
ABSTRACT
Pathogenic E. coli strains can be classified into two major groups, based on the presence of specific virulence factors: extraintestinal pathogenic E. coli (ExPEC) and diarrheagenic E. coli (DEC). Several case reports describe that DEC can cause bloodstream infections in some rare cases. This mainly concerns a few specific sequence types that express virulence factors from both ExPEC and DEC. In this study, we retrospectively analysed 234 E. coli blood isolates with whole genome sequencing (WGS). WGS was performed on an Illumina NovaSeq6000. Genotyping was performed using BioNumerics software. The presence of genes was determined with a minimum percentage sequence identity (ID) threshold of 95% and a minimum length for sequence coverage of 95%. Three of the 234 (1.28%) isolates were defined as DEC, 182 (77.78%) as ExPEC, and 49 (20.94%) did not carry pathotype-associated virulence genes. We identified 112 different virulence genes, 48 O-antigens, and 28 H-antigens 82 STs, among the 234 analyzed isolates. ST131 and ST88 were related to healthcare-associated infections. This study provides insight into the prevalence of virulence factors in a large set of E. coli blood isolates from the UZ Brussel. It illustrates high diversity in virulence profiles and highlights the potential of DEC to carry virulence factors associated with extraintestinal infections, making it possible for unusual pathotypes to invade and survive in the bloodstream causing bacteraemia. Diarrheagenic strains causing bacteremia are rare and presently underreported, but modern sequencing techniques will better underscore their importance.
Introduction
Escherichia coli (E. coli) is part of the commensal flora of the human intestine and rarely causes disease in healthy individuals. Nonetheless, there is a wide variety of pathogenic E. coli strains able to cause diarrhoea or extraintestinal infections. Based on the presence of specific virulence factors, we can classify pathogenic E. coli into two major groups: extraintestinal pathogenic E. coli (ExPEC) and diarrheagenic E. coli (DEC) [Citation1].
ExPEC strains can invade, survive and cause infection in tissues and fluids outside of the intestinal tract, typically the bloodstream and urinary tract. Two major pathotypes within this group are uropathogenic E. coli (UPEC) and meningitis- and sepsis-associated E. coli (MNEC). UPEC is the most common cause of both complicated and uncomplicated urinary tract infections through a set of specific virulence factors that aid in migration, adhesion, and persistence within the urinary tract. Type 1 pili, like the fimH mannose-binding pili, play a central role in the initial attachment to the epithelial cells of the urethra [Citation2]. Sequence type (ST) 131 is of particular interest, which is an emerging and virulent multidrug-resistant strain predominantly isolated from the urinary tract and bloodstream infections [Citation3]. MNEC is the leading cause of early-onset meningitis, accounting for more than 30% of neonatal infections. The K1 antigen is a shared characteristic among most MNEC serotypes, which promotes serum resistance, intracellular survival, and traversal of the blood-brain barrier [Citation2].
DEC strains can lead to a plethora of diarrhoeal syndromes and are usually not identified outside the intestine. DEC expresses adhesins, invasins, and cytotoxins that target the gastro-intestinal epithelium causing osmotic dysregulation or cell damage. There are five major types of DEC: Shiga toxin-producing E. coli (STEC), enterotoxigenic E. coli (ETEC), enteropathogenic E. coli (EPEC), enteroinvasive E. coli (EIEC), and enteroaggregative E. coli (EAEC). All STEC isolates encode stx1 and/or stx2, which are potent toxins that target endothelial cells in the gastrointestinal tract and renal glomeruli. ETEC strains are characterized by the production of heat-labile and/or heat-stable enterotoxins, which can dysregulate multiple ion channels in intestinal epithelial cells leading to fluid loss. The LEE-encoded virulence factors eae and tir are two virulence factors that can induce lesions within intestinal epithelial cells, characteristic for EPEC and part of STEC, qualified as “typical.” EIEC has a close relationship with Shigella, both carrying the pINV plasmid harbouring multiple genes like set2, sigA, and ipaH responsible for cellular fluid loss leading to watery diarrhea. Finally, EAEC is a heterogeneous group characterized by a “stacked brick” architecture and the presence of typical virulence factors like aggR and aaiC [Citation4].
