
ABSTRACT
The soil saprophyte, Burkholderia pseudomallei, is the causative agent of melioidosis, a disease endemic in South East Asia and northern Australia. Exposure to B. pseudomallei by either inhalation or inoculation can lead to severe disease. B. pseudomallei rapidly shifts from an environmental organism to an aggressive intracellular pathogen capable of rapidly spreading around the body. The expression of multiple virulence factors at every stage of intracellular infection allows for rapid progression of infection. Following invasion or phagocytosis, B. pseudomallei resists host-cell killing mechanisms in the phagosome, followed by escape using the type III secretion system. Several secreted virulence factors manipulate the host cell, while bacterial cells undergo a shift in energy metabolism allowing for overwhelming intracellular replication. Polymerisation of host cell actin into “actin tails” propels B. pseudomallei to the membranes of host cells where the type VI secretion system fuses host cells into multinucleated giant cells (MNGCs) to facilitate cell-to-cell dissemination. This review describes the various mechanisms used by B. pseudomallei to survive within cells.
GRAPHICAL ABSTRACT
Introduction
The soil saprophyte, Burkholderia pseudomallei, is the causative agent of melioidosis, a multifaceted disease endemic to the tropical belt, with hotspots in South East Asia and northern Australia [Citation1,Citation2]. Despite leading a typically environmental lifestyle, B. pseudomallei is able to infect exposed humans and animals causing disease and mortality [Citation3–5]. Diabetes is the leading risk factor for severe illness, with in excess of 40% of melioidosis sufferers having diabetes, or being diagnosed with diabetes at the time of hospital presentation [Citation6]. The International Diabetes Federation (IDF) estimates that in South East Asia and the western Pacific, around 10% of the adult population lives with diabetes, with over half of those being undiagnosed [Citation7,Citation8]. These rates are predicted to steadily increase over the next two decades. In addition, international travel has grown exponentially in recent years, with 120 million international arrivals into South East Asia alone in 2017 [Citation9]. This highlights a disproportionately susceptible population living in highly endemic areas, as well as millions of regional visitors at risk of exposure to B. pseudomallei. This review aims to highlight what is known about the pathogenicity and virulence of B. pseudomallei.
The Burkholderia sensu stricto group
The Burkholderia sensu lato (Burkholderia sl.) group, formerly classified as the genus Burkholderia and previous to that, Pseudomonas, has recently undergone a taxonomical reshuffle partly due to the increased availability of genome sequences from the Burkholderia species [Citation10,Citation11]. Burkholderia sl. encompasses six genera: Burkholderia sensu stricto (Burkholderia ss.), Paraburkholderia, Trinickia, Robbsia, Mycetohabitans, and Caballeronia [Citation11]. Most species of Burkholderia sl. are found in the environment, particularly the soil, but can occupy many ecological niches such as the rhizosphere, plants, and root nodules [Citation12]. Several species are capable of causing disease in plants such as onion rot (T. caryophylii), leaf spot and stripe disease (R. andropogonis), and rot of rice grains (B. glumae) [Citation13–16]. The Burkholderia ss. group consists of predominantly opportunistic pathogens, which fall into one of two species groups: B. cepacia complex (BCC) or B. pseudomallei complex (BPC) [Citation17,Citation18]. The Burkholderia cepacia complex consists of 22 closely related species that commonly infect immunocompromised and cystic fibrosis patients, with the most notable of these being B. cenocepacia and B. multivorans [Citation19–21]. Species of BCC commonly cause chronic pulmonary infection in cystic fibrosis, patients as well as nosocomial infection in immunocompromised individuals [Citation22,Citation23]. The most severe presentation of disease includes rapid and uncontrolled deterioration of patients with onset of septicaemia and necrotising pneumonia, commonly referred to as “cepacia syndrome” [Citation24]. As with most Burkholderia species, BCCs are intrinsically multi-drug resistant and capable of long-term persistence in the lung due to the establishment of in vivo biofilms [Citation25]. Multi-week courses of at least two intravenous antibiotics, such as tobramycin, meropenem, or ceftazidime, are commonly prescribed to cystic fibrosis sufferers with pulmonary exacerbations [Citation23,Citation26].
The Burkholderia pseudomallei complex (BPC) now contains eight highly related species: B. pseudomallei, B. mallei, B. thailandensis, B. humptydooensis, B. oklahomensis, B. singularis, B. mayonis, and B. savannae [Citation18,Citation27]. The most studied members of the BPC are Burkholderia pseudomallei and Burkholderia mallei, which cause melioidosis and glanders, respectively [Citation28,Citation29]. Burkholderia thailandensis, a close relative of B. pseudomallei, rarely causes human disease and is largely considered avirulent [Citation30,Citation31]. B. pseudomallei and B. mallei are classified as Tier 1 Select Agents by the Center for Disease Control and Prevention (CDC) with international guidelines recommending handling within a class II biosafety cabinet (BSC) in a biosafety level 3 (BSL3) facility, making genetic manipulation and characterisation difficult [Citation32]. Burkholderia mallei is the causative agent of both glanders (a naso-pulmonary syndrome) and farcy (cutaneous infection), which not only primarily infects solipeds such as horses and donkeys but can also cause fatal human disease upon exposure [Citation29]. Human cases are sporadic and most commonly associated with people working in proximity to infected animals [Citation33]. Glanders is endemic in parts of the Middle East, Asia, Africa, and Central and South America [Citation34]. Interestingly, B. mallei is the only Burkholderia species that is an obligate intracellular pathogen and can cause fulminant disease in less than a week if acute symptoms manifest [Citation35]. It is thought that B. mallei evolved from a single clone of B. pseudomallei after the loss of over 1000 genes. The remaining genes share >90% sequence homology with B. pseudomallei, supporting this claim [Citation36,Citation37].
Melioidosis
Melioidosis is a multi-syndrome illness, first described over a century ago by Alfred Whitmore in Rangoon, Myanmar [Citation28], and for many years, was referred to as Whitmore’s Disease. Although predominantly a tropical illness [Citation38], importation of cases from returned travellers has also been documented [Citation39–41]. Overall, both cases and deaths are thought to be highly under-reported, primarily due to lack of disease awareness, misdiagnosis, and lack of seeking healthcare. As such, an accurate global burden is difficult to compile [Citation2,Citation6,Citation38,Citation42]. Modelling has estimated that there are around 165,000 cases of human melioidosis per annum globally, with up to 89,000 thousand deaths [Citation2]. The highly endemic hotspots of northern Australia and north east Thailand have reported incidence rates of up to 50 cases per 100,000 people [Citation6,Citation43]. Surrounding countries in South East Asia, such as Laos, Cambodia, and Vietnam, also report high levels of incidence [Citation38,Citation44,Citation45]. In north east Thailand, melioidosis is the third most common cause of infectious disease death behind only AIDS and tuberculosis, with a mortality rate of around 40% [Citation6]. Mortality rates are lower in northern Australia at 10%, probably due to increased public disease awareness and better access to tertiary medical care [Citation46]. A systematic review of all melioidosis case reports, conducted in 2019, allowed for modelling to determine the global burden of melioidosis. The burden of symptomatic melioidosis worldwide is 4.6 million DALYs (Disability-adjusted life-year) annually, of which over 98.9% is attributed to years of life lost (YLL) [Citation47]. This burden from melioidosis exceeds what is reported for more prominent diseases such as dengue fever (1.95 million DALYs), leishmaniasis (0.7 million DALYs), and schistosomiasis (1.6 million DALYs) [Citation48]. Taking into consideration both global modelling and local incidence, melioidosis is highly prevalent worldwide and is a significant cause of death in regions where it is endemic.
Melioidosis appears to be a predominantly opportunistic infection, with only 20% of patients presenting with no known risk factors. The main risk factors for melioidosis include diabetes, hazardous alcohol use, and chronic lung or renal disease [Citation6,Citation43,Citation49]. In north east Thailand, the incidence rate of melioidosis for non-diabetics is 6.8 per 100,000, while incidence in diabetics is 145.7 per 100,000 and 84.4 per 100,000 in undiagnosed diabetics [Citation6].
Melioidosis has been referred to as the “great mimicker” due to the wide variety of clinical symptoms that can be observed in patients presenting to hospitals around the globe [Citation1,Citation50,Citation51]. Pneumonia is the predominant clinical presentation of melioidosis and can appear in both acute and chronic cases [Citation42,Citation52], this is followed by non-healing skin lesions [Citation43,Citation53,Citation54]. The breadth of clinical manifestations can lead to difficulty in prompt disease diagnosis, particularly in tropical regions that are also endemic for diseases such as malaria and tuberculosis, and as a result, melioidosis cases are commonly misdiagnosed [Citation39,Citation54,Citation55].
Treatment consists of two phases: an intravenous intensive initial phase followed by an oral eradication therapy [Citation56]. A minimum of 14 days of intravenous therapy is required with either ceftazidime or meropenem [Citation57,Citation58]. Following the intensive phase of therapy, a prolonged treatment with orally administered trimethoprim-sulfamethoxazole is required to allow for the successful eradication of infection and to prevent infection recrudescence or relapse [Citation57,Citation59–62].
Burkholderia pseudomallei
B. pseudomallei possesses one of the largest bacterial genomes (7.2 Mb), which is shared over two chromosomes and contains an extensive arsenal of virulence determinants, which enable it to both survive a range of harsh environments and cause a multitude of clinical presentations of disease [Citation63]. Due to its environmental niche, B. pseudomallei is intrinsically resistant to many host factors, anti-bacterial agents, and antibiotics [Citation64]. Ultimately, this not only makes B. pseudomallei infection difficult to treat and eradicate but also makes elucidating the pathogenesis and the roles of virulence factors extremely challenging due to the many redundancies within its genome.
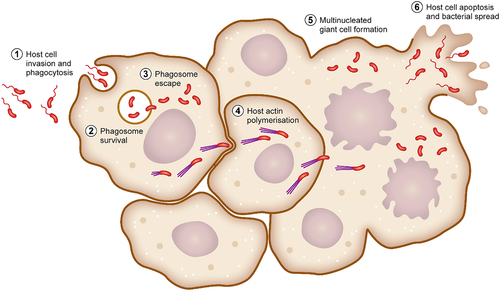
Following exposure to the bacterium, B. pseudomallei is able to attach to and invade host cells, as well as being phagocytosed into phagosomes by macrophages and neutrophils. B. pseudomallei resists host-cell killing mechanisms within the phagosome and then rapidly escapes into the cytoplasm. Rapid intracellular proliferation of bacteria then occurs as well as changes to primary bacterial energy production to allow for utilisation of available nutrients as well as subversion of the host through secretion of cytopathic toxins. B. pseudomallei then polymerises host-cell actin to propel itself to the host cell membrane where it induces host cell fusion into multinucleated giant cells (MNGCs), allowing it to spread throughout the body.
The host response to B. pseudomallei infection has recently been reviewed by Chomkatekaew et al. [Citation65]. Hence, this review will focus on the intracellular lifestyle of B. pseudomallei and describe the key determinants that affect the ability of the bacterium to survive in cells.
Adhesion to and invasion of host cells
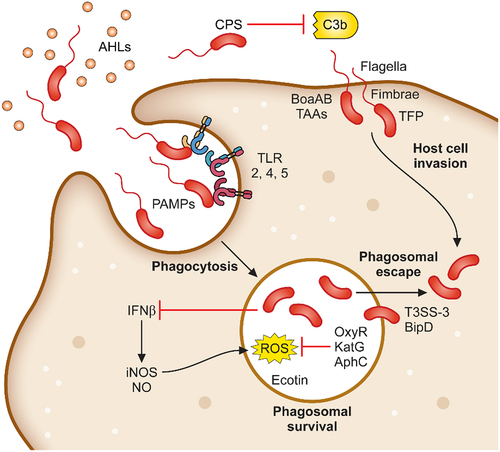
B. pseudomallei is able to infect virtually every cell type within a host [Citation66]. The bacterium is recognised by professional phagocytes, which subsequently phagocytose bacteria into a phagosome. B. pseudomallei also encodes for filamentous appendages and proteins, which facilitates tight binding to non-phagocytic cells and then subsequent invasion into the cell (). Exposure to B. pseudomallei can occur via inhalation, percutaneous inoculation, or accidental ingestion [Citation67]. It is thought that inhalation and incidental inoculation during occupational or recreational activity are the most frequent routes of bacterial acquisition. Breach of the epidermal layer may permit penetration by B. pseudomallei or contaminated matter (such as soil or surface water) into dermal tissue, extracellular matrices, endothelial cells, the bloodstream, and sufficiently traumatic, deeper organ tissues [Citation68–70]. Alternatively, after inhalation of aerosolised B. pseudomallei, bacterial cells are deposited along the upper and lower respiratory tract according to the droplet size as demonstrated by the BALB/c murine melioidosis model [Citation71], with bacteria in smaller droplets (1 µm) observed to colonise the lower respiratory epithelium, while those in larger droplets (12 µm) are primarily associated with nasal mucosa and nasal-associated lymphoid tissue (NALT) [Citation72]. Regardless of the initial colonisation site, B. pseudomallei infection rapidly spreads around the body with colonisation seen in distal organs within 72 hours [Citation72,Citation73]. After exposure, B. pseudomallei establishes physical contact with both phagocytic and non-phagocytic host cells and facilitates phagocytosis or cell invasion to establish an intracellular replicative niche.
