
ABSTRACT
African trypanosomes are vector-borne protozoa, which cause significant human and animal disease across sub-Saharan Africa, and animal disease across Asia and South America. In humans, infection is caused by variants of Trypanosoma brucei, and is characterized by varying rate of progression to neurological disease, caused by parasites exiting the vasculature and entering the brain. Animal disease is caused by multiple species of trypanosome, primarily T. congolense, T. vivax, and T. brucei. These trypanosomes also infect multiple species of mammalian host, and this complexity of trypanosome and host diversity is reflected in the spectrum of severity of disease in animal trypanosomiasis, ranging from hyperacute infections associated with mortality to long-term chronic infections, and is also a main reason why designing interventions for animal trypanosomiasis is so challenging. In this review, we will provide an overview of the current understanding of trypanosome determinants of infection progression and severity, covering laboratory models of disease, as well as human and livestock disease. We will also highlight gaps in knowledge and capabilities, which represent opportunities to both further our fundamental understanding of how trypanosomes cause disease, as well as facilitating the development of the novel interventions that are so badly needed to reduce the burden of disease caused by these important pathogens.
Introduction
African trypanosomes are protozoan parasites, transmitted either cyclically by tsetse flies or mechanically by other biting flies. Several species infect a range of mammals and cause disease, impacting upon both animal and human health. Animal disease is caused by multiple species, with T. congolense, T. vivax and T. brucei the main pathogens of cattle, sheep, goats, equids, and wild animals in sub-Saharan Africa, T. simiae and T. suis infecting pigs in the same region, and T. brucei evansi and T. vivax infecting cattle, equids, camels, and Asian buffalo across North Africa, Asia (T. b. evansi) and South America (T. b. evansi and T. vivax). T. brucei equiperdum causes a venereally transmitted form of trypanosomiasis in horses and donkeys, mostly in sub-Saharan Africa. Variants of T. brucei, T. b. gambiense and T. b. rhodesiense, also cause human infections and disease in sub-Saharan Africa. The economic and health impact of these pathogens is collectively enormous, with Animal Trypanosomiasis (AT) remaining widespread and causing millions of infections and deaths per year [Citation1–3]. There has been substantial progress in combating human African trypanosomiasis (HAT) in recent decades, in particular for T. b. gambiense, with an elimination program in place that aims to remove T. b. gambiense HAT as a disease of public health importance by 2030, an objective that seems achievable from recent progress [Citation4]. However, the methods used to control T. b. gambiense HAT (active case detection) will not eliminate T. b. rhodesiense HAT, due to the truly zoonotic nature and large animal reservoir of the latter pathogen [Citation5]. This outline serves to illustrate the point that trypanosomiasis is caused by a wide diversity of species or variants – and indeed AT can be caused by concurrent infections of multiple species. The genetic diversity within this complex of organisms has begun to be much better understood in the post-genomic era, which has underlined that the species are not only genetically divergent, but that there is also substantial genetic diversity within species (e.g. T. congolense Savannah, Forest and Kilifi subtypes). This inevitably means that genetic diversity translates to phenotypic diversity, and this includes virulence.
What do we mean by virulence in trypanosome infections? Virulence can be a very loosely used term in trypanosome literature, often applied to simple phenotypes such as parasite growth rate (including in vitro), but it is also used to refer to more complex traits such as host-specific infectivity or vector transmissibility. In reality, many of these phenotypes interact to determine the virulence of a trypanosome. However, virulence is also clearly an outcome of the interaction(s) of the trypanosome with the host, and host factors (for example, host species) can also shape and influence the virulence of trypanosomes in multiple ways. In this article, we define virulence as the ability to cause disease in the recipient host, i.e. the more virulent a trypanosome is, the more severe the disease is in the mammalian host. In this context, we aim to describe what is currently known about the spectrum of virulence diversity in trypanosomes, the variety of mechanisms that underpin virulence in trypanosomes, and the virulence factors that have thus far been identified in trypanosomes (see for overview). Additionally, we will outline the important current gaps in knowledge, and consider the opportunities that recent research presents for advancing understanding in this area. Finally, we will propose priorities for research on trypanosome virulence going forward.
Figure 1. Overview of virulence in African trypanosomes. (from left to right); Human infectivity: ApoL1 is the main component of human trypanosome lytic factor (TLF), a high-density lipoprotein subclass that confers protection against animal-infective trypanosomes through parasite lysis. The human-infective trypanosome species, T. b. rhodesiense and T. b. gambiense, have evolved mechanisms to evade ApoL1-mediated lysis, strongly influencing virulence in human hosts. For example, T. b. rhodesiense can express SRA, a protein that neutralises ApoL1 through direct interaction. Another mechanism is reduced ApoL1 uptake via an L210S mutation in the haptoglobin-haemoglobin receptor (HpHbr) that inactivates it. Coinfection: Infection with multiple species and/or strains can lead to multiple virulence phenotypes as described. For example, the presence of a less virulent strain can suppress the pathology associated with a more virulent strain of the same species in a coinfection setting. In addition, coinfection of multiple trypanosome species can impact differentiation dynamics. Immune response: The interaction of trypanosomes and the host immune response can greatly impact virulence phenotypes. Antigenic variation is undoubtedly a paradigm of trypanosome biology. Hydrodynamic flow of VSGs across the cell surface sweep bound antibodies to the cell posterior, where they are degraded following endocytosis. Furthermore, trypanosomes regularly switch the identity of the expressed VSG, leading to waves of parasitaemia with host antibodies eventually raised to the dominant VSG in the parasite population. A further parasite virulence phenotype associated with the host immune response is the ablation of B cell memory via killing of host B cells. Extravasation/sequestration: A key symptom of HAT is an ability of T. brucei to extravasate and enter extravascular tissues, in particular the brain, adipose tissue and the skin. A related virulence phenotype has also been described in animal-infective trypanosomes, albeit caused by intravascular sequestration rather than extravasation (e.g. strain and tissue specific sequestration in T. congolense). Secreted factors/EVs: Trypanosomes release a significant amount of metabolites, proteins and vesicles into the host environment, several of which have been characterised. In particular, virulence associated with secreted peptidases has been established, with oligopeptidase B (targeting atrial natriuretic factor), type 1 proglutamyl peptidase (targeting gonadotropin-releasing hormone and thyrotropin-releasing hormone) and prolyl oligpeptidase (type I and native collagen) all targeting host effectors. As-of-yet unidentified parasite-derived secretome components also target the maturation of host LPS-induced dendritic cells. Abbreviations: ApoL1: apolipoprotein L1; HpHbr: haptoglobin-haemoglobin receptor; SRA: serum resistance-associated protein; VSG: variant surface glycoprotein; OPB: oligopeptidase B; ANF: atrial natriuretic factor; PGP: proglutamyl peptidase; GnRH: gonadotropin-releasing hormone; TRH: thyrotropin-releasing hormone; POP: prolyl oligpeptidase; COL I: type I collagen; COL-N: native collagen; LPS: lipopolysaccharide; EVs: extracellular vesicles. Inset graph in immune response panel adapted from [Citation6].
![Figure 1. Overview of virulence in African trypanosomes. (from left to right); Human infectivity: ApoL1 is the main component of human trypanosome lytic factor (TLF), a high-density lipoprotein subclass that confers protection against animal-infective trypanosomes through parasite lysis. The human-infective trypanosome species, T. b. rhodesiense and T. b. gambiense, have evolved mechanisms to evade ApoL1-mediated lysis, strongly influencing virulence in human hosts. For example, T. b. rhodesiense can express SRA, a protein that neutralises ApoL1 through direct interaction. Another mechanism is reduced ApoL1 uptake via an L210S mutation in the haptoglobin-haemoglobin receptor (HpHbr) that inactivates it. Coinfection: Infection with multiple species and/or strains can lead to multiple virulence phenotypes as described. For example, the presence of a less virulent strain can suppress the pathology associated with a more virulent strain of the same species in a coinfection setting. In addition, coinfection of multiple trypanosome species can impact differentiation dynamics. Immune response: The interaction of trypanosomes and the host immune response can greatly impact virulence phenotypes. Antigenic variation is undoubtedly a paradigm of trypanosome biology. Hydrodynamic flow of VSGs across the cell surface sweep bound antibodies to the cell posterior, where they are degraded following endocytosis. Furthermore, trypanosomes regularly switch the identity of the expressed VSG, leading to waves of parasitaemia with host antibodies eventually raised to the dominant VSG in the parasite population. A further parasite virulence phenotype associated with the host immune response is the ablation of B cell memory via killing of host B cells. Extravasation/sequestration: A key symptom of HAT is an ability of T. brucei to extravasate and enter extravascular tissues, in particular the brain, adipose tissue and the skin. A related virulence phenotype has also been described in animal-infective trypanosomes, albeit caused by intravascular sequestration rather than extravasation (e.g. strain and tissue specific sequestration in T. congolense). Secreted factors/EVs: Trypanosomes release a significant amount of metabolites, proteins and vesicles into the host environment, several of which have been characterised. In particular, virulence associated with secreted peptidases has been established, with oligopeptidase B (targeting atrial natriuretic factor), type 1 proglutamyl peptidase (targeting gonadotropin-releasing hormone and thyrotropin-releasing hormone) and prolyl oligpeptidase (type I and native collagen) all targeting host effectors. As-of-yet unidentified parasite-derived secretome components also target the maturation of host LPS-induced dendritic cells. Abbreviations: ApoL1: apolipoprotein L1; HpHbr: haptoglobin-haemoglobin receptor; SRA: serum resistance-associated protein; VSG: variant surface glycoprotein; OPB: oligopeptidase B; ANF: atrial natriuretic factor; PGP: proglutamyl peptidase; GnRH: gonadotropin-releasing hormone; TRH: thyrotropin-releasing hormone; POP: prolyl oligpeptidase; COL I: type I collagen; COL-N: native collagen; LPS: lipopolysaccharide; EVs: extracellular vesicles. Inset graph in immune response panel adapted from [Citation6].](/cms/asset/99c711e6-b6db-455b-970f-0d430bd2467f/kvir_a_2150445_f0001_oc.jpg)
Evidence of virulence diversity of field isolates
Human trypanosomiasis
Disease caused by trypanosomiasis in humans is characterized by two stages. The first (the hemolymphatic stage or stage 1) is initiated by the deposition of trypanosomes into the skin by tsetse bite, from where parasites disseminate, initially via the lymphatics, to spread throughout the vascular system. This stage is typified by fever and lymphadenopathy, alongside malaise, weakness, and headaches. The second stage occurs when the parasites invade the brain (meningoencephalitic stage or stage 2) which is associated with motor and sensory dysfunction (including abnormal sleep patterns, giving rise to the colloquial name of sleeping sickness), and if untreated, eventually leads to seizures, coma, and death. The rate of progression from stage 1 to 2 disease is a classical metric of virulence in human infections, and here we will discuss the parasite-driven aspects of this.