Virulence genes are often transmissible between E. coli strains and are localized on chromosomes, plasmids, or phages. Horizontal gene transfer is probably the main driver for virulence and genetic diversity. It enables the transformation of non-pathogenic E. coli to specific pathotypes [Citation5]. Pathotypes are defined by the host’s clinical symptoms and a small set of pathotype-associated virulence genes [Citation6].
However, it should be mentioned that the plasticity of the E. coli genome has led to more virulent hybrid pathogenic strains carrying virulence factors of different pathotypes, making it sometimes possible for DEC strains to cause extraintestinal infections, mainly urinary tract infections [Citation7]. Reports of DEC strains causing bacteraemia are very rare. Because routine processing of blood isolates does not address genotyping, these hybrid strains inducing bacteremia are probably strongly underreported. Modern techniques such as whole genome sequencing (WGS) could help us to better identify these particular cases and provides information on the spread of specific bacterial clones responsible for bloodstream infections.
In the current research paper, we aimed to gain an insight into the prevalence of virulence factors present in E. coli causing bacteraemia in a relatively large set of samples collected between 1985 and 2018 in the University Hospital of Brussels. Based on these virulence factors, we aimed to look if bloodstream infections with hybrid or atypical pathotypes were occurring in our hospital. As relatively little is known about the clonal structure of E. coli causing bacteremia in Belgium, we also described sequence type (ST) using the Achtman typing scheme [Citation8] and compared core genome multilocus sequence type (cgMLST) data. Special attention was paid to a subgroup of 130 isolates for which a medical record was available, where an attempt was made to link ST to the source of infection (healthcare-acquired or community-acquired). Unusual or interesting cases are discussed in more detail with relevant additional data.
Material and methods
The UZ Brussel is a tertiary care centre with over 700 beds. Two hundred and thirty-four (234) non-duplicate clinical isolates were investigated. These E. coli strains were isolated from clinical blood samples randomly selected between 1985 and 2018. All isolates were stored at −80°C. The included isolates are distributed over the years as follows: 1985 (n = 10), 1990 (n = 11), 1995 (n = 12), 2000 (n = 34), 2005 (n = 33), 2010 (n = 33), 2015 (n = 51), and 2018 (n = 50).
The genomic DNA for whole genome sequencing was extracted using the Dneasy blood & tissue kit (Qiagen, Hilden, Germany) for 30 samples and DNA libraries were prepared via the KAPA Hyper Plus kit (Kapa Biosystems, Wilmington, MA, USA). All libraries were sequenced on a MiSeq instrument (Illumina, San Diego, CA, USA) using the v2 (2 × 250 bp) and v3 (2 × 300 bp) reagent kits. Of the other 203 samples, genomic DNA was extracted using the Maxwell RSC Cell DNA purification kit (Promega Corporation, Madison, USA). Fragmentation of 500 ng of genomic DNA was carried out using the NEBNext® Ultra™ II FS module. Sequencing libraries, with an insert size of on average 550 bp, were prepared using the KAPA Hyper Plus kit (Kapa Biosystems, Wilmington, USA) and a Pippin Prep (Sage Science, Beverly, MA, USA) size with the CDF1510 1.5% agarose dye-free cassette selection. To avoid PCR bias, the PCR amplifications step was omitted and a 500 ng input of genomic DNA was used. After equimolar pooling, libraries were sequenced on a Novaseq 6000 instrument (Illumina, San Diego, CA, USA) using the NovaSeq 6000 SP Reagent Kit (500 cycles) generating 2 × 250 bp reads. For this, the library was denatured and diluted according to the manufacturer’s instructions. A 1% PhiX control library was included in each sequencing run.
De novo assembly was performed using SPAdes genome assembler in BioNumerics v.8.1 (Applied Maths, BioMérieux, Sint-Martens-Latem, Belgium).
The E. coli functional genotyping plugin version 2.1 incorporated in BioNumerics v.8.1 was used to predict E. coli pathotypes, serotypes and virulence gene profiles (194 putative virulence genes; Blast minimum sequence identity 95%, Blast minimum length for coverage 95%).