Figure 1. Determinants required for B. pseudomallei attachment, invasion and phagosomal survival. B. pseudomallei uses a variety of mechanisms to initiate infection of host cells. Organelles and proteins such as the flagella, fimbriae, TFP and TAAs are used to attach to and directly infect host cells. In addition, B. pseudomallei is phagocytosed by macrophages. Following phagocytosis, B. pseudomallei induces expression of a plethora of proteins involved in phagosomal survival which as OxyR, KatG and AphC which protects against reactive oxygen species. Phagosomal escape is subsequently mediated by the type III secretion system which allows for rapid escape into the cytoplasm.
Pili
Pili, made up of pilin proteins, are filamentous appendages used by bacteria to mediate the initial stages of host cell adherence. Pili are able to retract and extend through filament polymerisation and depolymerisation to move along surfaces and come into close contact with host cells [Citation74]. A total of 13 pilin-like clusters are encoded in the prototype K96243 strain of B. pseudomallei [Citation63]. Deletion of pilA, which encodes a class A type IV pilus (TFP) in strain K96243, resulted in reduced adherence or invasion compared to the parental strain in three human epithelial cell lines, A549, BEAs2-B, and RPMI-2650 [Citation75]. Infection of the nematode, Caenorhabditis elegans, with the pilA mutant resulted in an 18-hour delay to death of 58 hours compared to 40 hours by the wild-type strain. Interestingly, this mutant showed no difference in its ability to cause disease in BALB/c mice compared to the wild-type strain by the intraperitoneal route, but a delay in death was observed when challenged by the intranasal route, with a lower bacterial challenge dose [Citation75]. In contrast, a pilA mutant generated from an alternative strain, B. pseudomallei strain 08, did not exhibit the same reduction in cellular adhesion or invasion against ME-180 human epithelial cells as was described by Essex-Lopresti et al. during their investigation of the K96243 strain. Rather, pilA was required by strain 08 for microcolony formation but only when grown at the lower temperature of 27°C [Citation76]. Deletion of a class B TFP cluster (BPSS2185-2198) resulted in attenuation in BALB/c mice when challenged intranasally [Citation77]. Further studies, where just the pilus subunit, BPSS2185, was deleted, resulted in a 50% decrease in adherence to A549 human lung epithelial cells [Citation78]. To fully understand the mechanism behind pilin-mediated attachment and/or adherence, further studies into the other pilin-like clusters are required.
Fimbriae
Another filamentous structure present on the outer surface of bacteria is the type I fimbriae, which bind to D-mannosylated sugar residues located on the cell surface glycoprotein receptors, expressed by intestinal epithelium and macrophages, thus facilitating internalisation of bacteria during intestinal infection [Citation79]. Deletion of the type I fimbriae in B. pseudomallei, fimA, resulted in a decrease in adherence of B. pseudomallei to both human and murine intestinal epithelial cells, as well as significantly decreased plaque formation. The mutant strain, ΔfimA was also required for full virulence in a chronic intestinal model of melioidosis, with 90% survival after 35 days [Citation80].
Autotransporters
Autotransporters (ATs) are outer membrane-anchored proteins that have been implicated in multiple pathogenesis pathways and can exist as monomeric (classical ATs) or trimeric structures (trimeric autotransporter adhesins, TAAs). B. pseudomallei encodes eleven ATs, two classical ATs and nine TAAs [Citation81]. Two B. pseudomallei TAAs, BoaA (BPSS0796) and BoaB (BPSL1705), act as adhesins, with deletion of boaA and boaB showing a decrease in adherence to A549, Hep2, and NHBE human respiratory epithelial cells, but only deletion of both TAAs in conjunction demonstrated a decrease in bacterial survival in J774A.1 murine macrophages [Citation82]. Six of the other encoded TAAs, bpaA, bpaB, bpaC, bpaD, bpaE, and bpaF play a role in either adherence to or invasion of A549 human lung epithelial cells [Citation83]. In the BALB/c mouse infection model, deletion of bpaC did not change the LD50 when challenged via an aerosol route of infection [Citation84]. However, previous studies showed a decrease in dissemination of ΔbpaC to the liver at 48 hours post-infection when challenged intranasally, suggesting a role during the early stages of in vivo infection [Citation83]. Another classical AT, BcaA, is involved in the invasion of non-phagocytic cells as a BcaA null-mutant exhibits a 50% reduction in invasive capacity of A549 human lung epithelial cells but exhibits no differences in phagocytic cells compared to the wild-type [Citation85].
Flagella
The flagellum of a bacterium not only allows for movement through the environment, but in many species, it is required for invasion of host cells. The B. pseudomallei flagellin subunit is encoded by fliC (BPSL3319). Deletion or inactivation of this gene results in a non-motile strain [Citation86–89]. Initial studies to identify protective antigens demonstrated that passive immunisation with anti-flagellin IgG provided 80% protection against an intraperitoneal challenge with 104 CFU of B. pseudomallei in a diabetic rat model [Citation90]. Interestingly, disruption of the flagellin gene (via Tn5 insertion) did not affect the virulence of B. pseudomallei mutant strains, MM35 and MM36, in the Syrian hamster and diabetic rat models of infection, leading to the hypothesis that the flagellum is not a virulence determinant per se but rather a protective antigen [Citation88]. This was verified by another study where challenge of BALB/c mice by the intraperitoneal route with an alternative B. pseudomallei flagellin mutant also demonstrated no change in LD50 [Citation89]. In contrast, deletion of fliC from the KHW strain of B. pseudomallei resulted in no attenuation during A549 human epithelial cell or C. elegans infection, but the fliC mutant did exhibit attenuation of infection in mice when challenged by the intranasal route, with decreased colonisation and mortality observed [Citation86]. This lack of in vitro attenuation shown by Chua et al. was attributed to the use of a centrifugation step during experimental procedures, as omitting this step resulted in decreased initial intracellular counts in both RAW264.7 murine macrophages and A549 human epithelial cells [Citation87], indicating that the flagellum is necessary for cell invasion and initiation of the intracellular lifecycle. The B. pseudomallei MM35-mutant strain lacking flagellum was also unable to attach to trophozoites of Acanthamoeba astronyxis [Citation91], postulating that the flagellum is likely to be important in the very early stages of adhesion.
A predominant number (88%) of Australian B. pseudomallei isolates encode a B. thailandensis-like flagellum and chemotaxis biosynthesis (BTFC) gene cluster. This BTFC cluster was replaced through horizontal gene transfer with a Yersinia-like fimbriae (YLF) cluster, with YLF B. pseudomallei isolates dominating in South East Asia (98% of isolates) [Citation92]. The BTFC fla2 locus encodes laterally positioned flagella that is used by B. thailandensis and BTFC-B. pseudomallei strains to rapidly move intracellularly independent of host-actin polymerisation [Citation93,Citation94]. Although deletion of fla2 does not affect swimming motility in B. thailandensis E264 [Citation94], whether the BTFC locus provides a virulence advantage over the YLF locus is yet unknown.
The flagellin (FliC) of B. pseudomallei is also involved in recognition by toll-like receptors (TLR) which are crucial for the host detection of pathogens and stimulation of the innate immune system. Purified FliC is recognised by TLR5 receptors and on stimulation of HEK-Blue™-hTLR5 and THP1-Dual™, cells activate NF-κB, in a TLR5-dependent manner, although this stimulation is not at the same level as purified flagellin from Salmonella Typhimurium [Citation95]. Amemiya et al. recently demonstrated that TLR5 recognises not only purified flagellin, but also the assembled filamentous flagella attached to B. pseudomallei, and deletion of fliC results in an elimination of TLR5 activation [Citation96]. These observations warrant further investigation into the activation of TRL5 by the flagella/flagellin, since ways to subvert this would be advantageous to the host.
Polysaccharides in B. pseudomallei
B. pseudomallei encodes numerous polysaccharide biosynthesis clusters, some of which have been well characterised. The nomenclatures of the various polysaccharide clusters in the literature can be confusing, as such shows each polysaccharide cluster found in B. pseudomallei and the various nomenclatures used to describe them in the literature. This review will use the nomenclature proposed by Reckseidler-Zeneto et al. for the capsular polysaccharides [Citation97] and Perry et al. for the O-antigen polysaccharides [Citation98]. An exopolysaccharide of the structure [-3)-2-O-acetyl-β-D-Galp-(1-4)-α-D-Galp-(1-3)-β-D-Galp-(1-5)-β-D-KDOp-(2-] has also been identified in B. pseudomallei, but its precise role in virulence is yet to be elucidated [Citation105,Citation106].
Table 1. Polysaccharides produced by B. pseudomallei.
Capsular polysaccharides (CPS) are tightly packed repeating polysaccharides that provide a barrier around bacterial cells and play a role in adhesion and pathogenicity. Four putative capsule polysaccharide (CPS) regions have been identified in B. pseudomallei with genes involved in sugar biosynthesis and transport () [Citation63,Citation97,Citation103]. The best characterised of these clusters is CPS I (BPSL2787-2810), which is present in all clinical isolates, indicating a conservation of this CPS amongst B. pseudomallei strains [Citation107]. The resulting product of this biosynthetic cluster is a high molecular weight unbranched polymer of [-3)-2-O-acetyl-6-deoxy-β-D-manno-heptopyranose-(1-] residues, also referred to as the type I O-PS in some papers () [Citation98,Citation103]. The CPS is thought to be a major virulence determinant, with initial studies using subtractive hybridisation, demonstrating that a CPS I-mutant had an LD50 similar to that of B. thailandensis [Citation99]. Further work with signature-tagged transposon mutants showed that CPS mutants were unable to establish infection in mice and had a significant increase in time to death [Citation89,Citation108]. During infection, capsule expression is induced in the presence of serum, resulting in a decrease in levels of complement C3b deposition, thus reducing the activation of the complement cascade [Citation100]. Deletion of the entire wcb capsule biosynthesis operon was successfully constructed by Warawa et al., indicating that capsule is not essential for bacterial viability. This mutant showed no significant difference from the wild-type strain when tested in vitro in J774A.1 murine macrophages, indicating that CPS I is not required for intracellular survival or replication of B. pseudomallei in a laboratory setting [Citation109]. However, this mutant displayed a 2-log increase in LD50 within an intranasal model using BALB/c mice. Decreased bacterial counts in the blood and spleen were observed, alongside decreased histopathology scores in the spleen and liver. This decrease in histopathology score is thought to be due to a reduced Th1 immune response to infection, which is usually stimulated by host recognition of the CPS I [Citation109]. Further investigations into single components of capsule biosynthesis were conducted by Cuccui et al., during which 18 of the 25 genes comprising the CPS I coding region were disrupted. Within this study, disruption of gmhA (BPSL2795), wcbJ (BPSL2798) and wcbN (BPSL2793) resulted in strains, which were unable to cause lethal disease following intranasal challenge. Immunization studies conducted with these strains prior to intranasal challenge using B. pseudomallei K96243 successfully increased time to death. However, immunization was not able to prevent recovery of B. pseudomallei from tissues of survivors, indicating that sterilizing immunity was not achieved [Citation101].