As described above, animal and human disease is caused by an assemblage of trypanosome species. The diversity within species can contribute to phenotypic divergence, and in particular can be critically linked to parasite virulence and disease outcome. This is exemplified by the variants (often termed subspecies) of T. brucei that are able to infect humans and cause disease; T. b. rhodesiense, T. b gambiense Group 1 and T. b. gambiense Group 2. In fact, the current status of T. b. gambiense Group 2 in the area of West Africa from where it was originally described is uncertain [Citation7] – and how these parasites evade lysis by human serum is detailed below. These human infective variants are genetically distinct from each other, and reflect multiple independent emergences of human infectivity as a trait [Citation8]. The genetic divergence also manifests as differences across multiple phenotypes:
T. b. gambiense Group 1 is primarily anthroponotic while T. b. rhodesiense and T. b. gambiense Group 2 are zoonotic.
T. b. gambiense Group 1 does not undergo sexual recombination and all data suggest this organism reproduces strictly clonally [Citation8–10] – indeed, genomic analysis demonstrated the Meselson effect in this organism whereby there is independent evolution and accumulation of mutations in the homologous chromosomes, which can only occur with long-term absence of sexual recombination [Citation8]. In contrast, evidence from both experimental and population genetics data indicates that T. b. rhodesiense and T. b. gambiense Group 2 are capable of frequent sexual recombination [Citation8,Citation11–15].
T. b. gambiense Group 1 and T. b. rhodesiense require treatment by different drugs, particularly in the meningoencephalitic stage 2 of infection when parasites have entered the brain [Citation16].
The nature of the disease caused is very different - T. b. gambiense Group 1 is classically described as leading to a chronic infection that takes many months to years to culminate in the neurological end stage of infection [Citation17], whereas T. b. rhodesiense is usually described as causing infections that tend to be much more acute [Citation18,Citation19], in some instances reaching the critical neurological complications in a matter of days [Citation20].
However, virulence is complex in human-infective trypanosomes, as there is a clear contribution of host genetic variation to disease progression and outcome – this has mostly been defined in T. b. gambiense Group 1, where there is evidently a spectrum of disease presentation, including long-term asymptomatic individuals and even apparent self-cure [Citation21]. This difference in outcome has been linked to differential immune response signatures, with high IL10 and low TNFα being associated with an increased risk of developing HAT, whereas increased IL8 was associated with individuals becoming seronegative [Citation22] – in contrast macrophage inhibitory factor (MIF) was shown to be elevated in infected people (HAT patients and latent infections) but to not contribute to pathology [Citation23]. Differential disease outcome has also been shown to be associated with different Apolipoprotein-1 genotypes (see section below on human serum resistance), with patients having G2 alleles of this gene showing improved disease outcome [Citation24], and genome-wide association studies have corroborated this observation [Citation25]. The analysis of host genetic factors is a continuing area of work, and how host genetic diversity may interact with parasite genetic diversity is a remaining question. Indeed, there have been attempts to link T. b. gambiense Group 1 genotype to disease presentation in HAT patients [Citation26], but given the remarkable clonality and high levels of homozygosity across multiple isolates [Citation8] it may be that this will prove difficult, and this homogeneity across T. b. gambiense Group 1 exemplifies why it may be a good model to explore the human genetic contribution to disease. Having said that, inoculation of T. b. gambiense Group 1 isolates into mice deriving from human patients that differed in clinical severity demonstrated that pathology in mice also broadly separated into three categories (“highly pathogenic,” “intermediate pathogenic,” and “low pathogenicity”) that mirrored the pattern of disease severity in the patients they were isolated from [Citation27]. Therefore, there may be a pathogen genetic factor underpinning at least some of the variation in disease outcome observed in T. b. gambiense Group 1 infections, perhaps sited in the highly variable subtelomeric regions of the trypanosome genome that are very difficult to assemble and which would not be covered or captured by low-resolution approaches, such as micro- or minisatellites, or by most genome approaches/assemblies. The substantial challenge of incorporating these regions in analysis is evidenced by one study currently being the only genome assembly that has managed to provide a complete picture of T. brucei subtelomeric regions [Citation28], despite the organism being the focus of multiple genome sequencing efforts for many years.
The population genetics of T. b. rhodesiense is much more complex than that of T. b. gambiense, deriving from the fact that T. b. rhodesiense is defined by the carrying of a single gene, the serum resistance associated (SRA) gene [Citation29] that confers resistance to the trypanolytic factors in human serum. T. b. rhodesiense are therefore essentially variants of T. brucei that carry the SRA gene, and the genetic diversity of T. b. rhodesiense reflects that of the underlying T. brucei population that it is a member of, with clonal expansions occurring when outbreaks occur [Citation8,Citation11,Citation14,Citation30]. As a result of T. b. rhodesiense being genetically diverse, there is unsurprisingly also evidence for variation in virulence within T. b. rhodesiense in human cases [Citation31]. This is perhaps best characterized by studies in Uganda and Malawi, where it was demonstrated that there were differing population dynamics of T. b. rhodesiense, with semi-stable clonal lineages in Uganda and frequent mating in Malawi [Citation14]. These differences likely correlate with their varying transmission intensity and population dynamics resulting in different levels of interactions and mating with the underlying non-human infective T. b. brucei population. This also correlated with differing clinical presentations, there being a more chronic form of disease in Malawi and acute disease in Uganda [Citation32]. Within Uganda, with the increased power to test associations afforded by the expansion of clonal lineages, it was possible to link genetically distinct T. b. rhodesiense isolated from different disease foci with differing severity of disease presentation [Citation33,Citation34], with circulating IFNγ levels correlating with progression to the neurological stage of disease [Citation33]. In a separate study, genomic analysis was carried out on Ugandan T. b. rhodesiense isolates deriving from human patients with differing clinical presentation. This analysis suggested that the genetic divergence of these pathogenically distinct isolates to some extent derived from introgression from West African T. b. brucei, with a region on chromosome 8 originating from West African T. b. brucei containing a gene(s) whose alleles underpin the virulence differences observed between the isolates [Citation35]. Interestingly, the virulence differences between these T. b. rhodesiense isolates variants also was recapitulated to some extent in murine infection models, with isolates of the strain that presented with more severe human disease (Z310) resulting in infections with substantially higher parasitemia and more severe symptoms than isolates from the strain derived from milder human infections (B17) [Citation35].
There have been relatively few attempts to directly examine the genetic determinants of parasite-driven differential pathology, exemplified by T. b. rhodesiense and T. b gambiense, in T. brucei. One approach has been to utilize a classical genetics approach, exploiting cloned progeny derived from a genetic cross between two strains of T. brucei (TREU927 and STIB247) that cause very different severity of infection in mice [Citation36]. Analysis of the inheritance pattern of induced pathology during infections with progeny in mice identified a locus on chromosome 3 that was linked to differential organomegaly (enlarged liver and spleen) [Citation37]. While the causative gene(s) in this locus remain to be identified, the phenotype observed involved differential arginase expression and alternative macrophage activation [Citation36], reminiscent of the parasite-induced arginase-mediated reduction of NO synthesis and consequent liver damage by T. brucei kinesin heavy chain 1 (TbKHC1 – see section below for further details) [Citation38]. With improved sequencing technologies enabling assembly and annotation of the previously difficult to assemble and highly repetitive subtelomeric regions [Citation28], in which many trypanosome virulence factors are located, it may be timely to revisit the utility of genetic crosses and exploit these resources as routes to identifying virulence factors that may be important for disease.
Animal trypanosomiasis
While HAT is caused by different variants of T. brucei, Animal Trypanosomiasis (AT) is caused by multiple species of trypanosome [Citation39], which are very genetically divergent indeed. These differences are such that traits defined in T. brucei as paradigms of African trypanosome biology, such as reliance upon high-rate glucose metabolism in the mammalian host, or the mechanistic utilization of variant surface glycoproteins (VSGs) in antigenic variation, have been shown to either not hold (reliance upon glucose metabolism is much reduced in T. congolense [Citation40]) or be achieved by very distinct mechanisms (the VSG content and family structure indicate that antigenic variation is achieved in T. congolense and T. vivax by different mechanisms to T. brucei [Citation41–45]). This means that animal disease is caused by a much greater spectrum of trypanosome genetic diversity, making it a very broad disease in terms of clinical severity, duration and presentation [Citation46]. This complexity is one reason why developing interventions such as drugs and vaccines against AT has been and remains such a challenge, as any product needs to be effective against multiple divergent pathogen species.