Assembled genomes were analysed using both the MLST Achtman scheme (7 MLST loci) and the core EnteroBase scheme (2.506 core loci) for E. coli/Shigella available in BioNumerics v.8.1. The assembly-based approach was used for allele calling. Minimal spanning trees (MSTs) were generated using the MLST Achtman allelic profiles and the core Enterobase allelic profiles as input data in BioNumerics. Branch lengths reflect the number of allele differences (AD) between the isolates in the connected nodes. For cgMLST clustering, the AD threshold was set at <5.
The quality of the sequence read sets, the de novo assemblies, and the assembly-based allele calls was verified using the quality statistics window in BioNumerics v.8.1.
Of 130 of the 234 patients, a medical record was available. These medical records were used to see if the sample collection was taken before (community-acquired infection) or after at least 48 hours of hospitalization (healthcare-acquired infection). A Fisher exact test with an alpha value of 0.05 was used to compare data.
Results
Pathotypes
Based on a pathogen-defining algorithm (Supplementary data 1), different pathotypes were found. One hundred and seventy-one of the 234 (73.01%) isolates carried one or more ExPEC-associated loci. Of these 171 isolates, 138 (80.07%) carried UPEC-specific loci. Thirteen of the 234 (5.56%) isolates carried UPEC-specific loci without the presence of ExPEC-specific loci. Forty-nine of the 234 (20.94%) isolates did not carry pathotype-associated virulence genes. Three of the 234 (1.28%) isolates carried virulence genes associated with DEC, which is highly unusual for bloodstream infections. These three unusual pathotypes belonged to ST38 (hybrid EAEC/ExPEC/UPEC), ST10 (hybrid EAEC/ExPEC), and ST13128 (EPEC).
Clinical and molecular characteristics of the atypical pathotypes
The first hybrid isolate (EAEC/ExPEC/UPEC) was isolated from a 76-year-old man in 2015 and belonged to ST38. The man presented to the emergency department of our hospital in poor condition with melaena, fever, tremor, loss of strength, general malaise, and pain all over the body. Both blood and urine cultures revealed an extended-spectrum beta-lactamase (ESBL) producing E. coli. The patient was successfully treated with meropenem. Virulence factors characteristic for UPEC (uropathogenic Escherichia coli), ExPEC (extraintestinal pathogenic Escherichia coli), and EAEC were identified. The EAEC pathotype-associated locus is aggR. The ExPEC pathotype-associated loci are papC, iutA, papA_F7-2 and kpsMII. The UPEC pathotype-associated loci are fyuA and chuA. Several acquired resistance genes were detected: mdf(a), dfrA1, aac(6’)-Ib-cr, blaOXA-1, sul2, blaCTX-M-15 and tet(A) resulting in a resistant phenotype for almost all clinically relevant antibiotics. Mutations were found in gyrA, parC, and parE leading to resistance to nalidixic acid and ciprofloxacin. The serotype was determined as O153/O178:H30.
The second hybrid isolate (EAEC/ExPEC) isolate was isolated from a one-year-old boy in 1990 and belonged to ST10. No further clinical information was available. Virulence factors characteristic of both EAEC and ExPEC were identified. The EAEC pathotype-associated locus is aggR. The ExPEC-associated loci are papC, iutA, papsA_fsiA_F16, and kpsMII. The acquired resistance gene blaTEM-1B was detected. No mutational resistance was found. The serotype was determined as O21:H2.
The last unusual pathotype (atypical EPEC) was isolated from a 65-year-old man in 2018 and belonged to the new ST13128. The nearest STs are ST10972, ST7912, ST20, ST3674 and ST561. The man presented to the emergency department of our hospital with abdominal pain. He was known for rectum carcinoma. Due to positive blood cultures, which revealed an E. coli, eight days of hospitalization were required. The bacteraemia originated probably from a polyp visualized on a colonoscopy. The patient was successfully treated with amoxicillin/clavulanic acid. Virulence factors characteristic of an atypical EPEC were identified. The atypical EPEC pathotype-associated locus is eae. The acquired resistance gene mdf(A) conferring resistance to macrolides was detected. No mutational resistance was found. The serotype was determined as O128:H2. All the above-mentioned clinical and functional genotypic characteristics are summarized in Supplementary data 2.