The lipopolysaccharide (LPS) is a glycolipid molecule decorating the outside of the cell wall of Gram-negative bacteria. The LPS consists of three components, the lipid A, the core region, and an O-antigen polysaccharide chain (O-PS) [Citation110]. The structure of the B. pseudomallei O-antigen was solved and found to be an unbranched heteropolymer of repeating structures of [-3)-β-D-glucopyranose-(1-3)-6-deoxy-α-L-talopyranose-(1-] and is referred to as type II O-PS [Citation98,Citation104]. The LPS biosynthesis cluster is found on Chromosome I (BPSL2672-BPSL2688) [Citation63]. A comprehensive study of 1327 isolates of environmental and clinical origin was investigated to determine the heterogeneity of LPS in B. pseudomallei isolates. Three serotypes of LPS have been observed in B. pseudomallei, smooth typical type A LPS, smooth atypical type B LPS, and rough LPS. The dominant serotype is smooth type A, which accounts for 97% of all screened isolates and consists of the characterised O-antigen moiety, type II O-PS [Citation111]. A variant of type B LPS, type B2, has now also been identified through genomic screening [Citation112]. A Tn5 transposon mutant of B. pseudomallei strain 1026b (insertion in wbiI, strain SRM117) deficient in type II O-PS was sensitive to killing in 30% non-human serum. Strain SRM117 also displayed a 10-fold reduction in virulence in both hamster and guinea-pig melioidosis models and 100-fold less virulence in an infant diabetic rat model [Citation102]. This decrease in virulence is also seen in BALB/c mice with a 4-log increase in LD50 in an intraperitoneal injection model [Citation89]. Further studies with SRM117 determined that deletion of the type II O-PS resulted in increased uptake into RAW264.7 murine macrophages. However, intracellular counts decreased during the early stages of infection, attributed to the activation of inducible nitric oxide synthase (iNOS), whereas macrophages infected by B. pseudomallei wild-type failed to activate iNOS [Citation113]. These results indicate that the LPS, particularly the O-PS moiety, plays an important role in suppressing the host response during early stages of infection in vitro and deletion results in attenuation in animal models of disease. TLR4 is an important component of this response due to its ability to detect the LPS of Gram-negative pathogens [Citation114]. B. pseudomallei can be recognised by both TLR4 and TLR2, and this has been demonstrated in vitro, but in vivo TLR2 is responsible for the detection of LPS and host response to infection [Citation115]. Further work has shown that only upon deletion of type II O-PS, does an increase in TLR4-dependent NF-κB activation occur [Citation116], perhaps explaining the in vivo host reliance on TLR2 activation. Differential signalling between murine and human cell line models has been shown with LPS-induced immune activation occurring solely through TLR4 within murine models both in vitro and in vivo, while additional TLR2 activation also occurs in human models [Citation117], highlighting the importance of more research into the immune response in different models of infection.
Phagosomal survival
Following the attachment and entry of B. pseudomallei into the host cell, the bacterium comes into contact with many host mechanisms that are utilised to kill and clear foreign material. Phagocytic cells employ and produce both reactive nitrogen intermediates (RNI) and reactive oxygen species (ROS) to control microbial infection [Citation118]. B. pseudomallei is a weak activator of beta-interferon (IFN-β), resulting in reduced iNOS production [Citation119]. iNOS can induce nitric oxide (NO) production and other reactive nitrogen species leading to the cytotoxic activity of macrophages against different microorganisms, intracellular parasites, and tumour cells [Citation120]. Infection of RAW264.7 murine macrophages with B. pseudomallei at a MOI of 10 showed no significant expression of iNOS compared to the high production observed when infected with either Escherichia coli or Salmonella typhi at a MOI of 0.1 [Citation121]. Furthermore, B. pseudomallei is susceptible to chemically generated NO in a macrophage-free system [Citation122], highlighting the importance of the subversion of iNOS expression and blocking of the NO production for the survival of B. pseudomallei in macrophages.
To protect against oxidative damage, B. pseudomallei expresses sigma factors, RpoS, and RpoE, and the transcriptional regulator OxyR, which can directly counteract oxidative stress through their downstream effectors. Sigma factors have been shown to mediate the response of B. pseudomallei to oxidative stress [Citation123]. In B. pseudomallei, inactivation of rpoS led to multiple effects including increased susceptibility to carbon starvation and oxidative stress with a rpoS mutant unable to induce MNGC formation [Citation123,Citation124]. RpoS also regulates genes involved in oxidative response including cysteine synthase B, 3-methyl-2-oxobutanoate hydroxymethyltransferase, several hypothetical stress response proteins, and succinyl-CoA: 3-ketoacid-coenzyme A transferase subunit A, with the latter shown to downregulate the expression of endogenous ROS [Citation125]. Additionally, deletion of sigma factor RpoE gene increased the susceptibility of B. pseudomallei against menadione and H2O2 and showed reduced intracellular survival in J774A.1 murine macrophages [Citation126]. Proteomic analysis of a rpoE mutant also showed differentially present proteins that are involved in oxidative and osmotic stress proteins in addition to chaperones, indicating the role it plays in the survival of B. pseudomallei under different adverse conditions [Citation127]. The transcriptional regulator, OxyR, regulates several genes associated with oxidative stress and within B. pseudomallei, it exerts a bi-functional role by repressing expression of the KatG catalase under normal growth and activating it under oxidative conditions [Citation128,Citation129]. The expression of OxyR is further regulated by the RpoS sigma factor [Citation130]. Inactivation of oxyR in B. pseudomallei increases the susceptibility to both H2O2 and paraquat, highlighting the essentiality of OxyR in protection against oxidative stress [Citation128]. Taken together, this emphasises the importance of the regulatory mechanisms in the intracellular survival and oxidative resistance of B. pseudomallei.
There are other genes involved in protection against oxidative stress including katG (BPSL2865) which encodes for catalase-peroxidase I in B. pseudomallei. Deletion of katG increases susceptibility to killing by different oxidative agents including H2O2, menadione, N-ethylmaleimide (NEM), and hypochlorite [Citation129]. Interestingly, the deletion of the katG gene led to increased resistance to organic hydroperoxide [Citation131]. This was identified to be due to the upregulation of alkyl hyroperoxidase reductase encoded by the ahpC gene. Overproduction of AhpC was shown to be protective against both reactive oxygen species and reactive nitrogen species [Citation131] explaining the resistance observed against tert-butyl hydroperoxide (t-BOOH) and demonstrating the role of AhpC in the intracellular survival of B. pseudomallei.
In addition to antioxidant effectors that can directly inactivate oxidants, B. pseudomallei is also armed with different proteins that can directly protect bacterial DNA. One of these proteins is the putative ferritin DPS-family DNA binding protein (dpsA/BPSL2863) encoded downstream of the katG gene in B. pseudomallei. The gene dpsA is regulated by both OxyR and RpoS, and under oxidative conditions, dpsA is co-transcribed with katG highlighting the multiple responses coordinated by the bacterium to resist oxidative stress [Citation129,Citation130]. It has been demonstrated that dpsA is important in the resistance of the bacterium against organic hydroperoxide with hypersensitivity to t-BOOH observed with the dpsA deletion mutant [Citation132]. Furthermore, B. pseudomallei also encodes for a spermidine acetyltransferase homologue that can act as a free radical scavenger protecting the DNA [Citation133]. This spermidine acetyltransferase, speG, is also indirectly regulated by RpoE under oxidative stress conditions [Citation126]. This multi-factorial approach allows B. pseudomallei to survive within the lysosomes by mounting a response, detoxifying the environment, and directly protecting the bacterial DNA.
Superoxide dismutases are enzymes that are able to convert superoxides, one of the most potent reactive oxygen intermediates, into the less damaging hydrogen peroxides for better clearance by other enzymes, including KatG [Citation129,Citation134]. Although not shown to participate in the resistance to intracellular superoxide, superoxide dismutase C (sodC) of B. pseudomallei has been shown to provide some protection against extracellular superoxides [Citation135]. Additionally, reduced replication in murine macrophages and attenuation in the BALB/c mice were observed upon deletion of sodC, demonstrating the importance of SodC in the detoxification of extracellular superoxides that may support intracellular growth of B. pseudomallei [Citation135]. These studies clearly demonstrate the ability of B. pseudomallei to adapt to and resist the phagosomal environment by modulating gene and protein expression. However, further studies are required to fully understand the roles of sigma factors and transcriptional regulators in the virulence of B. pseudomallei using in vivo infection models.
Phagosomal escape
B. pseudomallei has been shown to escape endocytic vesicles of HeLa, RAW264.7, and U937 cells within 15 minutes post-internalisation [Citation136,Citation137] (). The escape of the bacterium from the phagosome before the maturation into the phagolysosome is mediated by the type III secretion system (T3SS). This enables the bacteria to escape the lysosome and enter the host cytoplasm where replication and movement into adjacent cells can occur. The T3SS resembles a molecular syringe that delivers effector proteins into host cells. Three clusters of T3SS genes have been identified in B. pseudomallei strain K96243 [Citation63]. T3SS-1 of B. pseudomallei is homologous to the Hrp2 secretion system identified in Ralstonia solanacearum [Citation138,Citation139], whereas T3SS-2 has greater homology to the secretion systems of Xanthomonas spp [Citation138,Citation140]. T3SS-3 of B. pseudomallei has been characterised most extensively, and its components are named the Burkholderia secretion apparatus (Bsa). This secretion system has been observed to share high homology to the inv/spa/prg encoded T3SS of Salmonella Typhimurium and the ipa/mxi/spa T3SS of Shigella flexneri [Citation141]. Several structural, translocator, and effector proteins encoded by the T3SS-3 were identified to be essential in the escape of the bacterium from phagocytic vesicles.
Type III secretion systems (T3SS)
Deletion of genes encoding several structural proteins of the T3SS-3 has been shown to impair bacterial escape. A disruption in the bsaZ gene (BPSS1534), which encodes a structural component of the export apparatus homologous to Salmonella SpaS, showed a severe delay in phagosome escape as demonstrated by bacterial co-localization with the lysosomal-associated membrane protein 1 (LAMP-1) marker within J774.2 murine macrophages [Citation141] and transmission electron microscopy of RAW264.7 murine macrophages, which showed confinement of the bsaZ mutant to membrane-bound compartments [Citation142]. Insertional mutagenesis to inactivate bsaQ a structural component of the secretion apparatus resulted in delayed escape of the bacterium at 6 hours post-infection with the bsaQ mutant exhibiting greater co-localisation with LAMP-1-positive phagosomes compared to the wild-type [Citation143]. In addition to this phenotype, the secretion of BopE and BipD was absent in the supernatant indicating the inability of the bsaQ mutant to secrete effector proteins. BsaU (BPSS1539) is a part of the T3SS homologous to the InvJ of Salmonella spp., which is responsible for the control of the needle length [Citation144]. A bsaU transposon mutant showed significant impairment in vacuolar escape as visualised with LAMP-1 co-localisation staining [Citation145]. Intracellular replication and actin tail formation were shown to be functional, but the inability of the mutant to escape the phagosomes had led to the distension of the vesicles packed with B. pseudomallei bsaU mutant [Citation145].
The Bsa translocators, BipB, BipC, and BipD, sit at the tip of the Bsa needle complex forming a pore for the delivery of the bacterial effector proteins into host cells [Citation35]. As seen with the bsaZ mutant, severe impairment in vacuolar escape was seen with the bipD mutant, including reduced invasion of HeLa human epithelial cells [Citation146]. The bipD mutant was also shown to be almost exclusively co-localised with the LAMP-1-associated phagosomes in J774.2 murine macrophages, indicating the inability of the mutant to escape endocytic vesicles [Citation141]. In addition to the severe impairment in vacuolar escape, the bipD mutant was also unable to induce actin rearrangements and form cellular protrusions. Delay in vacuolar escape was also observed with the inactivation of BipB and BipC, which are homologous to SipB and SipC of Salmonella, respectively [Citation147,Citation148]. Both the bipB and bipC mutant also showed reduced intracellular survival with the bipB mutant and also display marked reduction in MNGC formation and in vivo virulence [Citation149]. This illustrates the importance of the B. pseudomallei Bsa translocators for escape from the phagosome and intracellular spread.