Typically in ruminants (cattle, sheep and goats) trypanosome infection manifests as a chronic wasting disease, with intermittent pyrexia, lymphadenopathy, weight loss and reduction in activity [Citation47]. Anemia is a consistent clinical sign in ruminants, and can be used as a diagnostic indicator of infection [Citation48] - albeit with the caveat that several co-endemic pathogens also cause anemia. As well as reduced production in terms of weight loss, trypanosome infections also result in reduced milk yield and fertility [Citation49–51], with abortions in some cases [Citation52,Citation53]. In contrast to the human disease, neurological symptoms are not a common feature of ruminant infections, although seem to be reported more frequently in T. vivax infections [Citation54,Citation55]. Donkeys and horses are also commonly infected with trypanosomes [Citation56–58]; in these hosts neurological disease is much more common, with data indicating that T. brucei (including T. b. evansi and T. b. equiperdum) may be implicated in being the primary causative agent of neurological complications [Citation59,Citation60]. Asian or water buffalo (Bubalus bubalis) are an important livestock species across Asia, as well as increasingly in South America, and the impact of T. b. evansi (as well as T. vivax in S. America) upon buffalo is important and currently underappreciated, where it causes a similar chronic production disease to that seen in cattle [Citation46], with occasional outbreaks of high mortality [Citation61]. T. suis, T. simiae and T. godfreyi all either only or preferentially infect pigs, causing either acute or chronic presentation, which is seemingly dependent upon age of infection [Citation62–65]. The extent of the impact of the latter species (as well as other species such as T. simiae Tsavo) on livestock has been poorly documented, partially due to lack of molecular tools to enable detection, but also due to a relative paucity of research focus. Similarly, the diversity of trypanosomes identified through molecular screening of tsetse is clearly greater than that of the traditionally described livestock pathogens T. brucei, T. congolense, and T. vivax, (for example the recently described “T. vivax-like” species [Citation66,Citation67], but the role that such trypanosomes play in causing disease in livestock is currently unclear.
This brief outline of pathology in animal trypanosomiasis [covered in more detail elsewhere - [Citation46,Citation47,Citation68] serves to illustrate the enormous complexity of the disease, and this can make identifying the contribution of parasite virulence to clinical severity difficult to ascertain – even before considering the added complication and contribution of coinfections (see section below) and host susceptibility/tolerance. However, it is clear that some of the variation in clinical presentation does derive from the parasite genotype. T. congolense groups broadly into three genetically distinct subtypes, Forest, Kilifi, and Savannah [Citation39,Citation44,Citation69], and evidence indicates that there is frequent genetic exchange in the field resulting in substantial genetic diversity, at least within T. congolense Savannah [Citation70–72], and furthermore suggestion that T. congolense Savannah and Forest may hybridize [Citation72]. It has been demonstrated that field isolates representative of the three subtypes differed in virulence in cattle infections – a Savannah isolate (Sam 28.1) causing more severe disease (higher parasitemia, lower packed cell volume and eventually death) than Forest (Dind.3.1) or Kilifi (K60.1A) subtypes, which either showed minimal clinical signs compared to control animals (Kilifi) or even apparent self-cure (or inability to detect parasites in the blood) in all five animals infected with Forest after 95 days of infection (with animals followed for 295 days post-infection) [Citation73]. This strain-specific pattern of virulence was mirrored in mice infections, with Sam 28.1 giving rise to lethal infections that lasted less than a week, whereas Dind.3.1 and K60.1A produced chronic infections with low parasitemia and low mortality rate (one death in each group of seven mice over 130 days of infection) [Citation74]. While the caveat of these studies is that there was a single isolate per subtype, the observations would fit with the increased representation of T. congolense Savannah isolates in AT field studies in the literature [Citation39] – the expansion of locations across the African subcontinent where T. congolense Kilifi (and to a lesser extent Forest) has been detected has coincided with the advent of more sensitive molecular diagnostics, and this suggests that these subtypes may be reasonably widespread but either cause limited severe disease or present with very low parasitemia in livestock, and are therefore picked up less frequently in surveillance efforts. However, there are really very significant knowledge gaps around T. congolense Kilifi and Forest, including the extent of the role they play in livestock disease, and these subtypes certainly warrant further investigation.
Virulence variation within T. congolense Savannah has also been demonstrated, with 31 field isolates derived from cattle in Zambia being tested by inoculation into two mice each (with all strains used at their fifth or sixth passage from cattle isolation). The isolates grouped into three categories – termed “extremely virulent,” “moderately virulent” or “low virulence,” as determined by parasitemia profile, survival time, prepatent period, and degree of anemia induced [Citation75]. How these virulence categories translate to clinical presentation in cattle is unclear and requires further work, but it is worth noting that highly virulent field strains of T. congolense Savannah have been isolated, which reproducibly give rise to very acute infections in cattle, resulting in death in 9–10 days unless treatment is provided [Citation76–78].
T.vivax broadly splits into two genetic groupings, East and West African [Citation45,Citation79,Citation80]. Several studies using different (low resolution) genetic markers indicate that South American strains are derived from West African T. vivax [Citation81–83]. While the population genomic analysis of Silva Pereira et al. [Citation45] robustly demonstrated grouping of South American T. vivax with Ugandan strains, Uganda is closely linked with West Africa (including via trade routes and transfer of livestock) by being on the edge of the Congo basin, and therefore it is possible that Ugandan T. vivax may be more representative of West African strains than those from elsewhere in East and Southern Africa (which were unable to be included in the analysis). This is backed up by a study that assessed cross-reactivity of sera from cattle inoculated with strains from West and East Africa, which showed that sera from cattle infected with Ugandan strains cross-reacted with that from cattle infected with West African strains, but not those infected with East African strains [Citation84]. Such data indicate that further genomic analysis is therefore required to fully resolve the continental picture of diversity for T. vivax. With respect to sexual recombination influencing genetic diversity, all evidence suggests that, like T. b. gambiense, T. vivax is clonal and does not undergo sexual recombination [Citation45,Citation85]. A feature of T. vivax infections of cattle (as opposed to T. congolense and T. brucei) is that self-cure is reasonably frequently reported (e.g. [Citation50,Citation86,Citation87]), and this has been suggested to potentially relate to the smaller VSG repertoire of this species [Citation42,Citation45,Citation50] and therefore “exhaustion” of available VSGs during infections, and/or a reduced amount of VSG protein on the cell surface that may result in greater exposure of other invariant antigens to the host immune response. However, while there are reduced levels of cellular VSG gene transcripts compared to T. brucei and T. congolense [Citation88], evidence that this translates to reduced levels of VSG protein at the cell surface is less clear [Citation50]. There is reported strain-specific virulence in T. vivax, perhaps the most notable being a reported hemorrhagic form of T. vivax infection that seems be more commonly reported in isolates deriving from East Africa [Citation50,Citation89–92] – this is associated with a hyperacute infection profile with very high and sustained parasitemia, fever, profound anemia and multiple hemorrhages of visceral and mucosal surfaces. The hemorrhagic stage is correlated with thrombocytopenia and dysregulation of the clotting cascade, as well as generation of autoantibodies that bind to and cause lysis of erythrocytes and platelets [Citation93]. Additionally, other highly virulent field strains (without the hemorrhagic presentation) have been isolated that give rise to experimental infections with very acute profile and short duration (9–10 days before rescue treatment is required) [Citation76,Citation77]. However, while we have good evidence for there being parasite-driven differences in virulence in T. vivax, a barrier to understanding the parasite factors that contribute to these differences is that very few T. vivax strains grow in mice and only one strain has been reproducibly cultured in vitro (Y486) [Citation94,Citation95] - although it should be noted that the culture of bloodstream form Y486 has only been successful to a limited extent. This limitation to ruminant in vivo experimental work for all but very few strains means that there has not been the ability to either assess translation of variable virulence in the murine model, or functionally assess potential mechanisms in vitro.
Trypanosome interactions with the host immune response
The host immune system and its interaction with the pathogen evidently is a major component of how virulence presents in the infected mammalian host. The details of immunology in trypanosome infections are well covered elsewhere [Citation68,Citation96–98], and in this section, we will aim to focus on aspects of the host immune response that are driven by the parasite (i.e. how parasite virulence influences elements of the host immune system).
The paradigmal trypanosome interaction with the host immune system is antigenic variation. Trypanosomes have developed an incredibly elaborate and extensive system of antigenic variation, which is driven by a large gene family of variant surface glycoproteins (VSGs), one of which is expressed in each cell through a monoallelic expression system that results in the parasite coat being covered in homodimers of the expressed VSG protein. The VSG N-terminal domains are the primary point of contact for the host adaptive immune response, and VSG epitope-specific antibodies are generated that clear parasites expressing the relevant VSG. However, the parasites regularly change the identity of the expressed VSG, meaning that within a population cells emerge that are not susceptible to the VSG-specific antibodies raised against epitopes on the previously expressed VSG. Through a combination of a very large VSG gene repertoire (2,000 in T. brucei – approximately 20% of the coding genome) and elaborate recombinatorial processes that massively amplify the potential encoded genetic VSG variation, antigenic variation in trypanosomes is a powerful tool that matches the host ability to generate antibodies, and is key to their ability to establish and maintain long-lasting chronic infections. The intricacies of antigenic variation, particularly in T. brucei, have been the subject of much research over many decades, and the mechanistic understanding is highly developed [Citation43,Citation99,Citation100]. While it is very evident that T. congolense and T. vivax also undergo antigenic variation, the structure and content of the VSG repertoire in these species is very different to that of T. brucei [Citation42,Citation101], and the degree of recombination-driven amplification of diversity also appears quite distinct. For example, while T. brucei massively multiplies antigenic diversity through recombination between VSGs that belong to one of two subfamilies (a-VSG and b-VSG), evidence suggests that T. vivax does not employ recombinational VSG switching [Citation45], with genes in four subfamilies corresponding to 174 phylotypes (where a phylotype is a clade of highly related VSGs based on amino acid alignment). T. congolense lies somewhere between, with a repertoire indicating recombination largely occurring within 15 phylotypes split between two subfamilies [Citation44,Citation101]. Currently, it is unclear if these repertoire differences translate to mechanistic differences in terms of how antigenic variation is expressed in T. congolense and T. vivax [Citation43]. Additionally, the implications of the VSG repertoire differences with respect to host-parasite interactions, such as the putative different effective repertoire sizes, remain to be elucidated.
The structure of the VSG coat and limited presentation of epitopes to the host response are one mechanism of immune evasion, but trypanosomes also elegantly exploit their motility as an immune evasion technique – the motility driven by the flagellum, combined with the free movement of VSGs across the cell surface, results in hydrodynamic pressures at the cell surface effectively sweeping bound antibodies to the cell posterior, where they are engulfed and removed by endocytosis in the flagellar pocket [Citation102]. This provides an extended time window for trypanosomes to switch VSG identity before antigen-specific antibodies reach a concentration threshold that can overcome the hydrodynamic flow effect. Initially described in T. brucei, this has since been shown to also occur in T. congolense and T. vivax [Citation103] – with species-specific differences in motility characteristics postulated to link to the differential infection biology of the parasites, such as extravascular tissue invasion for T. brucei, cellular adherence for T. congolense and intravascular circulation for T. vivax.