Virulence factors
We identified 112 different virulence genes among the 234 analysed isolates. These genes were classified based on their biochemical properties: adherence, protease, toxin, complement protease, regulation, type 3 translocated protein, survival, type 6 translocated protein, stress protein, secretion system, invasion, antiphagocytosis, and iron uptake in . Of these 112 virulence factors, 13 (terC, ompT, iss, sitA, irp2, fyuA, traT, kpsE, chuA, lutA, lucC, papC and yfcV) were detected in 50% or more of the 234 isolates. In contrast, 31 virulence factors were found in 1% or less of these isolates, highlighting high variability. The regulation virulence factor terC, responsible for tellurite resistance, was the only virulence factor isolated in all 234 isolates. Adherence-associated virulence factors were the most common: to this group belonged 47 different genes, of which 218 (93.16%) of the 234 isolates had at least one. A detailed list of genes can be found in Supplementary data 3.
Table 1. Virulence factors detected in the 234 E. coli blood isolates.
Multi-locus sequence typing
A total of 82 STs were identified among the 234 isolates when using the Achtman seven gene-MLST scheme. Among these STs, one was not described before (ST13128). Five dominant STs were observed: 26 (11.11%) ST73, 25 (10.68%) ST131, 21 (8.97%) ST69, 18 (7.69%) ST95, and 15 (6.41%) ST88. Eighty-two different O- and 28 different H-serotypes were identified and showed a correlation with ST. For example, 24/25 (96.00%) of the ST131 isolates had serotype O25:H4. Looking at the occurrence of these STs as a function of time, we see that ST131 is significantly (P = 0.0048) more prevalent in the period 2010–2018 (21/100 isolates) than in the period 1985–2005 (4/134 isolates) (Supplementary Excel file).
In some cases, the dominant STs and serotypes tend to show a correlation with virulence factor profiles, as they typically harboured very similar sets of virulence genes. Especially isolates within ST393, ST95 and ST62 seem to be very similar based on virulence genes since the nodes are close to each other on the minimum spanning tree (MST) (). However, this is not the case for all STs. In isolates within ST12 and ST88, there appears to be little similarity between virulence genes since the nodes are far apart on the MST ().
Figure 1. MST based on virulence allelic profiles of the 234 E. coli blood isolates. The analysis was carried out in Bionumerics. Nodes are colour-coded per ST. In brackets next to the ST is the number of strains with that ST. Branch length is directly proportional to the allelic difference.
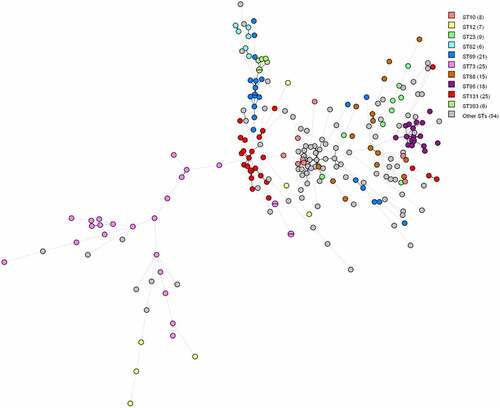
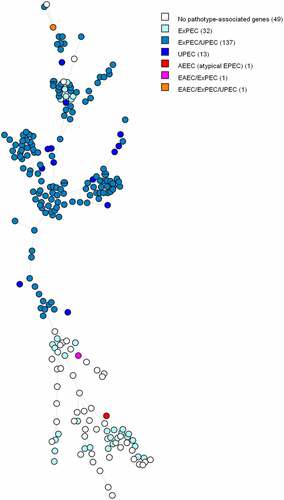
Based on the MST tree in , we suggest that the pathotype is correlated with the core genome since nodes harbouring identical pathotypes are often positioned close to each other. Except for a few outliers, ExPEC/UPEC and UPEC isolates tend to cluster together. We observe the same trend between ExPECs and isolates without pathotype-associated genes. The three DECs are far apart from each other on the figure, and therefore do not seem to be genetically associated.