T3SS effector proteins
In addition to the translocators, deletion of several genes that encode for effector proteins that subvert host functions has led to the delayed or impaired escape of B. pseudomallei in the phagosomes. BopC is an effector protein and was shown to bind to its cognate chaperone BPSS1517 [Citation150]. Characterisation of the bopC mutant demonstrated reduction in intracellular survival in addition to delayed bacterial escape from the vacuoles of J774A.1 murine macrophages. Similarly, inactivation of bopA, another Bsa effector also resulted in reduced bacterial survival in phagosomes [Citation151]. Increased co-localisation of the bopA mutant with the autophagosomal microtubule-associated protein light chain 3 (LC3) in RAW264.7 murine macrophages highlighted the impaired ability of this mutant to escape the phagosome [Citation151]. A T3SS-3 effector protein of B. pseudomallei, BPSS1385 (CHBP) has been shown to be a cycle inhibiting factor (Cif). Cifs are known to cause cytopathic effects in eukaryotic host cells, such as cell cycle arrest and cell death [Citation152]. Cifs and CHBP are deamidases, which specifically deamidate the Gln40 residue on host ubiquitin and ubiquitin-like proteins [Citation153,Citation154]. Treatment of macrophage cells such as differentiated bone marrow-derived macrophages and J774A.1 murine macrophages with recombinant CHBP resulted in rapid cell death, while leaving epithelial cells such as HeLa cells viable [Citation154]. Mutagenesis studies of BPSS1385 in B. pseudomallei have not yet been undertaken, so its exact role in vivo is yet to be elucidated. Taken together, this illustrates the different functions of the T3SS and its effector proteins in facilitating bacterial escape from the endocytic vesicles as well as manipulation of host cell. The importance of the T3SS in B. pseudomallei also makes it an attractive target for therapeutic intervention. This was demonstrated using small molecule ATPase inhibitors, targeting the T3SS ATPase bsaS, which effectively reduced bacterial escape from the phagolysosome, resulting in decreased intracellular bacteria [Citation155]. How effective these inhibitors are in vivo remains to be validated, however this highlights the potential for targeting the T3SS in B. pseudomallei.
Intracellular adaptation and replication
Bacterial phospholipases are membrane-bound or secreted hydrolases [Citation156]. In B. pseudomallei, three phospholipase C (Plc) are encoded, plc1 (BPSL2403), plc2 (BPSL0338), and plc3 (BPSS0067) [Citation63]. Deletion of plc2 reduced the intracellular survival, plaque formation, and cytotoxicity in HeLa human epithelial cells, indicating that plc2 is more associated with virulence and intracellular survival compared to plc1 and plc3 [Citation157,Citation158]. Further analysis of the plc2 mutant showed that the reduction of intracellular survival was not due to delayed escape [Citation158]. As mentioned previously, macrophages utilise ROS, NO, and proteases to kill internalised bacteria. For example, mature macrophages express elastase (matrix metalloproteinase 12), which enters the phagolysosomes to attach to bacterial cell walls disrupting membrane integrity and inducing bacterial cell death [Citation159]. B. pseudomallei encodes the serine protease inhibitor ecotin (eco/BPSL1054) that is active against elastase, trypsin, and chymotrypsin [Citation160]. This allows the bacteria to circumvent the macrophage proteolytic activity assisting intracellular survival. The deletion of ecotin resulted in reduced intracellular survival of the bacteria in J774A.1 murine macrophages and significant attenuation in the BALB/c model of infection [Citation160]. Additionally, several ecotin orthologues were shown to inhibit the complement pathway system [Citation161]. Taken together, it can be hypothesised that ecotin plays a role in both the inhibition of the normal macrophage degradative functions and the impairment of the immune response allowing bacterial survival.
Essential in vivo metabolic pathways
Several metabolic enzymes have also been implicated to be important in the survival of the bacterium inside the host cells (). Transposon mutagenesis has identified genes in the purine metabolism pathway to be important in intracellular survival, in particular phosphoribosylglycinamide formyltransferase (purN/BPSL0908) and phosphoribosylformyl-glycinamidine cyclo-ligase (purM/BPSL2818) [Citation145]. The inactivation of purN and purM from B. pseudomallei strain E8 resulted in decreased intracellular survival within HeLa cells, with the latter showing no intracellular replication. Furthermore, gene deletions that affected the histidine (hisF/BPSL3133) and para-aminobenzoate biosynthetic pathways (pabB/BPSL2825) also exhibited reduced intracellular survival in HeLa cells [Citation145]. The reason proposed in the study is that the metabolic intermediates of these biosynthetic pathways are limited inside the host cells, and therefore, any further disruption via mutagenesis leads to impaired intracellular survival and replication. Additionally, a deletion of asd (BPSS1704), which encodes the enzyme aspartate-β-semialdehyde dehydrogenase, important for amino acid biosynthesis, resulted in the generation of a diaminopimelate (DAP) auxotrophic strain that was unable to grow in rich media without supplementation and displayed reduced intracellular replication in both RAW264.7 murine macrophages and HeLa cells [Citation162]. Like other metabolic mutants, the absence of DAP in mammalian cells thus becomes detrimental to the survival of the auxotrophic bacterium in cells [Citation162].
Figure 2. The intracellular adaptation and spread of B. pseudomallei from cell-to-cell. Following phagosomal escape B. pseudomallei switches its metabolic pathways to allow for the greatest energy production within the cytoplasmic environment. Additionally, secretion of the toxin BLF1 and the T3SS effector CHBP modulate host cell processes, halting host protein synthesis. Host cell actin is polymerised by BimAC and B. pseudomallei cells rapidly propel to the cellular membrane upon which their T6SS–1 is expressed and Hcp1 causes membrane fusion and the formation of multinucleated giant cells (MNGCs). MNGC formation facilitates rapid spread of bacterial infection to neighbouring cells while escaping recognition by the immune response.
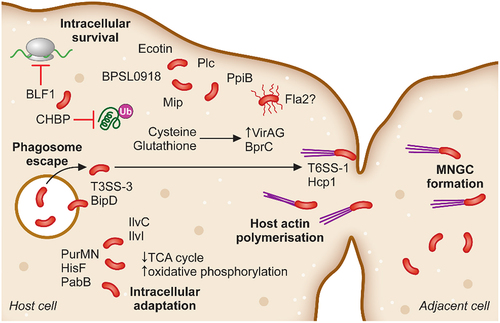
Other genes encoding for enzymes that participate in amino acid biosynthesis are equally important for B. pseudomallei survival and virulence. In particular, enzymes involved in the biosynthesis of the branched chain amino acids (BCAAs) leucine, isoleucine, and valine have been found to be essential for virulence. Disruption of ilvI (BPSL1196), encoding an acetolactate synthase catalytic subunit involved in 2-oxocarboxylic acid metabolism, was first discovered incidentally and investigated further due to the loss of virulence in a murine melioidosis model. The ilvI transposon mutant was auxotrophic for leucine, isoleucine, and valine. This gene assists the conversion of both pyruvate and 2-oxobutanoate into intermediate metabolites that form the skeleton for these BCAAs. The transposon mutant was unable to persist within mouse models or cause lethal infections to the same extent as wild-type B. pseudomallei strain 576. Interestingly, this gene is non-essential in rich media, but downstream components of the BCAA biosynthesis are necessary for survival (ilvC/BPSL1198) [Citation163,Citation164].
Energy metabolism during in vivo infection
Transcriptomic studies of B. pseudomallei following infection of Syrian hamsters showed an upregulation of genes involved in the intermediate steps of the glycolytic pathway (phosphoglycerate mutase, BPSL2902), inositol catabolism (BPSL1996, BPSL1997), glucosamine catabolism (glmS3), and tryptophan catabolism/synthesis (trpE, asnO, glmS3). Interestingly, genes involved in oxidative phosphorylation such as cyoA, cyoB, and fdsB, were highly induced, while genes involved in the TCA cycle (fumC, sucD and phbB), serine catabolism/synthesis (glyA, BPSL2219), and energy metabolism (ATP-synthase, NADH-dehydrogenase) were highly down regulated [Citation165]. The switch to alternative energy sources may be an adaptation for acute infection, which the Syrian hamster model represents, as inhibition of the TCA cycle enzyme, isocitrate lyase (aceA/BPSL2188), in Sprague-Dawley rats (a chronic model of melioidosis), results in the switching of infection from a persistent state to a lethal acute infection if untreated by antibiotics [Citation166]. This points to B. pseudomallei being metabolically flexible and able to utilise alternate energy sources during infection in vivo.
Toxins and their regulation
Toxins have also been shown to play a role in B. pseudomallei infection. The gene, BPSL1549, encodes for a toxin or Burkholderia Lethal Factor 1 (BLF1), which shows homology to E. coli toxin cytotoxic necrotizing factor 1 (CNF1) deamidase. This toxin is upregulated during virulence cues such as in the presence of human serum, taurine, and insulin. Purified BPSL1549 caused cell toxicity to J774.2 murine macrophages after 72 hours and a mutant strain, B. pseudomallei ΔBPSL1549 was severely attenuated in BALB/c mice and displayed a 100-fold increase in median lethal dose. BPSL1549 was shown to bind to the eukaryotic transcription initiation factor, eIF4A. BPSL1549 causes deamidation of Gln339 of eIF4A, which results in potent inhibition of host translation and protein synthesis [Citation167]. A putative toxin regulatory gene, BPSL1527 or tex, codes for a transcription accessory protein, which has been shown to be involved in toxin regulation in other pathogens. A deletion mutant, Δtex displayed reduced CFU numbers in spleen and lungs 48 hours post-infection and decreased survival in A549 epithelial cells and J774A.1 murine macrophages [Citation168]. It is not yet known if tex is directly involved in toxin regulation in B. pseudomallei as studies to determine this have not yet been published.
Folding proteins
Two members of the immunophilin superfamily, BPSS1823 (mip) and BPSL0918, have also been identified as important for intracellular survival of B. pseudomallei in J774A.1 murine macrophages [Citation169,Citation170]. These proteins are peptidyl-prolyl cis-trans isomerases and are involved in protein folding and chaperone activity [Citation171]. In addition, deletion of mip resulted in reduced motility as well as protease production and attenuation in a BALB/c mouse model [Citation170]. The proteins are likely to be playing a critical role in the folding of virulence factors required for B. pseudomallei to establish intracellular infection.
Cell-to-cell spread
Host actin polymerisation
B. pseudomallei is able to propel itself within a host cell by polymerising host cell actin [Citation172,Citation173] (). This is primarily mediated by the protein Burkholderia intracellular motility A (BimA), which was inferred through homology to other auto-secreted proteins in pathogens that polymerise host cell actin, in particular the YadA protein of Yersinia enterocolitica [Citation173]. Deletion of bimA results in loss of actin-based motility as shown in infected J774.2 murine macrophages, where bacterial cells cluster in the cytoplasm but exhibit a distinct lack of polar actin tails and cellular protrusions [Citation173]. Using monoclonal antibodies against BimA, Stevens et al. demonstrated that BimA localises to a singular pole of the bacterial cell with an F-actin tail occurring from the same pole. The polymerisation of actin into these actin tails is thought to propel B. pseudomallei towards the host cell membrane and cause cellular protrusion from which cell-to-cell spread is then mediated [Citation173]. BimA from B. pseudomallei is capable of polymerising actin in vitro independent of the Arp (actin-related protein) 2/3 complex, the protein complex that regulates and nucleates eukaryotic actin [Citation173]. However, the Arp 2/3 complex, as well as α-actinin, was found within the actin tail polymerised by BimA activity [Citation174]. Interestingly, recent work has shown that BimC, encoded directly upstream of bimA in the genome, is also required for actin tail formation in HeLa cells, but this deficiency was only partially restored by complementation [Citation175]. Although polar localisation of BimA still occurred in the ΔbimC-mutant strain, actin tail formation was absent. This indicates that BimC participates to some extent in BimA-mediated actin polymerisation [Citation175].