The VSG coat structure has long been posited to provide a near insurmountable barrier in terms of targeting the host immune response to underlying and conserved antigen epitopes, via vaccination for example. How this barrier functions as such, given there are invariant proteins whose structure suggested they should protrude above the protective VSG monolayer, has long been debated [Citation104]. However, the generation of the first model of a full T. brucei VSG structure provided insight that the C-terminal domain of the VSG is likely to be remarkably conformationally flexible, sufficiently so to enable VSGs to possibly shield invariant surface proteins [Citation105]. This is supported by efforts that have targeted invariant antigens in vaccination efforts providing at best partial protection [Citation106,Citation107]. Strategies have been implemented to try and bypass this structural barrier, such as using single-chain camel-derived nanobodies against invariant antigens [Citation108]. Overall, this strategy has also met with limited success, although some protective effect was demonstrated. However, recent data has demonstrated that vulnerabilities can be identified by targeting invariant antigens. Through a process of expressing recombinant versions of proteins predicted to be expressed on the cell surface of T. vivax, and immunization and challenge experiments in mice, the extracellular domain of one protein (“invariant flagellum from T. vivax,” IFX) resulted in reproducibly sterile protection against rechallenge [Citation109]. These remarkable data provide proof of principle that vaccination using surface-expressed proteins may be achievable, after decades of skepticism. The localization of IFX, between the flagellum and cell body, suggests it may play a role in flagellum structure or function, and this may provide a reason as to why it represents a vulnerability for the parasite, as due to its location it may not be subject to hydrodynamic clearance of bound antibodies. Whether this vulnerability also applies to T. congolense and T. brucei awaits further study. Additionally, the translation of successful immunization against IFX from the mouse model to a clinically relevant host species (goats) has been tried in pilot vaccination and challenge experiments, but did not result in protection [Citation109]. Therefore, significant hurdles clearly remain to be overcome in order to replicate the promising protection observed in mice in disease-relevant hosts such as cattle.
The host antibody response is clearly important in clearance of trypanosomes during infection [Citation110]. Debate continues about the role and efficacy of particular antibody isotypes; for example, the key isotype that conferred protection against T. vivax in IFX vaccinations studies was shown to be IgG2a [Citation109], but recent data demonstrated that activation-induced cytidine deaminase (AID)-deficient mice, which are incapable of somatic hypermutation and therefore cannot generate IgG antibodies, were more efficient at clearing challenge with T. b. evansi than their wild-type controls through IgM [Citation111] – consistent with previous studies showing the importance of IgM in controlling T. brucei infections in mice [Citation112]. Nguyen et al. [Citation111] interestingly hypothesized that the rapid onset of B cell follicle activation and isotype switching to IgG may in fact be driven by the trypanosome, as switching to the lower efficacy IgG would benefit parasite survival. These aspects of antibody response remain to be fully elucidated in the mouse model of T. brucei, let alone host species such as cattle, in which the mechanisms of antibody generation are very different and for which the antibodies can have some unique features that may impact upon antigen binding [Citation113,Citation114], and for T. congolense and T. vivax. If vaccine prospects for AT are to be achieved from candidates such as IFX, clarity on what constitutes an effective antibody response, and how this would be optimally induced, in the eventual host species and against the AT-relevant trypanosome species, will be needed.
Given the key role of antigen-specific antibodies, a notable parasite-driven phenotype is the destruction of host immune memory, with trypanosome infection of mice resulting in ablation of B cell memory via killing of B cells. This included the loss of memory to previously exposed non-trypanosome antigens [Citation115]; this was recently shown to specifically involve the loss of memory B cells from infected animals [Citation116]. This effect has also been shown to occur in mouse infections with T. congolense [Citation117], and observed disruptions to splenic architecture including lymphocyte-depleted germinal centers and depletion of splenic B cells in mice infected with T. vivax are consistent with the phenotype also occurring in infections with this species [Citation118,Citation119]. While the destruction of B cells has not been formally described in cattle infections, memory loss was observed in cattle immunized with irradiated T. brucei, infected with T. congolense and then re-challenged with homologous irradiated T. brucei, with the memory response against T. brucei being impaired in three of the five cattle [Citation120], suggesting that this process also occurs in cattle. The extent of any parasite-driven B cell destruction in human trypanosome infections is also not clear, although one study has demonstrated reduced anti-measles antibody levels in HAT patients [Citation121]. Both cattle and human data, although scanty, indicate that the phenomenon indeed may occur, but the extent of B cell memory loss may not be as extensive as in infected mice. The B cell destruction is known to be mediated by host natural killer (NK) cells and is perforin-mediated [Citation122], but the identity of any parasite ligand that may stimulate and drive this interaction is yet to be identified. While the impact of this parasite-driven phenomenon clearly benefits immune escape and survival of trypanosomes within infections, it also has potentially serious implications for the epidemiology of other infectious diseases in endemic areas. The impairment of immune memory in trypanosome infected animals or humans may mean hosts become more permissive for particular coinfections, and as suggested by other authors [Citation96], the trypanosome-mediated destruction of immune memory would in theory also disrupt vaccination-mediated protection, with consequent implications for disease control efforts. This latter suggestion is backed up by several observations of diminished antigen-specific antibody responses in trypanosome-infected Asian buffalo, goats, and cattle to vaccinations ranging from Pasteurella multocida, Bacillus anthracis, contagious bovine pleuropneumonia to foot and mouth disease virus [Citation96,Citation123–127]. This aspect of trypanosome infection biology deserves further attention, and in particular fuller understanding of the extent of immune memory loss in clinically relevant hosts, as this could be an important factor in both general disease susceptibility and epidemiology, and, for example, if efforts to generate a vaccine against AT bear fruit.
The symptomology of human trypanosomiasis is defined by the ability of T. brucei to extravasate and enter extravascular tissues, leading to encephalitis-related clinical signs. The description of adipose- and skin-resident trypanosomes in mouse and human infections with T. brucei sensu lato [Citation128–130] has focused attention on this aspect of T. brucei infection biology, with its obvious relevance for disease progression, transmission, diagnosis, parasite metabolism, and interactions with the host immune response. Indeed, the extravasation has been shown in a mouse model to be active (i.e. occurs prior to any vascular compromise induced by inflammation) and if the process is blocked by introducing antibodies against host molecules involved in cellular adhesion (E-selectin, P-selectin, ICAM2, CD36, and PECAM1) mouse survival was improved, indicating that extravasation is a key virulence phenotype in T. brucei infections [Citation131]. Notably, CD36 was shown to preferentially inhibit extravasation into adipose depots, indicating potential tissue-specific interaction in extravasation. Brain involvement in the mouse model has also been well defined in infections with T. vivax and T. congolense [Citation132,Citation133]. In the case of T. congolense, which binds to endothelial cells and is considered an intravascular parasite [Citation134], brain pathology was associated with trypanosome sequestration in brain vasculature and the consequent immune response; interestingly this effect was strain-specific (T. congolense 1/148 caused sequestration and pathology, while IL3000 did not), suggestive of a differentially expressed parasite virulence factor(s). While similar sequestration in cerebral capillaries and sequelae have been observed in cattle experimentally infected with T. congolense [Citation135,Citation136], neurological clinical signs associated with T. congolense infections are not frequently reported in the field in livestock [Citation18,Citation46]. It is not completely clear whether T. vivax readily extravasates or sequesters, and mouse data has either described mainly vascular lesions [Citation119] or used non-invasive bioluminescence imaging techniques that would not discriminate between intravascular and extravascular parasites [Citation132]. While neurological clinical signs have been reported from T. vivax livestock infections in the field [Citation54,Citation137], as with T. congolense it is also not a frequent clinical presentation. However, clearly a fuller exploration of tissue distribution in all three parasites, and in clinically relevant hosts as well as mice, is needed before the potentially important implications of tissue specificity and adaptation are understood.
The interaction of trypanosomes with the immune response is clearly multifaceted, and we have chosen here to focus on key parasite-driven aspects. The following sections also contain multiple examples of parasite biology whose interaction with the hosts, including with the immune response, also determine virulence and infection outcome. The examples outlined above particularly serve to illustrate gaps in our knowledge – many of these derive from the need to translate findings from either T. brucei to T. congolense and T. vivax, or from in vitro or mouse models to clinically relevant hosts.
Human infectivity
Another defense mechanism elicited by the mammalian host is the presence of apolipoprotein L1 (ApoL1) in normal human serum. ApoL1 is a component of the trypanosome lytic factor-1 and −2 (TLF1 and TLF2) which is a subclass of high-density lipoprotein (HDL) [Citation138–140]. Human ApoL1 lyses exclusively animal infective trypanosomes through the formation of pH-dependent ionic pores in the lysosomal membrane. This causes the inflow of chloride ions from the cytoplasm leading to lysosomal swelling [Citation141–144], and ultimately, parasite death. Permeabilization of the mitochondrial membrane has also been reported [Citation145]. However, the human infective forms of the parasite, T. b. rhodesiense and T. b. gambiense, are resistant to these TLFs. T. b rhodesiense evades ApoL1 lysis by the possession of serum resistance-associated (SRA) protein [Citation146], which neutralizes the ApoL1 toxin by direct interaction. However, some variants of ApoL1, variants G1 and G2, are able to avoid this deactivation resulting in the killing of T. b. rhodesiense [Citation142]. These variants are primarily found in African Americans and West Africans [Citation147], potentially contributing to the absence of T. b. rhodesiense infections in west Africa, and factor in the spectrum of disease severity in T. b. rhodesiense patients (see “Evidence of virulence diversity of field isolates” section), with the G2 allele being associated with less severe disease in a genetic analysis of T. b. rhodesiense patients in Malawi [Citation148]. T. b. gambiense, consisting of two groups, Group 1 and Group 2, are both resistant to ApoL1 lysis. While T. b. gambiense Group 1 stably avoids TLF lysis, T. b. gambiense Group 2 shows variable TLF resistance in a way seemingly similar to T. b. rhodesiense but which does not involve SRA, and thus, remains to be fully elucidated [Citation149,Citation150]. T. b. gambiense Group 1 on the other hand, uses the specific glycoprotein (TgsGP) to inhibit ApoL1-mediated lysosomal damage by membrane stiffening when it interacts with lipids [Citation151]. Other mechanisms employed by T. b. gambiense Group 1 to escape ApoL1 killing include reduced sensitivity to ApoL1 by cysteine proteases [Citation152], reduced uptake of ApoL1 due to an L210S substitution in the haptoglobin-hemoglobin receptor, resulting in inactivation [Citation151,Citation153], and increased digestion of ApoL1 [Citation142]. There have been reports of atypical infections of humans with species of trypanosome not normally infective to humans, including T. lewisi and T. b. evansi, and very rarely T. b. brucei, T. vivax and T. congolense – while sporadic and clearly rare, instances of such infections either are increasing or are being detected more often [Citation154]. While often the basis for human infectivity in such infections has not been able to be fully investigated, T. b. evansi infections have been identified to occur both in an individual lacking APoL1 due to null mutations [Citation155], but also recently in a patient with no observable ApoL1 deficiency [Citation156], suggesting there remain aspects yet to be explained in this intensively studied and important host-parasite interaction.