Figure 2. MST based on cgMLST analysis of 234 E. coli blood isolates. The analysis was carried out in Bionumerics. Nodes are coloured per pathotype. In brackets next to the pathotype is the number of strains belonging to that pathotype. Branch length is directly proportional to the allelic difference.
Healthcare-acquired infections
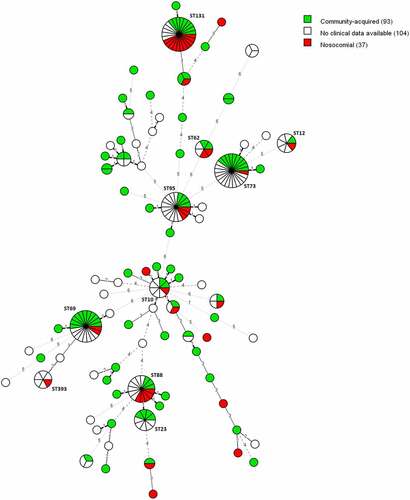
In the subset of 130 isolates for which a medical record was available, 37 (28.46%) of the positive blood samples were collected for the first time at least 48 hours after hospitalization. These infections are defined as healthcare-acquired. Eleven (29.73%) of these 37 healthcare-acquired infections were caused by ST131 and five (13.51%) by ST88, which caused both significantly (P = 0.0069 and P = 0.0416) more healthcare-acquired infections than community-acquired infections ().
Figure 3. MST of STs using the Achtman seven-gene MLST scheme for 234 E. coli blood isolates. The analysis was carried out in Bionumerics. The tree is coloured based on the infection type of the isolates (green: community-acquired, red: healthcare-acquired). Thirty-five nodes are colored red and 93 nodes are colored green. Branch length and width (dotted versus non-dotted) are directly proportional to the allelic difference (ranging from one for the shortest branches to seven for the longest branches).
Clustering
Four clusters, consisting of two isolates per cluster were identified based on the cgMLST analysis of the 234 E. coli blood isolates. ().
Figure 4. MST based on cgMLST analysis of 234 E. coli blood isolates. The analysis was carried out in Bionumerics. A fixed threshold of < 5 allelic differences was used for clustering isolates, highlighting four different clusters. Branch length is directly proportional to the allelic difference.
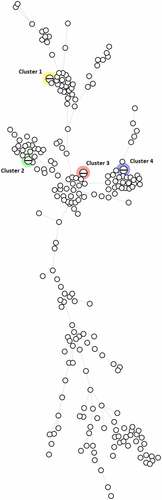
The two isolates from the first cluster belonged to ST73. Both isolates were isolated from two different patients in September 2005. No further medical data was available.
The two isolates from the second cluster belonged to ST69. The first sample was collected on 8 March 2018 and the second sample on 9 March 2018. However, both samples were taken 48 hours before admission to the hospital, suggesting a community-acquired infection. The clinical focus of the first patient’s bacteraemia was probably the urinary tract, and that of the second patient was probably cholangitis.
The third cluster consisted of two ST95 isolates. The samples were collected on 4 May 2018 and 14 June 2018. In both cases, the urinary tract was the clinical focus of the bacteraemia and the sample was collected within 48 hours of hospitalization. These two patients lived within walking distance of each other. However, it is not clear whether they had direct contact with each other.
The last cluster comprised two ST131 isolates. Both samples were collected on the same date in 2018 within 48 hours of hospitalization, pointing to a healthcare-acquired infection. Both patients were also hospitalized in the same unit, suggesting transmission between the environment and patients or healthcare workers and patients.
Discussion
The conventional understanding that E. coli strains causing diarrhoea are distinct from those that cause extraintestinal disease according to their repertoire of pathotype-associated genes conflicts with our findings. We report a hybrid EAEC/ExPEC ST10, a hybrid EAEC/ExPEC/UPEC ST38, and an atypical EPEC ST13128 causing bacteraemia.