Multinucleated giant cell formation (MNGC)
Following the movement of B. pseudomallei through the cytoplasm, fusion of the host cell membranes occurs and MNGCs are formed (). This phenomenon has also been demonstrated in bone biopsies from human infections as well as in post-mortems [Citation176], indicating that this biological process occurs both within the laboratory and during natural infection. B. pseudomallei-infected RAW264.7 murine macrophages are induced to form MNGCs during infection with increased expression of calcitonin receptor (CTR), cathespin K (CTSK), and tartrate-resistant acid phosphatase (TRAP), markers which are hallmarks of functional osteoclasts, natural MNGCs formed from the fusion of mononuclear cells [Citation177,Citation178], although further work showed that B. pseudomallei-induced MNGCs are only osteoclast-like [Citation177]. Studies showed that lfpA (lactonase family protein A; BPSS2074) was responsible for the increase in expression of osteoblast markers, CTR, CTSK, and TRAP seen in RAW264.7 murine macrophages. Additionally, lfpA is required for optimal virulence in animal models, as deletion of IfpA results in a 4.5-fold-decrease in LD50 and a delayed time-to-death in Syrian hamsters and BALB/c mice [Citation177]. The formation of MNGCs by the host has been attributed to the type VI secretion system (T6SS) of B. pseudomallei, in particular the Cluster 1 T6SS [Citation179].
Type VI secretion systems (T6SS)
T6SS are essential components for virulence in a variety of pathogens including Vibrio cholerae and Francisella tularensis [Citation180,Citation181], as they are contact-dependent secretion apparatuses, which can inject toxins and other effectors into eukaryotic cells [Citation182]. B. pseudomallei encodes six T6SS clusters termed clusters 1 through to 6 (). There are currently two nomenclatures for these T6SS based on work by Schell et al. and Shalom et al. which were both published in 2007. This review will use the Schell et al. nomenclature whereby clusters 1-6 are as follows: T6SS-1 (tss-5), T6SS-2 (tss-4), T6SS-3 (tss-6), T6SS-4 (tss-3), T6SS-5 (tss-2), and T6SS-6 (tss-1) () [Citation190,Citation191]. These clusters share high levels of homology with those found in B. mallei and B. thailandensis, but interestingly, T6SS-4 is absent in B. thailandensis and T6SS-5 is absent in B. mallei, while T6SS-6 is truncated in B. mallei [Citation190] indicating that there may be a specific role for these clusters in B. pseudomallei. Expression of T6SS in B. pseudomallei is tightly controlled, with only T6SS-6 showing detectable levels of expression in nutrient broth [Citation179], with further studies demonstrating a 12-fold increase in expression of T6SS-1 following invasion into macrophages [Citation191]. Deletion of the inner tubule of the contractile sheath, composed of polymerised hexameric haemolysin-coregulated protein (Hcp) rings, of each T6SS cluster determined that only deletion of hcp1 of T6SS-1 was essential for disease in the Syrian Golden hamster model [Citation179], indicating that T6SS-1 is the major virulence-associated T6SS of B. pseudomallei. This mutant, Δhcp1, also displayed decreased intracellular counts and was less cytotoxic to RAW264.7 murine macrophages. This was corroborated by fluorescence microscopy, which showed a defect in intracellular spread with the inability to form MNGCs [Citation179]. Hcp proteins, in addition to being a structural component of the T6SS, can act as chaperones for effector proteins, although further experimental work needs to be conducted to determine this in B. pseudomallei [Citation192,Citation193]. Toesca et al. also demonstrated that the tip of the T6SS needle, VgrG, is also required for MNGC formation, in particular the C-terminal domain, which mediates host membrane fusion [Citation183] and that T6SS-1 is localised to the bacterial cell pole [Citation184]. T6SS-1 in B. pseudomallei was recently reviewed by Lennings et al. [Citation194].
Table 2. Type VI secretion systems (T6SS) of B. pseudomallei. B. pseudomallei encodes for six T6SS clusters as determined by homology to cluster of orthologous genes (COG) in other Gram-negative pathogens. Two nomenclature systems for B. pseudomallei T6SS were published in 2007, Schell et al. and Shalom et al. [Citation190,Citation191]. Both nomenclatures are listed as well as the known functions of these clusters in B. pseudomallei.
Activation of the T6SS-1 is mediated by the VirAG two-component system, which senses host intracellular signals, thus activating transcription of genes encoding components of the T6SS-1 such as hcp1, tssAB, and vgrG. The T3SS regulator protein, BprC, mediates expression of tssAB but not of hcp1 and virAG [Citation195]. Work by Wong et al. determined that exposure of B. pseudomallei to cysteine and reduced glutathione, present in the cytoplasm of host cells, activated the two-component system sensor kinase VirA, driving expression of T6SS-1 [Citation185]. Expression of T6SS-1 components is detectable at 3 hours post-infection and increases until 6 hours post-infection [Citation195].
The precise roles of the remaining five T6SS clusters requires further investigation, with only Cluster 1 shown to be essential for virulence in the mouse model [Citation179]. Studies in B. thailandensis have shown that T6SS-6 is important for contact-dependent bacterial competition, with deletion of clpV (encoding an AAA+ ATPase required for recycling the contracted sheath), resulting in a strain unable to persist in mixed population biofilms with Pseudomonas putida [Citation186]. A MarR transcription factor (TctR) was found to negatively regulate the expression of T6SS-2 but positively regulates T6SS-6, but ΔtctR was indistinguishable from wild-type in a Syrian hamster infection model [Citation187], indicating that TctR does not play a role in regulating the T6SS-1, which is essential for disease. T6SS-2 is expressed under oxidative stress conditions in B. thailandensis and has been observed to be regulated by OxyR. Overexpression of T6SS-2 was observed in the B. thailandensis ΔoxyR mutant [Citation188]. This strain is also required for export of zinc and manganese chelators into the extracellular space for combating oxidative stress [Citation189,Citation196]. These studies indicate that each of the T6SSs in B. pseudomallei may be important for survival in different environmental niches, investigation into this has been limited to date, with focus mainly on the intracellular environment.
Another protein important for B. pseudomallei intracellular spread and MNGC formation is the cyclophilin ppiB. The ppiB gene is essential for establishment of disease in BALB/c mice [Citation197]. It was shown that deletion of ppiB resulted in major disruption of the bacterial proteome ultimately leading to a 67% reduction in MNGC formation [Citation197], likely due to disruption of T6SS assembly in vitro and in vivo, although further work needs to be done to corroborate this.
In conclusion, B. pseudomallei has maintained numerous strategies to successfully establish a niche in humans and animals. B. pseudomallei employs many mechanisms for cell entry, vacuole escape, and subsequent cell-to-cell spread. It subverts the immune response to allow for bacterial proliferation, spreads to all systems in the host causing a multitude of symptoms, and can rapidly develop into septic pneumonia which can be fatal.
Author’s perspective
Burkholderia pseudomallei remains a resilient environmental pathogen capable of causing major morbidity in tropical regions of the world. Although studies have now unravelled mechanisms by which B. pseudomallei infects and replicates in the host, there is still much left to understand, such as its ability to persist, hide away effectively, and then remerge to cause infection years later. Unique mechanisms for virulence in B. pseudomallei, such as Arp2/3-independent actin polymerisation by BimA [Citation173] and multiple functional homologues of virulence factors such as the autotransporters, boaA and boaB [Citation82], mean careful manipulation and characterisation is required to fully understand the molecular mechanisms of pathogenicity. Additionally, different studies suggest variable roles of classical virulence factors. An example is the flagellum, in B. pseudomallei KHW strain, the absence of the flagella results in attenuation in BALB/c mice [Citation86]; in contrast, in strain 1026b, loss of flagella had no impact on the virulence using the Syrian hamster model [Citation88]. These instances highlight the differences between strains and infection models used between different laboratories to investigate these molecular mechanisms. Furthermore, only a small fraction of the genome has been investigated using genetic and biochemical methods, although high-throughput techniques such as transposon insertion site sequencing (TraDIS) have revealed a number of genes important for pathogenesis. To fully understand the pathogenicity of B. pseudomallei, more investment into genetic tools and characterisation of genes important for pathogenesis and persistence is required. With no currently licensed vaccine and treatment compliance moderate at best, particularly in endemic areas, more work is needed to find novel intervention points, which could be used in conjunction with current antibiotic therapies. This review has highlighted many virulence factors and lifestyle adaptations, which are required by B. pseudomallei to cause infection and survive within the host. Selection of some of these virulence determinants for therapeutic intervention studies, with the focus on synthesis of novel inhibitors, should support the development of new strategies to help melioidosis patients and those living in endemic areas.
Acknowledgements
CLM and JB are supported by an Australian Government Research Training scholarship. JB is the recipient of a DMTC PhD Top-Up Scholarship. Figures created using Biorender.com.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Cheng AC, Currie BJ. Melioidosis: epidemiology, pathophysiology, and management. Clin Microbiol Rev. 2005;18(2):1945–1965.
- Limmathurotsakul D, Golding N, Dance DA, et al. Predicted global distribution of Burkholderia pseudomallei and burden of melioidosis. Nat Microbiol. 2016;1:15008.
- Choy JL, Mayo M, Janmaat A, et al. Animal melioidosis in Australia. Acta Trop. 2000;74(2–3):153–158.
- Limmathurotsakul D, Thammasart S, Warrasuth N, et al. Melioidosis in animals, Thailand, 2006–2010. Emerg Infect Dis. 2012;18(2):325.
- Webb JR, Rachlin A, Rigas V, et al. Tracing the environmental footprint of the Burkholderia pseudomallei lipopolysaccharide genotypes in the tropical “Top End” of the Northern Territory, Australia. PLoS Negl Trop Dis. 2019;13(7):e0007369.
- Limmathurotsakul D, Wongratanacheewin S, Teerawattanasook N, et al. Increasing incidence of human melioidosis in Northeast Thailand. Am J Trop Med Hyg. 2010;82(6):1113.
- Sun H, Saeedi P, Karuranga S, et al. IDF diabetes Atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. 2022;183:109119.
- Ogurtsova K, Guariguata L, Barengo NC, et al. IDF diabetes Atlas: global estimates of undiagnosed diabetes in adults for 2021. Diabetes Res Clin Pract. 2022;183:109118.
- Tourism Highlights U. International tourism trends 2017. 2018.
- Yabuuchi E, Kosako Y, Oyaizu H, et al. Proposal of Burkholderia gen. nov. and transfer of seven species of the genus Pseudomonas homology group II to the new genus, with the type species Burkholderia cepacia (Palleroni and Holmes 1981) comb. nov. Microbiol Immunol. 1992;36(12):1251–1275.
- Mannaa M, Park I, Seo Y-S. Genomic features and insights into the taxonomy, virulence, and benevolence of plant-associated Burkholderia species. Int J Mol Sci. 2019;20(1):121.
- Coenye T, Vandamme P. Diversity and significance of Burkholderia species occupying diverse ecological niches. Environ Microbiol. 2003;5(9):719–729.
- Ballard R, Palleroni N, Doudoroff M, et al. Taxonomy of the aerobic pseudomonads: Pseudomonas cepacia, P. marginata, P. alliicola and P. caryophylli. Microbiology. 1970;60(2):199–214.
- Urakami T, Ito-Yoshida C, Araki H, et al. Transfer of Pseudomonas plantarii and Pseudomonas glumae to Burkholderia as Burkholderia spp. and description of Burkholderia vandii sp. nov. Int J Syst Evol Microbiol. 1994;44(2):235–245.
- Palleroni N, Krieg N, Holt J. Bergey’s manual of systematic bacteriology. Baltimore: The Willian and Wilkins Co; 1984.
- Goto K. New bacterial diseases of rice-bacterial brown stripe and bacterial grain rot. Ann Phytopathol Soc Jpn. 1956;21:46–47.
- Beukes CW, Palmer M, Manyaka P, et al. Genome data provides high support for generic boundaries in Burkholderia Sensu Lato. Front Microbiol. 2017;8.
- Hall CM, Baker AL, Sahl JW, et al. Expanding the Burkholderia pseudomallei complex with the addition of two novel species: Burkholderia mayonis sp. nov. and Burkholderia savannae sp. nov. Appl Environ Microbiol. 2021;88(1):e01583–21.
- Sousa SA, Feliciano JR, Pita T, et al. Burkholderia cepacia complex regulation of virulence gene expression: a review. Genes (Basel). 2017;8(1):43.
- Somprasong N, Yi J, Hall CM, et al. Conservation of resistance-nodulation-cell division efflux pump-mediated antibiotic resistance in Burkholderia cepacia complex and Burkholderia pseudomallei complex species. Antimicrob Agents Chemother. 2021;65(9):e00920–21.
- LiPuma JJ. Update on the Burkholderia cepacia complex. Curr Opin Pulm Med. 2005;11(6):528–533.
