okwumabua O, Persaud JS, Reddy PG. Cloning and characterization of the gene encoding the glutamate dehydrogenase of Streptococcus suis serotype 2. Clin Diagn Lab Immunol. 2001 Mar;8(2):251–257.
- Zhang A, Mu X, Chen B, et al. IgA1 protease contributes to the virulence of Streptococcus suis. Vet Microbiol. 2011 Mar 24 148;(2–4)436–439. 10.1016/j.vetmic.2010.09.027
- Li Q, Fei X, Zhang Y, et al. The biological role of MutT in the pathogenesis of the zoonotic pathogen Streptococcus suis serotype 2. Virulence. 2021 Dec;12(1):1538–1549. DOI:10.1080/21505594.2021.1936770
- Li Q, Zhang Y, Dechao D, et al. Characterization and functional analysis of PnuC that is involved in the oxidative stress tolerance and virulence of Streptococcus suis serotype 2. Vet Microbiol. 2018 Mar;216:198–206.
- Wang Q, Hassan BH, Lou N, et al. Functional definition of NrtR, a remnant regulator of NAD(+) homeostasis in the zoonotic pathogen Streptococcus suis. Faseb J. 2019 May;33(5):6055–6068. DOI:10.1096/fj.201802179RR