Previous studies already pointed out the potential of both EAEC ST10 and EAEC ST38 to cause extraintestinal infections, mainly urinary tract infections [Citation9,Citation10]. In 2014, Chattaway et al. identified eight multidrug-resistant EAEC isolates from urine specimens and one from a blood culture. EAEC ST38 was the most common ST. They described ST38 as an emerging UPEC/EAEC hybrid strain that probably originated from the gut and independently acquired both phenotypes [Citation9]. Alghoribi et al. also described ST38 as an evolving multidrug-resistant strain with both EAEC and UPEC virulence factors. They highlight that plasmid-mediated carriage of the EAEC transport regulator gene aggR is a key factor of emerging ST38. Accordingly, they screened 15 ST38 isolates for aggR and found that four were positive. They noticed no clear differentiation between the amounts of virulence factors present in aggR-positive or aggR-negative strains [Citation10]. ST10 is one of the most common STs in the E. coli MLST database [Citation11]. Olesen et al. reported in 2012 an outbreak of a single clonal EAEC ST10 causing 19 urinary tract infections in Copenhagen. They highlighted the potential of ST10 to exhibit both EAEC and ExPEC characteristics. All the ST10 isolates had highly homogeneous virulence profiles. Notably, all outbreak isolates carried one or more of the EAEC pathotype-associated genes: aggR, aatA and aaiC. In contrast, the only detected ExPEC-defining gene was iutA. However, the outbreak isolates all contained fyuA, traT, sat, and pic, which are all associated with ExPEC [Citation12]. In our EAEC ST10 case, the strain did not carry virulence genes coding for toxins, yet its enterotoxigenic nature could be related to the acquisition of plasmid-borne toxin-coding virulence factors. Error-prone repair genes on aggA-bearing aggregative plasmids, which are harboured by many EAEC ST10 isolates, could play an important role [Citation11].
The MLST of our atypical bacteraemia-related EPEC strain has not been previously described in Enterobase (https://enterobase.warwick.ac.uk/), therefore it was determined as ST13128. The serotype was identified as O128ab:H2, a serotype often related to the EPEC pathotype-associated locus eae. In 2013, Buvens et al. described a case of a DEC with serotype O128ab:H2 causing bacteremia leading to sepsis. The strain acquisitioned both stx1 and st2 genes defining this strain as a STEC. In this case, the infection led to haemolytic-uraemic syndrome (HUS), highlighting the potential of STEC to also cause severe extraintestinal infections [Citation13].
This research showed a high variation in genomic characteristics of E. coli strains causing bloodstream infections. The most prevalent genes are terC, ompT, iss, sitA, irp2 and fyuA. The virulence factor terC, responsible for tellurite resistance, frequent in pathogenic bacteria, was the only virulence factor isolated in all 234 isolates. The iss gene, responsible for increased serum survival, was found in 191 (81.62%) of the 234 isolates. This gene is required for the synthesis of a capsule, necessary to protect the bacterium from complement proteins. The presence of capsule-expressing genes, like K-antigens, also seems to play an important role in extra-intestinal infections as 160 (68.38%) of all the 234 isolates carried one or more genes coding for a capsule. Interestingly, the gene coding for the K1-antigen (kpsMII_K1), a virulence factor that is strongly associated with neonatal meningitis, was present in 41 (17.52%) of the 234 isolates and only occurred in UPEC and ExPEC/UPEC isolates. ST95 and ST62 were found to be statistically associated (P < 0.001) with the occurrence of this gene. Both ST95 and ST62 are frequently isolated from cases of neonatal meningitis [Citation14–16]. The genes irp2 and fyuA are both responsible for iron uptake, an important virulence characteristic that 89.32% of the isolates harboured. Adherence-associated virulence factors were the most common: to this group belonged 47 different genes, of which 218 (93.16%) of the 234 isolates had at least one gene and 186 (79.49%) carried irp2. Except for the pathotype-associated genes, we found genes that were specifically present in the ExPEC and DEC groups. This is, for example, the case for virulence genes coding for toxins, which were found in 80.87% of ExPEC and UPEC isolates, but was not found in any of the three DECs.