- Pegues CF, Pegues DA, Ford DS, et al. Burkholderia cepacia respiratory tract acquisition: epidemiology and molecular characterization of a large nosocomial outbreak. Epidemiol Infect. 1996;116(3):309–317.
- Lord R, Jones AM, Horsley A. Antibiotic treatment for Burkholderia cepacia complex in people with cystic fibrosis experiencing a pulmonary exacerbation. Cochrane Database Syst Rev. 2020;2020(4). DOI:10.1002/14651858.CD009529.pub4.
- Isles A, Maclusky I, Corey M, et al. Pseudomonas cepacia infection in cystic fibrosis: an emerging problem. J Pediatr. 1984;104(2):206–210.
- Leitão JH, Sousa SA, Ferreira AS, et al. Pathogenicity, virulence factors, and strategies to fight against Burkholderia cepacia complex pathogens and related species. Appl Microbiol Biotechnol. 2010;87(1):31–40.
- Trust U. Antibiotic treatment for cystic fibrosis. UK Cystic Fibrosis Trust Antibiotic Working Group; 2009.
- Sahl JW, Vazquez AJ, Hall CM, et al. The effects of signal erosion and core genome reduction on the identification of diagnostic markers. MBio. 2016;7(5):e00846–16.
- Whitmore A. An account of a glanders-like disease occurring in Rangoon. Epidemiol Infect. 1913;13(1):1–34.
- Whitlock GC, Mark Estes D, Torres AG. Glanders: off to the races with Burkholderia mallei. FEMS Microbiol Lett. 2007;277(2):115–122.
- Brett P, Deshazer D, Woods D. Characterization of Burkholderia pseudomallei and Burkholderia pseudomallei-like strains. Epidemiol Infect. 1997;118(2):137–148.
- Brett PJ, DeShazer D, Woods DE. Note: Burkholderia thailandensis sp. nov., a Burkholderia pseudomallei-like species. Int J Syst Evol Microbiol. 1998;48(1):317–320.
- Chosewood LC, Wilson DE. Biosafety in microbiological and biomedical laboratories. US Department of Health and Human Services, Public Health Service, Centers of Disease Control and Prevention, National Institutes of Health ; 2009.
- Van Zandt KE, Greer MT, Gelhaus HC. Glanders: an overview of infection in humans. Orphanet J Rare Dis. 2013;8(1):1–7.
- Elschner MC, Neubauer H, Sprague LD. The resurrection of glanders in a new epidemiological scenario: a beneficiary of “global change”. Curr Clin Microbiol Repo. 2017;4(1):54–60.
- Galyov EE, Brett PJ, DeShazer D. Molecular insights into Burkholderia pseudomallei and Burkholderia mallei pathogenesis. Annu Rev Microbiol. 2010;64:495–517.
- Godoy D, Randle G, Simpson AJ, et al. Multilocus sequence typing and evolutionary relationships among the causative agents of melioidosis and glanders, Burkholderia pseudomallei and Burkholderia mallei. J Clin Microbiol. 2003;41(5):2068–2079.
- Nierman WC, DeShazer D, Kim HS, et al. Structural flexibility in the Burkholderia mallei genome. Proc Nat Acad Sci. 2004;101(39):14246–14251.
- Dance DA. Melioidosis as an emerging global problem. Acta Trop. 2000;74(2–3):115–119.
- Le Tohic S, Montana M, Koch L, et al. A review of melioidosis cases imported into Europe. Eur J Clin Microbiol Infect Dis. 2019;38(8):1395–1408.
- Aardema H, Luijnenburg EM, Salm EF, et al. Changing epidemiology of melioidosis? A case of acute pulmonary melioidosis with fatal outcome imported from Brazil. Epidemiol Infect. 2005;133(5):871–875.
- Benoit TJ, Blaney DD, Gee JE, et al. Melioidosis cases and selected reports of occupational exposures to Burkholderia pseudomallei—United States, 2008–2013. Morbidity Mortality Weekly Rep. 2015;64(5):1–9.
- Currie BJ. Melioidosis: an important cause of pneumonia in residents of and travellers returned from endemic regions. Eur Respir J. 2003;22(3):542–550.
- Currie BJ, Ward L, Cheng AC. The epidemiology and clinical spectrum of melioidosis: 540 cases from the 20 year Darwin prospective study. PLoS Negl Trop Dis. 2010;4(11):e900.
- Dance D. Melioidosis: the tip of the iceberg? Clin Microbiol Rev. 1991;4(1):52–60.
- Dance DA, Luangraj M, Rattanavong S, et al. Melioidosis in the Lao People’s Democratic Republic. Trop Med Infect Dis. 2018;3(1):21.
- Parameswaran U, Baird RW, Ward LM, et al. Melioidosis at Royal Darwin Hospital in the big 2009–2010 wet season: comparison with the preceding 20 years. Med J Aust. 2012;196(5):345–348.
- Birnie E, Virk HS, Savelkoel J, et al. Global burden of melioidosis in 2015: a systematic review and data synthesis. Lancet Infect Dis. 2019;19(8):892–902.
- World Health O. Global health estimates: leading causes of DALYs. World Health Organization Retrieved. 2021;23.
- Limmathurotsakul D, Chaowagul W, Chierakul W, et al. Risk factors for recurrent melioidosis in northeast Thailand. Clin Infect Dis. 2006;43(8):979–986.
- Yee K, Lee M, Chua C, et al. Melioidosis, the great mimicker: a report of 10 cases from Malaysia. J Trop Med Hyg. 1988;91(5):249–254.
- White N. Melioidosis. Lancet. 2003;361(9370):1715–1722.
- Meumann EM, Cheng AC, Ward L, et al. Clinical features and epidemiology of melioidosis pneumonia: results from a 21-year study and review of the literature. Clinl Infect Dis. 2012;54(3):362–369.
- Churuangsuk C, Chusri S, Hortiwakul T, et al. Characteristics, clinical outcomes and factors influencing mortality of patients with melioidosis in southern Thailand: a 10-year retrospective study. Asian Pac J Trop Med. 2016;9(3):256–260.
- In: Currie BJ, editor. Melioidosis: evolving concepts in epidemiology, pathogenesis, and treatment. In: Seminars in respiratory and critical care medicine . Thieme Medical Publishers; 2015;36(01):111–125.
- Garg R, Shaw T, Vandana KE, et al. Melioidosis in suspected recurrent tuberculosis: a disease in disguise. J Infect Developing Countries. 2020;14(03):312–316.
- Lipsitz R, Garges S, Aurigemma R, et al. Workshop on treatment of and postexposure prophylaxis for Burkholderia pseudomallei and B. mallei infection, 2010. Emerg Infect Dis. 2012;18(12):e2.
- Dance D. Treatment and prophylaxis of melioidosis. Int J Antimicrob Agents. 2014;43(4):310–318.
- Cheng AC, Fisher DA, Anstey NM, et al. Outcomes of patients with melioidosis treated with meropenem. Antimicrob Agents Chemother. 2004;48(5):1763–1765.
- Currie BJ, Fisher DA, Howard DM, et al. Endemic melioidosis in tropical northern Australia: a 10-year prospective study and review of the literature. Clinl Infect Dis. 2000;31(4):981–986.
- Chusri S, Hortiwakul T, Charoenmak B, et al. Outcomes of patients with melioidosis treated with cotrimoxazole alone for eradication therapy. Am J Trop Med Hyg. 2012;87(5):927–932.
- Chetchotisakd P, Chierakul W, Chaowagul W, et al. Trimethoprim-sulfamethoxazole versus trimethoprim-sulfamethoxazole plus doxycycline as oral eradicative treatment for melioidosis (MERTH): a multicentre, double-blind, non-inferiority, randomised controlled trial. Lancet. 2014;383(9919):807–814.
- Fisher DA, Harris PN. Melioidosis: refining management of a tropical time bomb. Lancet. 2014;383(9919):762–764.
- Holden MT, Titball RW, Peacock SJ, et al. Genomic plasticity of the causative agent of melioidosis, Burkholderia pseudomallei. Proc Natl Acad Sci U S A. 2004;101(39):14240–14245.
- Schweizer HP. Mechanisms of antibiotic resistance in Burkholderia pseudomallei: implications for treatment of melioidosis. Future Microbiol. 2012;7(12):1389–1399.
- Chomkatekaew C, Boonklang P, Sangphukieo A, et al. An evolutionary arms race between Burkholderia pseudomallei and host immune system: what do we know? Front Microbiol. 2021;11. DOI:10.3389/fmicb.2020.612568.
- Jones AL, Beveridge TJ, Woods DE. Intracellular survival of Burkholderia pseudomallei. Infect Immun. 1996;64(3):782–790.
- Wiersinga WJ, Currie BJ, Peacock SJ. Melioidosis. N Engl J Med. 2012;367(11):1035–1044.
- Katz M, Smith S, Conway L, et al. Melioidosis in a patient with type 1 diabetes mellitus on an insulin pump. Endocrinol Diabetes Metab Case Rep. 2018;2018. DOI:10.1530/EDM-18-0062.
- Soffler C, Bosco-Lauth AM, Aboellail TA, et al. Pathogenesis of percutaneous infection of goats with Burkholderia pseudomallei: clinical, pathologic, and immunological responses in chronic melioidosis. Int J Exp Pathol. 2014;95(2):101–119.
- Bodilsen J, Langgaard H, Nielsen HL. Cutaneous melioidosis in a healthy Danish man after travelling to South-East Asia. BMJ Case Rep. 2015;2015. DOI:10.1136/bcr-2014-207340.
- Thomas RJ, Davies C, Nunez A, et al. Particle-size dependent effects in the BALB/c murine model of inhalational melioidosis. Front Cell Infect Microbiol. 2012;2:101.
- Thomas RJ. Particle size and pathogenicity in the respiratory tract. Virulence. 2013;4(8):847–858.
- Tan G-Y, Liu Y, Sivalingam SP, et al. Burkholderia pseudomallei aerosol infection results in differential inflammatory responses in BALB/c and C57Bl/6 mice. J Med Microbiol. 2008;57(4):508–515.
- Craig L, Forest KT, Maier B. Type IV pili: dynamics, biophysics and functional consequences. Nature Rev Microbiol. 2019;17(7):429–440.
- Essex-Lopresti AE, Boddey JA, Thomas R, et al. A type IV pilin, PilA, contributes to adherence of Burkholderia pseudomallei and virulence in vivo. Infect Immun. 2005;73(2):1260–1264.
- Boddey JA, Flegg CP, Day CJ, et al. Temperature-regulated microcolony formation by Burkholderia pseudomallei requires pilA and enhances association with cultured human cells. Infect Immun. 2006;74(9):5374–5381.
- Nandi T, Holden MTG, Didelot X, et al. Burkholderia pseudomallei sequencing identifies genomic clades with distinct recombination, accessory, and epigenetic profiles. Genome Res. 2015;25(1):129–141.
- Okaro U, Mou S, Lenkoue G, et al. A type IVB pilin influences twitching motility and in vitro adhesion to epithelial cells in Burkholderia pseudomallei. Microbiology. 2022;168(3):001150.
- Avalos Vizcarra I, Hosseini V, Kollmannsberger P, et al. How type 1 fimbriae help Escherichia coli to evade extracellular antibiotics. Sci Rep. 2016;6(1):1–13.
- Sanchez-Villamil JI, Tapia D, Borlee GI, et al. Burkholderia pseudomallei as an enteric pathogen: identification of virulence factors mediating gastrointestinal infection. Infect Immun. 2020;89(1):e00654–20.
- Adler NRL, Stevens JM, Stevens MP, et al. Autotransporters and their role in the virulence of Burkholderia pseudomallei and Burkholderia mallei. Front Microbiol. 2011;2:151.
- Balder R, Lipski S, Lazarus JJ, et al. Identification of Burkholderia mallei and Burkholderia pseudomallei adhesins for human respiratory epithelial cells. BMC Microbiol. 2010;10(1):1–19.
- Campos CG, Byrd MS, Cotter PA. Functional characterization of Burkholderia pseudomallei trimeric autotransporters. Infect Immun. 2013;81(8):2788–2799.
- Lafontaine ER, Balder R, Michel F, et al. Characterization of an autotransporter adhesin protein shared by Burkholderia mallei and Burkholderia pseudomallei. BMC Microbiol. 2014;14(1):1–14.