Parasite metabolism and virulence
Parasite metabolism is crucial to enable generation of sufficient ATP to persist in the host bloodstream. It is well established that African trypanosomes rely on host carbohydrates in the form of glucose for ATP production. However, metabolic enzymes and their products can also impact host gene expression and metabolism in ways that maximize parasite survival, modulate host immunity, and directly contribute to virulence phenotypes. The role of parasite metabolism in mediating host immune responses has been studied in several pathogenic protozoan parasite species, including Trypanosoma cruzi and Leishmania spp., in addition to African trypanosomes [Citation157–159]. These parasites all release a significant number of proteins and metabolites into their host environment (the former detailed by studies of the secretome [Citation160–162]), although relatively few studies have detailed the impacts of metabolism on host-pathogen dynamics, and thus, virulence, during infection. Nonetheless, there is clear evidence that parasite-derived metabolites and proteins impact host immune responses with implications for parasite virulence [Citation161,Citation163].
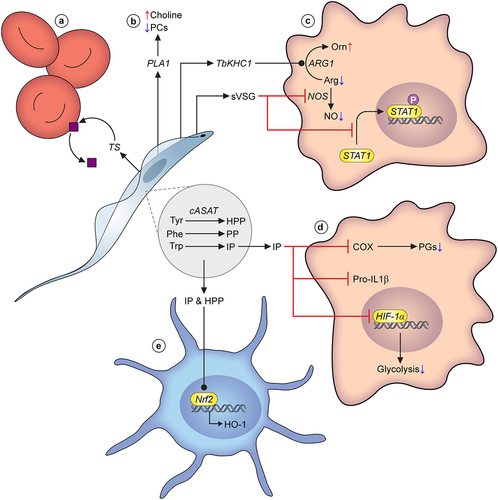
Nitric oxide (NO) is a key host effector molecule in the defense against trypanosome infection and NO exhibits cytostatic and cytolytic properties. To counter the effects of NO, Kinesin Heavy Chain (TbKHC)-1 is a protein secreted by T. brucei under both in vitro and in vivo conditions, and has been shown to induce arginase-1 activity in host myeloid cells, even those from uninfected mice [Citation38]. Arginase-1 converts L-arginine to L-ornithine and urea, and its activity leads to reduction in the synthesis of NO. Presumably, increased competition for L-arginine (an important substrate for NO synthesis) leads to this reduction. Indeed, it has previously been shown that L-arginine bioavailability is an important determinant of NO production and parasite killing [Citation164]. Recombinant TbKHC1 was shown to trigger IL-10 and arginase-1 expression mediated by a C-type lectin (SIGN-R1; CD209b) receptor. Importantly, host survival time is significantly prolonged in TbKHC1 KO-infected mice, compared to wild-type controls [Citation38].
TbKHC1 secretion is not the only form of host NO modulation. Earlier work highlighted that soluble VSG (sVSG), a form of VSG released by trypanosomes, modulates host gene expression in macrophages [Citation165]. Importantly, the timing of sVSG exposure in relation to that of IFN-γ is crucial. Whereas IFN- γ exposure followed by sVSG exposure leads to the expression of TNF-α, IL-6, and IL12p40, treatment of macrophages with sVSG prior to IFN-γ led to a reduction in IFN-γ-induced responses, including reduced NO synthase expression and NO secretion [Citation165]. Further work showed that the glycosylinositolphosphate moiety of the sVSG is crucial for these host modulatory effects [Citation165,Citation166].
Metabolism of fatty acids also impacts virulence. In particular, phospholipase A1 (PLA1) activity is thought to correlate with pathogenesis [Citation167] and indeed, PLA1 activity in plasma and tissue fluid from experimentally infected rabbits correlates with parasitemia [Citation168]. It is thought PLA1 (and potentially PLA2) activity is responsible for the severe changes seen in plasma lipids in infected animals, in particular a reduction in phosphocholines (phosphatidylcholine) accompanied by increased levels of choline, indicative of phospholipase action [Citation169,Citation170]. Interestingly, the phospholipase activity from non-pathogenic trypanosome species such as Trypanosoma lewisi is relatively low compared to that of pathogenic species, suggesting a correlation between PLA1 action and virulence/pathogenesis [Citation167].
Trypanosomiasis is also associated with significant perturbations in serum/plasma levels of amino acids [Citation170]. In particular, there is depletion of the aromatic amino acids L-tryptophan, L-tyrosine and L-phenylalanine [Citation171–174]. Concomitantly, T. brucei excretes biologically relevant levels of aromatic ketoacids, specifically indolepyruvate (IP), hydroxyphenylpyruvate (HPP) and phenylpyruvate (PP) [Citation171,Citation172,Citation175,Citation176]. These aromatic ketoacids are generated through degradation of aromatic amino acids by the cytosolic aspartate aminotransferase (cASAT) [Citation163,Citation177]. This protein, as well as the reactions it catalyzes, are essential to the parasite [Citation163,Citation178], but the products of these reactions possess several important immunomodulatory properties.
The most studied excretory aromatic keto acid, indolepyruvate (IP), has been implicated in several virulence roles [Citation163,Citation179]. IP is a product of L-tryptophan metabolism through cASAT action [Citation163]. Firstly, IP treatment of host cells leads to reduced glycolytic capacity by interfering with the transcription factor hypoxia-inducible factor-1α (HIF-1α) [Citation163]. Furthermore, this study showed that IP inhibits the induction of pro-IL-1β, a potent pro-inflammatory cytokine. More recent work on IP has highlighted the modulation of host eicosanoid production associated with this trypanosome-derived metabolite [Citation179], specifically the downregulation of a class of eicosanoids called prostaglandins (PGs). In this study, Diskin and colleagues further showed that IP acts as a direct inhibitor of cyclooxygenase (COX) activity, an upstream mediator of PG production, and this effect is replicated in human macrophages [Citation179]. Thus, IP is a powerful modulator of host activity, in particular that of the pro-inflammatory and innate immune response to infection.
Whilst the aforementioned studies were in large part carried out in murine trypanosomiasis models, recent evidence shows that the immunomodulatory properties of IP (in addition to HPP) are replicated in primary human dendritic cells [Citation180], with HO-1 induction through Nrf2 activation, suppressed production of pro-inflammatory cytokines, reduced CD4+ T cell activation and modulation of host cell metabolism, including downregulation of glycolytic capacity [Citation180]. To our knowledge, there are no reports on any immunomodulatory effects of PP, another aromatic ketoacid excreted at high levels by African trypanosomes. Unlike IP, PP has no effect on the ability of LPS to induce IL-1β [Citation163], but other roles cannot be ruled out.
In murine models, trypanosome infection is associated with global host metabolic disturbances, including in the bloodstream, but also in other anatomical locations such as the gut and brain [Citation170,Citation181,Citation182]. These changes are the result of both host and parasite metabolism. The main glycolytic end-product from trypanosomes is pyruvate, which accumulates to high levels in the host plasma [Citation170]. There are also increased plasma concentrations of lactate and these are, to an extent, indicative of upregulated glycolysis in host cells [Citation170]. T. brucei does not encode lactate dehydrogenase (LDH) [Citation183], and, therefore, cannot generate lactate via fermentation of glucose [Citation184]. However, glucose-derived L-lactate is excreted from T. brucei at low levels, likely via methylglyoxal detoxification [Citation183,Citation185]. It should be noted that procyclic form T. brucei, as well as bloodstream form T. lewisi, also excrete L-lactate, the latter able to do so via lactate dehydrogenase [Citation186,Citation187]. It is currently unknown whether the livestock trypanosomes T. congolense and T. vivax generate L-lactate via fermentation, although LDH is not annotated in their respective genomes. It is plausible that both host and parasite-derived lactate likely contribute to metabolic acidosis, a significant contributor to pathology. As the disease progresses, the host can enter a ketotic state, characterized by increased levels of D-3-hydroxybutyrate, where lipids are metabolized for energy [Citation170]. This is partially due to competition for the main energy source, glucose [Citation188–190]. Ultimately, hypoglycemia can play a role in host survival [and has been noted in cattle infections, e.g. [Citation54]], and, therefore, parasite glycolytic rates have the potential to impact upon parasite virulence.
Several other important metabolic processes have been shown to impact upon virulence, including proteases such as serine peptidase 2 (ISP2) [Citation191] and the cysteine proteases Cathepsin L and Cathepsin B [Citation192]. In addition, increased levels of O- and N-acetylated glycoproteins have been detected in T. brucei-infected plasma, which are likely T. brucei derived [Citation193]. Whilst the underlying mechanisms remain to be elucidated, it is clear that these proteins are involved in trypanosome-mediated attenuation of the immune response [Citation191–193]. Finally, T. brucei, like other pathogens, exhibits an ability for metabolic mimicry, where T. brucei derived inositol phosphate glycans released from GPI anchors are able to affect the host in the same way as insulin, an important hormone for glucose regulation [Citation194].