In line with Paramita et al., we observed a correlation between ST and virulence genes, as strains within the same ST and serotype often harboured the same set of virulence genes [Citation17]. We noticed that some genomic characteristics, like ST, are related to healthcare-acquired infections or their occurrence over time. In particular, ST131 occurred significantly (P = 0.0048) more often in the period after 2010 than before 2010. This finding is consistent with the literature, where ST131 was described for the first time in 2008 and has spread globally since then, becoming one of the most common multidrug-resistant E. coli lineages [Citation3].
Different E. coli strains can spread both inside and outside a healthcare setting. In our study we describe four clusters, each consisting of two isolates, with identical (allelic difference of <5) strains based on cgMLST. For three of these cases, additional information was available (date of collection, admission department, place of residence, …) which weakly suggested that isolates within the same cluster could be associated with each other. In one of these cgMLST clusters (), we found two ST131 strains, both isolated on the same day in two patients hospitalized for more than 48 hours in the same unit, suggesting healthcare-acquired transmission between the environment and patients or healthcare workers and patients. Nevertheless, cgMLST typing showed that most isolates are unrelated to each other, confirming the sporadic character of E. coli bacteraemia.
Conclusion
This study provides insight into the prevalence of virulence factors in a large set of E. coli blood isolates from the UZ Brussel. It illustrates high diversity in virulence profiles and highlights the potential of DEC to carry virulence factors associated with extraintestinal infections, making it possible for unusual pathotypes to invade and survive in the bloodstream causing bacteraemia. Although these E. coli pathotypes are considered non-invasive, some of them cause bacteremia without being identified. We believe that the prevalence of diarrheagenic strains causing bacteremia is underreported, and that the increased access to WGS analysis will lead to a better understanding of the role of virulence factors in bacteraemic E. coli clinical isolates, in particular hybrid pathotypes. We also highlight the potential of WGS-based surveillance as an early warning system for emerging lineages, like E. coli ST131.
Author contributions
RV, TD, DP and FC designed the study and wrote the study protocol. RV and FC performed the DNA-extraction and purification. TJ and BC performed the WGS. RV, IW, AMC and DD performed the data analysis. RV drafted the first version of the manuscript. All authors had full access to all data, aided in finalizing the text and had final responsibility for the submission for publication.
Data availability
The datasets generated and/or analysed during the current study are available in the National Library of Medicine repository. BioProject: PRJNA854358
Ethics approval
The Medical Ethics Committee of UZ Brussel/VUB reviewed and approved the documents concerning the project from the ethical, legal and medical science points of view (reference: 1432021000569)
Supplemental Material
Download Zip (163.9 KB)Acknowledgements
We would like to thank the lab technicians and bioinformaticians of the Brussels Interuniversity Genomics High Throughput core and clinical microbiology laboratory.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/21505594.2022.2147735.
Additional information
Funding
References
- Kaper J, Nataro J, Mobley H. Pathogenic Escherichia coli. Nat Rev Microbiol. 2004;2:123–140.
- Sarowska J, Futoma-Koloch B, Jama-Kmiecik A, et al. Virulence factors, prevalence and potential transmission of extraintestinal pathogenic Escherichia coli isolated from different sources: recent reports. Gut Pathog. 2019 Feb;21(11:10): PMID: 30828388; PMCID: PMC6383261. DOI:10.1186/s13099-019-0290-0
- Nicolas-Chanoine MH, Bertrand X, Madec JY. Escherichia coli ST131, an intriguing clonal group. Clin Microbiol Rev. 2014 Jul;27(3):543–574. DOI:10.1128/CMR.00125-13. PMID: 24982321; PMCID: PMC4135899.
- Gomes TA, Elias WP, Scaletsky IC, et al. Diarrheagenic Escherichia coli. Braz J Microbiol. 2016 Dec;47(Suppl 1):3–30. DOI:10.1016/j.bjm.2016.10.015. Epub 2016 Nov 5. PMID: 27866935; PMCID: PMC5156508.
- Kado CI J. Horizontal gene transfer: sustaining pathogenicity and optimizing host-pathogen interactions. Mol Plant Pathol. 2009 Jan;10(1):143–150. DOI:10.1111/j.1364-3703.2008.00518.x. PMID: 19161360; PMCID: PMC6640513.