- Campos CG, Borst L, Cotter PA. Characterization of BcaA, a putative classical autotransporter protein in Burkholderia pseudomallei. Infect Immun. 2013;81(4):1121–1128.
- Chua KL, Chan YY, Gan YH. Flagella are virulence determinants of Burkholderia pseudomallei. Infect Immun. 2003;71(4):1622–1629.
- Chuaygud T, Tungpradabkul S, Sirisinha S, et al. A role of Burkholderia pseudomallei flagella as a virulent factor. Trans R Soc Trop Med Hyg. 2008;102 Suppl 1:S140–4.
- DeShazer D, Brett PJ, Carlyon R, et al. Mutagenesis of Burkholderia pseudomallei with Tn5-OT182: isolation of motility mutants and molecular characterization of the flagellin structural gene. J Bacteriol. 1997;179(7):2116–2125.
- Wikraiphat C, Charoensap J, Utaisincharoen P, et al. Comparative in vivo and in vitro analyses of putative virulence factors of Burkholderia pseudomallei using lipopolysaccharide, capsule and flagellin mutants. FEMS Immunol Med Microbiol. 2009;56(3):253–259.
- Brett PJ, Mah D, Woods DE. Isolation and characterization of Pseudomonas pseudomallei flagellin proteins. Infect Immun. 1994;62(5):1914–1919.
- Inglis TJJ, Robertson T, Woods DE, et al. Flagellum-mediated adhesion by Burkholderia pseudomallei precedes invasion of Acanthamoeba astronyxis. Infect Immun. 2003;71(4):2280–2282.
- Tuanyok A, Auerbach RK, Brettin TS, et al. A horizontal gene transfer event defines two distinct groups within Burkholderia pseudomallei that have dissimilar geographic distributions. J Bacteriol. 2007;189(24):9044–9049.
- French CT, Toesca IJ, Wu T-H, et al. Dissection of the Burkholderia intracellular life cycle using a photothermal nanoblade. Proc Nat Acad Sci. 2011;108(29):12095–12100.
- Maloy JP. Characterization of an intracellular flagellar system in pathogenic Burkholderia. Species: University of California; 2017.
- Koosakulnirand S, Phokrai P, Jenjaroen K, et al. Immune response to recombinant Burkholderia pseudomallei FliC. PLoS One. 2018;13(6):e0198906.
- Amemiya K, Dankmeyer JL, Bernhards RC, et al. Activation of toll-like receptors by live gram-negative bacterial pathogens reveals mitigation of TLR4 responses and activation of TLR5 by Flagella. Front Cell Infect Microbiol. 2021;11:1089.
- Reckseidler-Zenteno SL, Viteri D-F, Moore R, et al. Characterization of the type III capsular polysaccharide produced by Burkholderia pseudomallei. J Med Microbiol. 2010;59(12):1403–1414.
- Perry MB, MacLean LL, Schollaardt T, et al. Structural characterization of the lipopolysaccharide O antigens of Burkholderia pseudomallei. Infect Immun. 1995;63(9):3348–3352.
- Reckseidler SL, DeShazer D, Sokol PA, et al. Detection of bacterial virulence genes by subtractive hybridization: identification of capsular polysaccharide of Burkholderia pseudomallei as a major virulence determinant. Infect Immun. 2001;69(1):34–44.
- Reckseidler-Zenteno SL, DeVinney R, Woods DE. The capsular polysaccharide of Burkholderia pseudomallei contributes to survival in serum by reducing complement factor C3b deposition. Infect Immun. 2005;73(2):1106–1115.
- Cuccui J, Milne TS, Harmer N, et al. Characterization of the Burkholderia pseudomallei K96243 capsular polysaccharide I coding region. Infect Immun. 2012;80(3):1209–1221.
- DeShazer D, Brett PJ, Woods DE. The type II O‐antigenic polysaccharide moiety of Burkholderia pseudomallei lipopolysaccharide is required for serum resistance and virulence. Mol Microbiol. 1998;30(5):1081–1100.
- Sarkar-Tyson M, Thwaite J, Harding S, et al. Polysaccharides and virulence of Burkholderia pseudomallei. J Med Microbiol. 2007;56(8):1005–1010.
- Knirel YA, Paramonov NA, Shashkov AS, et al. Structure of the polysaccharide chains of Pseudomonas pseudomallei lipopolysaccharides. Carbohydr Res. 1992;233:185–193.
- Nimtz M, Wray V, Domke T, et al. Structure of an acidic exopolysaccharide of Burkholderia pseudomallei. Eur J Biochem. 1997;250(2):608–616.
- Masoud H, Ho M, Schollaardt T, et al. Characterization of the capsular polysaccharide of Burkholderia (Pseudomonas) pseudomallei 304b. J Bacteriol. 1997;179(18):5663–5669.
- Burtnick MN, Heiss C, Roberts RA, et al. Development of capsular polysaccharide-based glycoconjugates for immunization against melioidosis and glanders. Front Cell Infect Microbiol. 2012;2:108.
- Atkins T, Prior R, Mack K, et al. Characterisation of an acapsular mutant of Burkholderia pseudomallei identified by signature tagged mutagenesis. J Med Microbiol. 2002;51(7):539–553.
- Warawa JM, Long D, Rosenke R, et al. Role for the Burkholderia pseudomallei capsular polysaccharide encoded by the wcb operon in acute disseminated melioidosis. Infect Immun. 2009;77(12):5252–5261.
- Sperandeo P, Dehò G, Polissi A. The lipopolysaccharide transport system of gram-negative bacteria. Biochim Biophys Acta (BBA)-Mol Cell Biol Lipids. 2009;1791(7):594–602.
- Anuntagool N, Wuthiekanun V, White NJ, et al. Lipopolysaccharide heterogeneity among Burkholderia pseudomallei from different geographic and clinical origins. Am J Trop Med Hyg. 2006;74(3):348–352.
- Tuanyok A, Stone JK, Mayo M, et al. The genetic and molecular basis of O-antigenic diversity in Burkholderia pseudomallei lipopolysaccharide. PLoS Negl Trop Dis. 2012;6(1):e1453.
- Arjcharoen S, Wikraiphat C, Pudla M, et al. Fate of a Burkholderia pseudomallei lipopolysaccharide mutant in the mouse macrophage cell line RAW 264.7: possible role for the O-antigenic polysaccharide moiety of lipopolysaccharide in internalization and intracellular survival. Infect Immun. 2007;75(9):4298–4304.
- Beutler B, Hoebe K, Du X, et al. How we detect microbes and respond to them: the Toll‐like receptors and their transducers. J Leukoc Biol. 2003;74(4):479–485.
- Wiersinga WJ, Wieland CW, Dessing MC, et al. Toll-like receptor 2 impairs host defense in gram-negative sepsis caused by Burkholderia pseudomallei (Melioidosis). PLoS Med. 2007;4(7):e248.
- Sengyee S, Yoon SH, Paksanont S, et al. Comprehensive analysis of clinical Burkholderia pseudomallei isolates demonstrates conservation of unique lipid a structure and TLR4-dependent innate immune activation. PLoS Negl Trop Dis. 2018;12(2):e0006287.
- Weehuizen TA, Prior JL, van der Vaart TW, et al. Differential Toll-like receptor-signalling of Burkholderia pseudomallei lipopolysaccharide in murine and human models. PLoS One. 2015;10(12):e0145397.
- Bogdan C, Röllinghoff M, Diefenbach A. Reactive oxygen and reactive nitrogen intermediates in innate and specific immunity. Curr Opin Immunol. 2000;12(1):64–76.
- Utaisincharoen P, Anuntagool N, Limposuwan K, et al. Involvement of beta interferon in enhancing inducible nitric oxide synthase production and antimicrobial activity of Burkholderia pseudomallei-infected macrophages. Infect Immun. 2003;71(6):3053–3057.
- MacMicking J, Xie Q-W, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15(1):323–350.
- Utaisincharoen P, Tangthawornchaikul N, Kespichayawattana W, et al. Burkholderia pseudomallei interferes with inducible nitric oxide synthase (iNOS) production: a possible mechanism of evading macrophage killing. Microbiol Immunol. 2001;45(4):307–313.
- Miyagi K, Kawakami K, Saito A. Role of reactive nitrogen and oxygen intermediates in gamma interferon-stimulated murine macrophage bactericidal activity against Burkholderia pseudomallei. Infect Immun. 1997;65(10):4108–4113.
- Subsin B, Thomas MS, Katzenmeier G, et al. Role of the stationary growth phase sigma factor RpoS of Burkholderia pseudomallei in response to physiological stress conditions. J Bacteriol. 2003;185(23):7008–7014.
- Utaisincharoen P, Arjcharoen S, Limposuwan K, et al. Burkholderia pseudomallei RpoS regulates multinucleated giant cell formation and inducible nitric oxide synthase expression in mouse macrophage cell line (RAW 264.7). Microb Pathog. 2006;40(4):184–189.
- Chutoam P, Charoensawan V, Wongtrakoongate P, et al. RpoS and oxidative stress conditions regulate succinyl-CoA: 3-ketoacid-coenzyme a transferase (SCOT) expression in Burkholderia pseudomallei. Microbiol Immunol. 2013;57(9):605–615.
- Korbsrisate S, Vanaporn M, Kerdsuk P, et al. The Burkholderia pseudomallei RpoE (AlgU) operon is involved in environmental stress tolerance and biofilm formation. FEMS Microbiol Lett. 2005;252(2):243–249.
- Thongboonkerd V, Vanaporn M, Songtawee N, et al. Altered proteome in Burkholderia pseudomallei rpoE operon knockout mutant: insights into mechanisms of rpoE operon in stress tolerance, survival, and virulence. J Proteome Res. 2007;6(4):1334–1341.
- Loprasert S, Sallabhan R, Whangsuk W, et al. The Burkholderia pseudomallei oxyR gene: expression analysis and mutant characterization. Gene. 2002;296(1–2):161–169.
- Loprasert S, Whangsuk W, Sallabhan R, et al. Regulation of the katG‐dpsA operon and the importance of KatG in survival of Burkholderia pseudomallei exposed to oxidative stress. FEBS Lett. 2003;542(1–3):17–21.
- Jangiam W, Loprasert S, Smith DR, et al. Burkholderia pseudomallei RpoS regulates OxyR and the katG‐dpsA operon under conditions of oxidative stress. Microbiol Immunol. 2010;54(7):389–397.
- Loprasert S, Sallabhan R, Whangsuk W, et al. Compensatory increase in ahpC gene expression and its role in protecting Burkholderia pseudomallei against reactive nitrogen intermediates. Arch Microbiol. 2003;180(6):498–502.
- Loprasert S, Whangsuk W, Sallabhan R, et al. DpsA protects the human pathogen Burkholderia pseudomallei against organic hydroperoxide. Arch Microbiol. 2004;182(1):96–101.
- Ha HC, Sirisoma NS, Kuppusamy P, et al. The natural polyamine spermine functions directly as a free radical scavenger. Proc Nat Acad Sci. 1998;95(19):11140–11145.
- Fridovich I. Superoxide radical and superoxide dismutases. Annu Rev Biochem. 1995;64(1):97–112.
- Vanaporn M, Wand M, Michell SL, et al. Superoxide dismutase C is required for intracellular survival and virulence of Burkholderia pseudomallei. Microbiology. 2011;157(8):2392–2400.
- Harley V, Dance D, Drasar B, et al. Effects of Burkholderia pseudomallei and other Burkholderia species on eukaryotic cells in tissue culture. Microbios. 1998;96(384):71–93.
- Harley V, Dance D, Tovey G, et al. An ultrastructural study of the phagocytosis of Burkholderia pseudomallei. Microbios. 1998;94(377):35–45.
- Rainbow L, Hart CA, Winstanley C. Distribution of type III secretion gene clusters in Burkholderia pseudomallei, B. thailandensis and B. mallei. J Med Microbiol. 2002;51(5):374–384.
- Winstanley C, Hales B, Hart C. Evidence for the presence in Burkholderia pseudomallei of a type III secretion system-associated gene cluster. J Med Microbiol. 1999;48(7):649–656.
- Attree O, Attree I. A second type III secretion system in Burkholderia pseudomallei: who is the real culprit? Microbiology. 2001;147(12):3197–3199.
- Stevens MP, Wood MW, Taylor LA, et al. An Inv/Mxi-Spa-like type III protein secretion system in Burkholderia pseudomallei modulates intracellular behaviour of the pathogen. Mol Microbiol. 2002;46(3):649–659.