Recent evidence has revealed that T. brucei is capable of invading adipose tissue [Citation130], a site abundant in glycerol. Indeed, T. brucei is capable of proliferation in glycerol-based medium [Citation195], and these findings may also contribute to our understanding of trypanosome virulence in vivo. Furthermore, imbalances in plasma lipid bioavailability have also been detected in plasma samples derived from experimental model and human infections [Citation196–198].
Whilst the majority of studies on trypanosome metabolism and its impact on virulence have focused on the relevant model for human infection, T. brucei, comparatively few studies have investigated the relevant species for Animal Trypanosomiasis - T. congolense and T. vivax. Recent studies, however, have shown that the former differs from T. brucei in key metabolic areas, such as glycolysis and lipid metabolism, and this may impact metabolic phenotypes associated with virulence [Citation40]. For example, it was hypothesized some time ago that free fatty acids released from autolyzing trypanosomes can significantly impact pathogenesis and virulence [Citation199]. The differences in fatty acid metabolism between T. congolense and T. brucei that have been observed recently could underpin differences in virulence between the species [Citation40]. Furthermore, differences in metabolite uptake and excretion may also play an important role in differing virulence between African trypanosome species, but these are as yet unstudied.
Whilst the genetic basis of differential virulence in livestock trypanosomes has not been elucidated, there is clear evidence that strain-dependent variation in virulence exists in T. congolense [Citation73–75], and it is likely that differential metabolism underlies at least some aspects of this variation. T. vivax is unique amongst African trypanosomes in that it encodes a proline racemase not found in T. brucei or T. congolense [Citation200]. This enzyme was subsequently shown to be a potent B-cell mitogen and thus, can be considered a virulence factor underpinning hypergammaglobulinemia, a symptom observed during acute stages of T. vivax infection in mice [Citation200]. Furthermore, unlike T. brucei, adhesion to host cells is an important aspect of T. congolense and T. vivax bloodstream form biology, as well as pathogenesis [Citation134,Citation201,Citation202]. In both T. vivax and T. congolense, it is established that trans-sialidases are involved in host cell attachments, and are also a key mediator of anemia, and thus, virulence [Citation203–206]. Trans-sialidases are both expressed on the parasite surface and secreted into the extracellular environment, and they are responsible for desialylation of red blood cells, leading to erythrophagocytosis and anemia. There is evidence that trypanotolerant N’Dama (African taurine) cattle exhibit reduced anemia compared to susceptible indicine cattle [Citation207], and concomitant evidence that trans-sialidases purified from T. vivax desialylated indicine but not African taurine RBCs [Citation203], indicating a correlation between trans-sialidase host-specificity and parasite virulence.
Comparative proteomics analysis of T. vivax strains with contrasting virulence revealed differential expression of several metabolic enzymes [Citation208]. In that study, virulence and pathogenesis were interpreted as capacity to multiply and capacity to produce disease/mortality, respectively. Protein expression profiles of two strains (high virulence and moderate pathogenicity vs low virulence and high pathogenicity) were compared. Amongst the significant differentially expressed proteins, there were also important glycolytic enzymes, pyruvate kinase, and glycerol kinase, expressed at higher levels in a T. vivax strain eliciting significantly more severe clinical pathogenesis, suggesting that glycolytic metabolism may play a role in driving symptoms [Citation208].
It is worth noting that in vivo experiments focused on dissecting host–pathogen interactions and virulence have been carried out almost exclusively on rodent models in contrast to clinically relevant models such as, in the case of animal trypanosomiasis, cattle [Citation95]. Ruminants such as cattle exhibit markedly divergent blood biochemistry from non-ruminants such as rodents and humans [Citation209], and this has the potential to impact both parasite and host metabolism during infection. For example, ruminant blood contains reduced levels of glucose and substantially increased levels of small volatile fatty acids (e.g. propionic acid and butyric acid) [Citation210–212] compared to human or mouse blood. Thus, given that, as an example, glycolysis is a cornerstone of trypanosome metabolism, host metabolic differences may play an important role in influencing host–pathogen interactions and virulence ( summarizes our current understanding of how trypanosome metabolism influences infection severity and outcome).
Figure 2. Parasite metabolism and virulence. A) Trans-sialidases released by T. vivax cleave sialic acid moieties from glycoproteins on the erythrocyte cell surface, leading to erythrophagocytosis and eventually, anaemia. B) All three species of pathogenic African trypanosomes are known to release phospholipases that degrade phosphocholine-bound lipids. They are considered significant virulence factors, and their action results in a build up of choline in the host bloodstream. C) T. brucei secretes multiple factors that modulate macrophage ability to generate nitric oxide (NO), including TbKHC1, and soluble VSG (sVSG). The latter stimulates arginase-1 activity, leading to increased usage of the available arginine pool to generate ornithine, reducing substrate availability for NO production through nitric oxide synthase (NOS). Simultaneously, sVSG has an inhibitory effect on NOS. sVSG also interferes with the phosphorylation of STAT1, an important transcription factor that drives pro-inflammatory responses. D) Parasite amino acid metabolism and its effect on host responses has been studied to some degree in T. brucei. In particular the fate of hydroxyphenylpyruvate (HPP), phenylpyruvate (PP) and indolepyruvate (IP), products of cASAT-catalysed conversions of L-tyrosine, L-phenylalanine and L-tryptophan, respectively. IP is a potent modulator of pro-inflammatory responses in macrophages. Firstly, IP interferes with HIF-1α, leading to a reduction in glycolytic capacity of macrophages. Secondly, IP inhibits induction of pro-IL-1, a potent pro-inflammatory cytokine. Finally, more recent work has established that IP is a direct inhibitor of cyclooxygenase (COX), leading to reduced prostaglandin (PG; mediators of inflammation) production. E) Trypanosome-derived IP as well as HPP can impact upon dendritic cells, by stimulating Nrf2-mediated hemeoxygenase-1 (HO-1) induction, again leading to a reduced pro-inflammatory response. Many other metabolic factors are known to be excreted by trypanosomes, but their molecular interactions with the host environment remain to be established, and they are therefore not included in this overview figure.
Quorum sensing
Although trypanosomes are single-celled parasites, individuals within the population show the ability to act co-operatively to restrict parasite numbers. This has a direct impact on virulence and parasite transmissibility. Virulence is affected because with unlimited population growth, hosts lethally and rapidly succumb [Citation213,Citation214]. Correspondingly, the prolongation of host viability increases the probability of transmission – the essential requirement for any parasitic lifestyle. This is particularly the case for African trypanosomes where transmission is restricted by the poor vectorial capacity and relative scarcity of tsetse flies in comparison to, for example, mosquito vectors for parasites such as Plasmodium [Citation215–217]. In addition to the direct consequences of uncontrolled parasite proliferation on host survival, the co-operative behavior of trypanosomes acts to promote transmission by driving the generation of non-proliferative transmission adapted developmental forms of the bloodstream parasites – so-called stumpy forms [Citation218]. These forms, specific to T. brucei at least as a morphologically distinct entity, dominate the peak of acute and chronic parasitaemias in experimental infections and predominate in tissue reservoirs such as the skin and adipose tissue when quantitated using the molecular marker defining this form, PAD1 [Citation129,Citation130,Citation219].
Stumpy generation is a quorum sensing phenomenon whereby parasite numbers are detected and responded to by individuals within the population – a characteristic described in many species of social microbe. Evidence of inter-parasite communication controlling the production of stumpy forms was initially provided by the analysis and modeling of parasites in animal infections [Citation220], but definitive evidence emerged with the successful culture of parasites with the developmental competence to generate stumpy forms [Citation221,Citation222]. These are representative of tsetse-transmitted trypanosomes in the field and are termed pleomorphic [Citation223], and are distinguished from so-called monomorphic forms that arise through laboratory passage, or in parasite subspecies that have lost the capacity for tsetse transmission, and are instead spread either by mechanical transmission by other biting flies (T. b. evansi) or by venereal transmission between equids (T. b. equiperdum) [Citation224,Citation225].
The molecular details that generate the quorum sensing signal and how this is detected and transduced to effect development in the parasites have been recently unraveled (). The signal that induces the parasite to undergo cell cycle arrest and stumpy formation (classically described as an ill-defined “stumpy induction factor”, SIF) is oligopeptides in the environment of the parasite [Citation226]. These are generated by proteolytic enzymes or peptidases that are released by the parasite in the mammalian host, apparently through an unconventional protein secretion pathway [Citation227]. This allows a density-dependent signal to be generated because as parasite numbers increase, the abundance of the released peptidases correspondingly increases and, though their activity in the blood or tissues, produce oligopeptides to activate the developmental signaling pathway [Citation228]. Two peptidases have been found to provide a major contribution to the generation of the QS signal, Oligopeptidase B and metallocarboxypeptidase 1, and the individual or combined deletion of both peptidases by gene knockout increases parasite virulence through reduced stumpy formation [Citation227]. Other parasite-derived peptidases are also likely to contribute, however, complementing the dominant activities of TbOPB and TbMCP1, or pre-processing larger polypeptide substrates so that they can act as substrates for these enzymes, which show specificity for substrates limited in size [Citation229,Citation230]. Host peptidases in the parasite’s environment could also provide a signal to augment stumpy formation; this would not be dependent upon parasite numbers directly, although immune responses to the parasite population may involve proteolytic activities [Citation231]. Although untested, the immune response against the parasite could also contribute to promote quorum sensing if parasite-specific antibodies proximal to the parasite could provide substrates for trypanosome-released peptidases, potentially contributing to the altered stumpy formation of intact versus immunocompromised mice [Citation232]. Importantly, the generation of oligopeptides by parasite-derived peptidases allows stumpy formation at high parasite numbers in the blood and also low parasite numbers where trypanosomes are constrained within tissues such as the skin and adipose, such that local accumulation of their activities and products can occur [Citation226,Citation227]. This resolves the perceived conundrum that stumpy formation in rodents involves large parasite numbers, whereas in livestock hosts the circulating parasite population might be relatively low but stumpy forms are prevalent [Citation233].