- Rasko DA, Rosovitz MJ, Myers GS, et al. The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J Bacteriol. 2008 Oct;190(20):6881–6893. DOI:10.1128/JB.00619-08. Epub 2008 Aug 1. PMID: 18676672; PMCID: PMC2566221.
- Croxen MA, Law RJ, Scholz R, et al. Recent advances in understanding enteric pathogenic Escherichia coli. Clin Microbiol Rev. 2013 Oct;26(4):822–880. DOI:10.1128/CMR.00022-13. PMID: 24092857; PMCID: PMC3811233.
- Wirth T, Falush D, Lan R, et al. Sex and virulence in Escherichia coli: an evolutionary perspective. Mol Microbiol. 2006 Jun;60(5):1136–1151. DOI:10.1111/j.1365-2958.2006.05172.x. PMID: 16689791; PMCID: PMC1557465.
- MA C, Jenkins C, Ciesielczuk H, et al. Evidence of evolving extraintestinal enteroaggregative Escherichia coli ST38 clone. Emerg Infect Dis. 2014 Nov;20(11):1935–1937. DOI:10.3201/eid2011.131845. PMID: 25340736; PMCID: PMC4214294.
- Majed F, Alghoribi TM, Gibreel GF, et al. Mathew Upton Antibiotic-resistant ST38, ST131 and ST405 strains are the leading uropathogenic Escherichia coli clones in Riyadh, Saudi Arabia. J Antimicrob Chemother. 2015 Oct;70(10):2757–62. DOI:10.1093/jac/dkv188. Epub 2015 Jul 16. PMID: 26183183.
- Okeke IN, Wallace-Gadsden F, Simons HR, et al. Multi-locus sequence typing of enteroaggregative Escherichia coli Isolates from Nigerian children uncovers multiple lineages. PLoS One. 2010;5:e14093. DOI:10.1371/journal.pone.0014093
- Olesen B, Scheutz F, Andersen RL, et al. Enteroaggregative Escherichia coli O78:h10, the cause of an outbreak of urinary tract infection. J Clin Microbiol. 2012 Nov;50(11):3703–3711. DOI:10.1128/JCM.01909-12. Epub 2012 Sep 12. PMID: 22972830; PMCID: PMC3486220.
- Buvens G, De Rauw K, Roisin S, et al. Verocytotoxin-producing Escherichia coli O128ab:h2 bacteremia in a 27-year-old male with hemolytic-uremic syndrome. J Clin Microbiol. 2013 May;51(5):1633–1635. DOI:10.1128/JCM.03025-12. Epub 2013 Mar 6. PMID: 23467596; PMCID: PMC3647934.
- Berman H, Barberino MG, Moreira ED Jr, et al. Distribution of strain type and antimicrobial susceptibility of Escherichia coli isolates causing meningitis in a large urban setting in Brazil. J Clin Microbiol. 2014 May;52(5):1418–1422. DOI:10.1128/JCM.03104-13. Epub 2014 Feb 12. PMID: 24523478;. PMCID: PMC3993653.s.n.
- Mora A, López C, Dabhi G, et al. Extraintestinal pathogenic Escherichia coliO1:k1:h7/NM from human and avian origin: detection of clonal groups B2 ST95 and D ST59 with different host distribution. BMC Microbiol. 2009;9(1):132. DOI:10.1186/1471-2180-9-132
- Cummins ML, Reid CJ, Djordjevic SF. Plasmid lineages in Escherichia coli ST95: implications for host range, antibiotic resistance, and zoonoses. mSystems. 2022 Feb 22;7(1):e0121221. DOI:10.1128/msystems.01212-21. Epub 2022 Jan 25. Erratum in: mSystems. 2022 Apr 26;7(2):e0021022. PMID: 35076267; PMCID: PMC8788324. : s.n.
- Paramita RI, Nelwan EJ, Fadilah F, et al. Genome-based characterization of Escherichia coli causing bloodstream infection through next-generation sequencing. PLoS One. 2020;15:e0244358.