- Burtnick MN, Brett PJ, Nair V, et al. Burkholderia pseudomallei type III secretion system mutants exhibit delayed vacuolar escape phenotypes in RAW 264.7 murine macrophages. Infect Immun. 2008;76(7):2991–3000.
- Muangsombut V, Suparak S, Pumirat P, et al. Inactivation of Burkholderia pseudomallei bsaQ results in decreased invasion efficiency and delayed escape of bacteria from endocytic vesicles. Arch Microbiol. 2008;190(6):623–631.
- Kubori T, Sukhan A, Aizawa S-I, et al. Molecular characterization and assembly of the needle complex of the Salmonella Typhimurium type III protein secretion system. Proc Nat Acad Sci. 2000;97(18):10225–10230.
- Pilatz S, Breitbach K, Hein N, et al. Identification of Burkholderia pseudomallei genes required for the intracellular life cycle and in vivo virulence. Infect Immun. 2006;74(6):3576–3586.
- Stevens MP, Friebel A, Taylor LA, et al. A Burkholderia pseudomallei type III secreted protein, BopE, facilitates bacterial invasion of epithelial cells and exhibits guanine nucleotide exchange factor activity. J Bacteriol. 2003;185(16):4992–4996.
- Srinon V, Muangman S, Imyaem N, et al. Comparative assessment of the intracellular survival of the Burkholderia pseudomallei bopC mutant. J Microbiol. 2013;51(4):522–526.
- Kang W-T, Vellasamy KM, Chua E-G, et al. Functional characterizations of effector protein BipC, a type III secretion system protein, in Burkholderia pseudomallei pathogenesis. J Infect Dis. 2015;211(5):827–834.
- Suparak S, Kespichayawattana W, Haque A, et al. Multinucleated giant cell formation and apoptosis in infected host cells is mediated by Burkholderia pseudomallei type III secretion protein BipB. J Bacteriol. 2005;187(18):6556–6560.
- Muangman S, Korbsrisate S, Muangsombut V, et al. BopC is a type III secreted effector protein of Burkholderia pseudomallei. FEMS Microbiol Lett. 2011;323(1):75–82.
- Cullinane M, Gong L, Li X, et al. Stimulation of autophagy suppresses the intracellular survival of Burkholderia pseudomallei in mammalian cell lines. Autophagy. 2008;4(6):744–753.
- Yao Q, Cui J, Zhu Y, et al. A bacterial type III effector family uses the papain-like hydrolytic activity to arrest the host cell cycle. Proc Nat Acad Sci. 2009;106(10):3716–3721.
- Cui J, Yao Q, Li S, et al. Glutamine deamidation and dysfunction of ubiquitin/NEDD8 induced by a bacterial effector family. Science. 2010;329(5996):1215–1218.
- Yao Q, Cui J, Wang J, et al. Structural mechanism of ubiquitin and NEDD8 deamidation catalyzed by bacterial effectors that induce macrophage-specific apoptosis. Proc Nat Acad Sci. 2012;109(50):20395–20400.
- Gong L, Lai S-C, Treerat P, et al. Burkholderia pseudomallei type III secretion system cluster 3 ATPase BsaS, a chemotherapeutic target for small-molecule ATPase inhibitors. Infect Immun. 2015;83(4):1276–1285.
- Sinha AK, Dutta A, Chandravanshi M, et al. An insight into bacterial phospholipase C classification and their translocation through Tat and Sec pathways: a data mining study. Meta Gene. 2019;20:100547.
- Korbsrisate S, Tomaras AP, Damnin S, et al. Characterization of two distinct phospholipase C enzymes from Burkholderia pseudomallei. Microbiology. 2007;153(6):1907–1915.
- Srinon V, Withatanung P, Chaiwattanarungruengpaisan S, et al. Functional redundancy of Burkholderia pseudomallei phospholipase C enzymes and their role in virulence. Sci Rep. 2020;10(1):1–12.
- Houghton AM, Hartzell WO, Robbins CS, et al. Macrophage elastase kills bacteria within murine macrophages. Nature. 2009;460(7255):637–641.
- Ireland PM, Marshall L, Norville I, et al. The serine protease inhibitor Ecotin is required for full virulence of Burkholderia pseudomallei. Microb Pathog. 2014;67:55–58.
- Nagy ZA, Szakács D, Boros E, et al. Ecotin, a microbial inhibitor of serine proteases, blocks multiple complement dependent and independent microbicidal activities of human serum. PLoS Pathog. 2019;15(12):e1008232.
- Norris MH, Propst KL, Kang Y, et al. The Burkholderia pseudomallei δasd mutant exhibits attenuated intracellular infectivity and imparts protection against acute inhalation melioidosis in mice. Infect Immun. 2011;79(10):4010–4018.
- Atkins T, Prior RG, Mack K, et al. A mutant of Burkholderia pseudomallei, auxotrophic in the branched chain amino acid biosynthetic pathway, is attenuated and protective in a murine model of melioidosis. Infect Immun. 2002;70(9):5290–5294.
- Moule MG, Hemsley CM, Seet Q, et al. Genome-wide saturation mutagenesis of Burkholderia pseudomallei K96243 predicts essential genes and novel targets for antimicrobial development. MBio. 2014;5(1):e00926–13.
- Tuanyok A, Tom M, Dunbar J, et al. Genome-wide expression analysis of Burkholderia pseudomallei infection ina hamster model of acute melioidosis. Infect Immun. 2006;74(10):5465–5476.
- van Schaik EJ, Tom M, Woods DE. Burkholderia pseudomallei isocitrate lyase is a persistence factor in pulmonary melioidosis: implications for the development of isocitrate lyase inhibitors as novel antimicrobials. Infect Immun. 2009;77(10):4275–4283.
- Cruz-Migoni A, Hautbergue GM, Artymiuk PJ, et al. A Burkholderia pseudomallei toxin inhibits helicase activity of translation factor eIF4A. Science. 2011;334(6057):821–824.
- Moule MG, Spink N, Willcocks S, et al. Characterization of new virulence factors involved in the intracellular growth and survival of Burkholderia pseudomallei. Infect Immun. 2015;84(3):701–710.
- Norville IH, Breitbach K, Eske-Pogodda K, et al. A novel FK-506-binding-like protein that lacks peptidyl-prolyl isomerase activity is involved in intracellular infection and in vivo virulence of Burkholderia pseudomallei. Microbiology. 2011;157(9):2629–2638.
- Norville IH, Harmer NJ, Harding SV, et al. A Burkholderia pseudomallei macrophage infectivity potentiator-like protein has rapamycin-inhibitable peptidylprolyl isomerase activity and pleiotropic effects on virulence. Infect Immun. 2011;79(11):4299–4307.
- Fischer G, Aumüller T. Regulation of peptide bond cis/trans isomerization by enzyme catalysis and its implication in physiological processes. In: Reviews of physiology, biochemistry and pharmacology. Springer; 2003. p. 105–150.
- Kespichayawattana W, Rattanachetkul S, Wanun T, et al. Burkholderia pseudomallei induces cell fusion and actin-associated membrane protrusion: a possible mechanism for cell-to-cell spreading. Infect Immun. 2000;68(9):5377–5384.
- Stevens MP, Stevens JM, Jeng RL, et al. Identification of a bacterial factor required for actin‐based motility of Burkholderia pseudomallei. Mol Microbiol. 2005;56(1):40–53.
- Breitbach K, Rottner K, Klocke S, et al. Actin‐based motility of Burkholderia pseudomallei involves the Arp 2/3 complex, but not N‐WASP and Ena/VASP proteins. Cell Microbiol. 2003;5(6):385–393.
- Srinon V, Chaiwattanarungruengpaisan S, Korbsrisate S, et al. Burkholderia pseudomallei BimC is required for actin-based motility, intracellular survival, and virulence. Front Cell Infect Microbiol. 2019;9:63.
- Wong K, Puthucheary S, Vadivelu J. The histopathology of human melioidosis. Histopathology. 1995;26(1):51–55.
- Boddey JA, Day CJ, Flegg CP, et al. The bacterial gene lfpA influences the potent induction of calcitonin receptor and osteoclast‐related genes in Burkholderia pseudomallei‐induced TRAP‐positive multinucleated giant cells. Cell Microbiol. 2007;9(2):514–531.
- Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289(5484):1504–1508.
- Burtnick MN, Brett PJ, Harding SV, et al. The cluster 1 type VI secretion system is a major virulence determinant in Burkholderia pseudomallei. Infect Immun. 2011;79(4):1512–1525.
- Miyata ST, Kitaoka M, Brooks TM, et al. Vibrio cholerae requires the type VI secretion system virulence factor VasX to kill Dictyostelium discoideum. Infect Immun. 2011;79(7):2941–2949.
- Rigard M, Bröms JE, Mosnier A, et al. Francisella tularensis IglG belongs to a novel family of PAAR-like T6SS proteins and harbors a unique N-terminal extension required for virulence. PLoS Pathog. 2016;12(9):e1005821.
- Sana TG, Lugo KA, Monack DM. T6SS: the bacterial“fight club” in the host gut. PLoS Pathog. 2017;13(6):e1006325.
- Toesca IJ, French CT, Miller JF. The Type VI secretion system spike protein VgrG5 mediates membrane fusion during intercellular spread by pseudomallei group Burkholderia species. Infect Immun. 2014;82(4):1436–1444.
- Schwarz S, Singh P, Robertson JD, et al. VgrG-5 is a Burkholderia type VI secretion system-exported protein required for multinucleated giant cell formation and virulence. Infect Immun. 2014;82(4):1445–1452.
- Wong J, Chen Y, Gan Y-H. Host cytosolic glutathione sensing by a membrane histidine kinase activates the type VI secretion system in an intracellular bacterium. Cell Host Microbe. 2015;18(1):38–48.
- Schwarz S, West TE, Boyer F, et al. Burkholderia type VI secretion systems have distinct roles in eukaryotic and bacterial cell interactions. PLoS Pathog. 2010;6(8):e1001068.
- Losada L, Shea AA, DeShazer D. A MarR family transcriptional regulator and subinhibitory antibiotics regulate type VI secretion gene clusters in Burkholderia pseudomallei. Microbiology. 2018;164(9):1196–1211.
- Si M, Zhao C, Burkinshaw B, et al. Manganese scavenging and oxidative stress response mediated by type VI secretion system in Burkholderia thailandensis. Proc Nat Acad Sci. 2017;114(11):E2233–42.
- Si M, Wang Y, Zhang B, et al. The type VI secretion system engages a redox-regulated dual-functional heme transporter for zinc acquisition. Cell Rep. 2017;20(4):949–959.
- Schell MA, Ulrich RL, Ribot WJ, et al. Type VI secretion is a major virulence determinant in Burkholderia mallei. Mol Microbiol. 2007;64(6):1466–1485.
- Shalom G, Shaw JG, Thomas MS. In vivo expression technology identifies a type VI secretion system locus in Burkholderia pseudomallei that is induced upon invasion of macrophages. Microbiology. 2007;153(Pt 8):2689–2699.
- Silverman JM, Agnello DM, Zheng H, et al. Haemolysin coregulated protein is an exported receptor and chaperone of type VI secretion substrates. Mol Cell. 2013;51(5):584–593.
- Lim YT, Jobichen C, Wong J, et al. Extended loop region of Hcp1 is critical for the assembly and function of type VI secretion system in Burkholderia pseudomallei. Sci Rep. 2015;5(1):1–10.
- Lennings J, West TE, Schwarz S. The Burkholderia type VI secretion system 5: composition, regulation and role in virulence. Front Microbiol. 2019;9:3339.
- Chen Y, Wong J, Sun GW, et al. Regulation of type VI secretion system during Burkholderia pseudomallei infection. Infect Immun. 2011;79(8):3064–3073.
- DeShazer D. A novel contact-independent T6SS that maintains redox homeostasis via Zn2+ and Mn2+ acquisition is conserved in the Burkholderia pseudomallei complex. Microbiol Res. 2019;226:48–54.
- Bzdyl NM, Scott NE, Norville IH, et al. Peptidyl-Prolyl Isomerase ppiB is essential for Proteome Homeostasis and virulence in Burkholderia pseudomallei. Infect Immun. 2019;87(10):e00528–19.