Figure 3. Quorum sensing in Trypanosoma brucei. Schematic pathway for the quorum sensing signalling pathway in Trypanosoma brucei. Slender form parasites release several peptidases into their environment, with two peptidases, Oligopeptidase B and Metallocarboxypeptidase I being important contributors to the generation of the quorum sensing signal, oligopeptides. Environmental oligopeptides can be transported into recipient parasites by the TbGPR89 surface transporter that is expressed on slender cells but not stumpy forms. In an unknown mechanism, transported oligopeptides stimulate a signal transduction cascade that promotes stumpy formation through the action of gene regulators (RNA binding proteins). Molecules that act to inhibit stumpy formation (slender retainers) are inactivated. At least one kinase, TbDYRK, acts on both control arms, inhibiting slender retainers and promoting stumpy formation. Other molecules, annotated as “Hypothetical proteins” in TryTrypdb (https://tritrypdb.Org/tritrypdb/app) have been identified that control stumpy formation but their positions in the regulatory pathway are unknown.
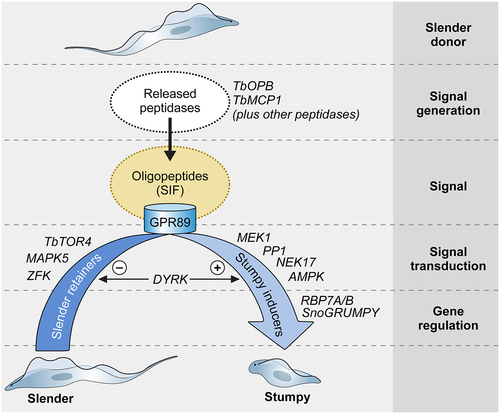
The presence of oligopeptidases activates a developmental signaling response. The signal is transported by a surface molecule, TbGPR89, specific to slender cells as the signal-receiving population. Interestingly, not all oligopeptides operate equally effectively, with tripeptides being more active than dipeptides, and with particular amino acid combinations being more effective than others [Citation226] - suggesting a specificity code. The quorum sensing signaling pathway has many components, originally identified via a genome-wide RNAi screen designed to isolate parasites unresponsive to a cell permeable mimic of the quorum sensing signal [Citation234]. These molecules include protein phosphatases and protein kinases as well as RNA binding proteins acting as predicted gene regulators and, more recently, a long non-coding RNA regulator [Citation235]. Several hypothetical proteins of unknown function are also implicated [Citation236]. In combination, these components drive stumpy formation, although an analysis of their respective dependency relationships indicated that the signal transduction pathway was not a simple linear hierarchy [Citation237]. Perhaps more than one signal input contributes to ensure appropriate activation of the developmental response, or perhaps there is regulatory input or feedback from other molecular components, including those not yet uncovered? Molecular inhibitors of stumpy formation have also been identified [Citation238–241], as has at least one molecule that seems to act on both stimulatory and inhibitory arms of the process [Citation242]. This reflects the complexity and stringent regulation of quorum sensing, which is necessary because stumpy formation represents a terminal developmental step unless the parasites are transmitted to tsetse.
The gene expression response to the quorum sensing signal has been analyzed by single-cell RNA sequencing [Citation243]. This identified the trajectory of the transition in terms of gene expression from the slender to the stumpy forms. In particular, parasites were observed to transition in the G1 phase of their cell cycle with no “intermediate form” transcriptome identified, despite the description of these morphologically transitional forms [Citation223]. Analysis of a parasite line defective in quorum sensing through its deletion of a component of the QS signaling pathway identified early transcript-level changes in gene expression as parasite initiate the developmental QS response [Citation243], providing a route to pinpoint the molecular commitment events that define the initiation of the decision to progress toward stumpy formation.
The generation of stumpy forms is a unique innovation to T. brucei with limited evidence for morphological development in either T. congolense or T. vivax. Nonetheless, both these species exhibit density-dependent growth arrest, accumulating as G1-arrested forms at higher parastiaemias [Citation244,Citation245]. In T. congolense, gene expression changes that accompany this arrest have been analyzed, which predicts changes in the expression of some surface proteins [Citation246]. The genomes of both T. congolense and T. vivax also encode orthologues of many of the regulators of quorum sensing identified in T. brucei, and at least one of these (TcIL3000.0.19510) can complement a T. brucei null mutant for TbHYP2 (Tb927.9.4080) to restore stumpy formation, demonstrating functional equivalence [Citation245]. Thus, despite the absence of morphological development, it appears both T. congolense and T. vivax exhibit quorum sensing to regulate their virulence in mammalian hosts and, potentially, as an adaptation for tsetse uptake.
Secreted factors and EVs
The importance of released peptidases and their role in generating the signal that promotes the development transition to stumpy forms has reemphasized that trypanosomes are not passive entities in their hosts but instead behave interactively to support their survival and transmission and, potentially, to contribute to virulence.
Several studies have analyzed the secretome of bloodstream form trypanosomes. Secreted proteases, and T. brucei Cathepsin-L (TbCatL) in particular, have been implicated mediating extravasation of T. brucei cells via perturbation of intracellular calcium levels in brain endothelial cells [Citation247], and secreted TbCatL has also been shown to induce spontaneous depolarization events in isolated cardiomyocytes, also via perturbation of intracellular calcium levels [Citation248], which may contribute to cardiac pathology observed in human and animal infections. Secreted peptidases have been proposed to be involved in trypanosome pathogenicity by hydrolyzing host hormone peptides, hence affecting their functions [Citation24950Citation250, . Other studies have also described peptidases in the blood of infected mice and rodents where they remained catalytically active [Citation251–253]. Various studies have looked at these secreted or released peptidases and their substrates both in vitro and in vivo. For example, oligopeptidase B (OPB), which cleaves Arg/Lys containing peptides smaller than 30 amino acid residues, is released into the bloodstream of rats infected with trypanosomes. This cleaves available host hormones such as atrial natriuretic factor resulting in hormonal deregulation linked with trypanosome infections [Citation249,Citation253]. Similarly, type 1 pyroglutamyl peptidase (PGP) and prolyl oligopeptidase (POP) are also released into the blood of rats during trypanosome infections and remain catalytically active. PGP cleaves host gonadotropin-releasing hormone (GnRH) and thyrotropin-releasing hormone (TRH) by removing the N-terminal pyroglutamic acid-blocking groups [Citation252] while POP, which hydrolyses Pro/Ala containing substrates at the carboxyl end, hydrolyses type 1 collagen and native collagen [Citation251]. POP has also been demonstrated to hydrolyze substance P, oxytocin, vasopressin, and angiotensin in vitro [Citation251]. In all, the expression of peptidases, their secretion/release into the host bloodstream and their activity in the host plasma have been reported in African trypanosomes, with interest in these molecules enhanced with the recent discovery of their involvement in the generation of the quorum-sensing signal for stumpy differentiation [Citation227,Citation228].
Numerous studies have looked at the secretome of the different species of trypanosomes, including in the different life cycle forms [Citation160,Citation161,Citation254,Citation255–258]. These studies have identified proteins that are involved in different functions, including folding and degradation, nucleotide, carbohydrate and amino acid metabolism, protein synthesis, and transport [Citation258]. These proteins may interact with the host’s immune system, and by so doing, contribute to immunopathology and also the survival of the parasites. This was demonstrated by Garzon et al. [Citation257], where they found a reduction in the secretion of host immune molecules and an impairment in the maturation of lipopolysaccharide (LPS)-induced dendritic cells in the presence of T. b. gambiense secretome. As highlighted above, among the identified secreted proteins in previous studies were peptidases. Different classes of peptidases are expressed in trypanosome parasites; serine, cysteine, metallopeptidases, threonine, and aspartyl with peptidases from each of these classes being characterized in T. brucei.
Exosomes are a subset of extracellular vesicles and are formed from the budding of the late endosomes. They are cup-shaped, approximately 20–100 nm in diameter [Citation259], and are secreted through fusion with the plasma membrane. In a variety of pathogens, exosomal secretion has been proposed to be involved in cell-to-cell communication, cell-to-host communications, and has also been implicated in pathogenicity and cell differentiation [Citation260–262]. Protein release through exosomes has been described in trypanosomes when the secretome of the parasite was analyzed, revealing the release of leaderless proteins as well as exosome associated proteins such as Rab proteins, clathrin heavy chain, enolase, and heat-shock protein 70 [Citation254]. Extracellular vesicular secretion has also been reported in T. cruzi [Citation259], and in Leishmania it was recently demonstrated that genes encoding drug resistance can be passed between Leishmania cells via exosomes [Citation263] – highlighting the potential importance of this phenomenon for multiple phenotypes. Molecules involved in this type of secretion include the components of the ESCRT complex (Endosomal Sorting Complex Required for Transport) [Citation262,Citation264,Citation265].
Parasite-parasite interactions and coinfections
The extracellular release of factors into their environment open the possibility for coinfecting trypanosome strains and species to interact with one another through collaboration or competition, either directly or indirectly. Different trypanosomes species co-circulate in sub-Saharan Africa, with transmission by the same vector species [Citation266]. As a consequence, coinfections have been frequently reported both in livestock infection and in trapped tsetse flies [Citation267–269]. In several studies, the coinfection between different strains and species have been found to alter the infection dynamics and/or virulence in the host [Citation57]. This was exemplified in concomitant infections of livestock with virulent and less virulent strains of T congolense in livestock, where the presence of a less virulent strain suppressed the pathology associated with a more virulent strain [Citation270]. This was not linked to shared antigens or immune cross reactivity between the parasites in the infection, since removing one of the strains with trypanocides before inoculating the second strain eliminated the response. Competitive suppression has also been observed between strains of T. brucei in rodent infections, with a more virulent strain being suppressed by a less virulent strain, enabling extended host survival and reduced pathology [Citation271]. These phenomena are reminiscent of the interactions between closely related Theileria species in livestock, where the impact of the more pathogenic Theileria parva was ameliorated by infection of the less virulent Theileria mutans, to the extent that infection with the less virulent species was proposed as a potential anti-virulence approach to disease control, potentially more valuable than anti-parasitic drugs [Citation272]. In many cases, however, the coinfection of different parasite species generates significantly worse outcomes [Citation273,Citation274].
Interactions between strains in coinfection are also observed with respect to quorum-sensing signals. As highlighted earlier, T. congolense does not generate morphologically stumpy forms but the parasite exhibits arrest in G1 in a density-dependent manner [Citation245]. To explore whether the parasites could detect the QS signals, mice with T. congolense infections were superinfected with T. brucei and the ability of the latter to generate stumpy forms compared with mice infected with T. brucei alone. The T. brucei in the coinfection were found to accelerate their stumpy formation reflective of the overall parasite load, rather than the contribution of T. brucei to the infection alone. Furthermore, the response of the parasites was dependent on their intact QS signaling pathway since null mutants for a component of the signaling path proliferated in the coinfection setting regardless of the existing T. congolense parasites [Citation245]. Overall, this demonstrated that T. brucei responds to the presence of T. congolense and that this is mediated via a T. congolense derived QS signal, although whether there was a signal from T. brucei to T. congolense could not be explored in the absence of suitable molecular markers for transmission stages in the latter species.
These experiments established that different trypanosome strains and species can detect and respond to one another in a coinfection and that this can alter their potential for virulence and transmission. Such interactions in the field have the potential to select for evolutionary strategies that provide an advantage in a coinfection scenario for parasites that reduce their sensitivity to the QS signal, or occupy niches where they are less susceptible to manipulation by a coinfecting strain [Citation218]. As discussed elsewhere, there are also potential implications for therapeutic strategies [Citation275] or with species-specific vaccination [Citation109], since this has the potential to perturb a coinfection equilibrium to allow the emergence of parasites less sensitive to QS signals and so more likely to be virulent when in a monoinfection setting.
Future perspectives: identification of gaps, priorities, and opportunities
In this article, we have aimed to summarize current knowledge with respect to parasite factors that influence the severity of clinical signs and disease outcome in the mammalian host. The complexity of disease caused by trypanosomes, with the involvement of multiple host and multiple trypanosome species, means that this is inevitably an ambitious undertaking, and covers multiple aspects of trypanosome infection biology. However, the advances in the last decades by many researchers mean that we now have an impressively detailed understanding of trypanosome factors that influence host-parasite interactions. This is particularly true for T. brucei, and for infections in mice, where the understanding is particularly advanced – the detailed knowledge we now have of the differentiation process from long slender to stumpy life cycle stages being a prime example. In recent years, efforts in T. brucei have also begun to significantly shift toward working with pleomorphic, differentiation-competent strains (e.g. T. brucei EATRO 1125 AnTat 1.1) and away from the heavily laboratory-adapted monomorphic Lister 427, a move that will provide data more relevant to field infections. However, the science reviewed in this article highlights that we have identified relatively few virulence factors responsible for even well characterized phenotypes (see ), and has also highlighted areas where knowledge in general is much less developed. These gaps in understanding represent opportunities for the trypanosome research community going forward, and in the following section we aim to outline research needs and opportunities.
Table 1. Virulence phenotypes and state of current knowledge.
While the ability to work with T. brucei advances at a spectacular pace, with examples such as precise and scalable gene silencing and editing techniques that facilitate high throughput phenotypic screens and highly detailed functional analysis [Citation234,Citation279–281], the rate of progress in terms of advancement in knowledge, data, and capabilities lags significantly behind for T. congolense and T. vivax. Partly, this is due to the sheer difference in scale in terms of amount of research investment into these pathogens, as evidenced by the stark disparity in the number of research outputs on T. congolense and T. vivax compared to T. brucei over the past decades [Citation282]. However, there has been a refocusing on T. congolense in recent years that has seen the generation of foundational datasets and capabilities [Citation40,Citation44,Citation72,Citation133,Citation283–285], and combined with renewed interest from funders in AT, this is contributing to a revival of research on this organism. Although not to the same extent, there are also tools and resources available for T. vivax that enable work on this pathogen that was not previously possible [Citation45,Citation88,Citation94], which is already leading to notable, paradigm-challenging studies [Citation109]. However, there are key basic capabilities where improvement would be transformative. The ability to culture bloodstream form T. congolense and T. vivax in vitro is still limited, with a very small number of strains for T. congolense having been successfully adapted to in vitro culture, and only one for T. vivax – with a requirement in T. vivax for mammalian feeder cells for mid- to long-term culture. Development of media formulations that supported axenic growth of multiple strains for both species would be a substantial breakthrough that would accelerate meaningful functional studies, and provide the ability to dissect the genetic diversity present in both species. Additionally, while genetic modification is clearly possible in both species [Citation94,Citation283], in T. vivax this is currently restricted to the epimastigote life cycle stage, and requires differentiation through metacyclics to bloodstream forms to obtain the relevant mammalian life cycle stage. The ability to directly genetically modify T. vivax bloodstream form cells would be a major step forward, albeit this may depend on the prior development of an appropriate in vitro culturing medium. It is clear that the multiple fundamental differences between these three species mean that if such basic capabilities are improved, there are many opportunities for identifying novel and important aspects of trypanosome cell and infection biology.
As is evident from many of the advances outlined in this article, the mouse model has been and remains hugely influential and useful in providing key insights into the infection biology of African trypanosomes. The mouse model provides experimental tractability and scalability that can make it an incredibly powerful experimental tool. However, ultimately for most traits there is a need or desire to assess translation to the clinically relevant host model. It should be stressed that this is not always simply a matter of assessing clinical relevance, but that it can also be because analyzing a trait in such hosts provides genuine scientific insight and interest. While this is clearly a challenge with the human disease in terms of both ethical reasons and the small and reducing number of clinical cases (although the extravascular skin populations of T. brucei are a recent example of laboratory observations proving important and useful in HAT [Citation128,Citation129]), for AT there is substantial scope for analysis in clinically relevant hosts such as cattle. Several facilities are now available that enable experimental infection of cattle, and there are multiple phenotypes observed either in vitro or in the mouse model that it would be of significant interest to translate to the bovine model – whether for reasons of host size and scale, physiology, bovine-specific aspects of the immune response, or to demonstrate potential clinical relevance. Example phenotypes where the interest in this translation to the cow are obvious include antigenic variation (where modelling has indicated host size is likely to play a substantive role in VSG-population dynamics [Citation286]), parasite-mediated B cell memory loss [Citation115], cell adherence and tissue-specific sequestration [Citation133], coinfection and interaction between trypanosome species [Citation245], and the validation of vaccine candidates [Citation109]. However, this is to name but a few phenotypes where analysis in the bovine host would prove informative; it is abundantly clear that many aspects of our understanding of trypanosome infection biology would greatly benefit from assessment in the bovine or other clinically relevant host species.
Similar to increased research on the clinically relevant hosts, there is equally a lot of value to be gained from more assessment of the extent to which laboratory findings are recapitulated in the field. Likewise, findings in the field also have the obvious potential to stimulate meaningful laboratory and experimental work, and there could perhaps be better integration of laboratory and field approaches across many aspects of trypanosome biology. The tissue distribution of trypanosomes in the skin and adipose, and the findings that have resulted from the original studies describing these phenomena [Citation129,Citation130], is a prime example of the mutually beneficial advances that can be gained across basic and applied approaches from combining field and laboratory experimental work. But as noted above, there are multiple aspects of trypanosome biology where integration of field approaches with laboratory approaches would enhance our understanding – for example, the increasing interest in coinfections, where experimental approaches provide the ability to control for multiple confounders and gain mechanistic insights, but properly designed field approaches enable the deconvolution of the interaction of trypanosomes (or mechanism) with host, pathogen, and environmental factors.
There also remain some fundamental gaps in our field knowledge of trypanosome virulence. For example, almost all T. congolense studies (laboratory or field) focus on T. congolense Savannah. We know astonishingly little about the other subtypes of T. congolense, Forest and Kilifi, other than that they are detected in cattle and wildlife across sub-Saharan Africa, a very few assessments of relative virulence of selected strains in cattle and mice, and the generation of a limited amount of genome data (for Forest). But the extent of the disease caused in cattle and other livestock attributed to these subtypes, and the intricacies of their epidemiology (e.g. tsetse transmission, interaction with other trypanosome species and other subtypes of T. congolense) are questions that are ripe for answering – similar questions pertain to several of the understudied trypanosomes, such as T. simiae, T. suis and T. vivax-like. This lack of knowledge feeds into some of the capability challenges outlined above (e.g. limited capacity for in vitro culture), but is likely also exacerbated by confirmation bias in many field study designs (i.e. targeted surveillance will only detect what is tested for). Improved diagnostics, and in particular the increasing power of sequence capture or enrichment approaches, should enable a better disentangling of such questions.
In summary, reflecting the complexity of disease caused by multiple and genetically distinct trypanosome species that infect a diversity of host species, virulence in trypanosomes is complicated and multifactorial. While we have made substantial progress in some areas and have a good understanding of virulence factors and how they exert their effect, particularly with respect to T. brucei, there remain substantial gaps in knowledge. However, these represent opportunities going forward to further our understanding of how these fascinating and important pathogens interact with the mammalian host and cause disease. This has the potential to reveal yet more scientifically novel aspects of trypanosome biology, as well as lead to the design of novel interventions that are still sorely lacking for both human and animal trypanosomiasis.
Acknowledgements
LM and PS received support from the UK Biotechnology and Biological Sciences Research Council (BBSRC; BB/W000296/1; BB/S00243X/1), and the Roslin Institute was supported through core funding from the BBSRC (BS/E/D/20002173). KM received support from the Wellcome Trust (221717/Z/20/Z). KM and LM received support through a collaborative award from the Wellcome Trust (206815/Z/17/Z). MT was supported through a PhD studentship from the Darwin Trust of Edinburgh.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Giordani F, Morrison LJ, Rowan TG, et al. The animal trypanosomiases and their chemotherapy: a review. Parasitology. 2016;143(14):1862–29.
- Shaw AP, Wint GR, Cecchi G, et al. Mapping the benefit-cost ratios of interventions against bovine trypanosomosis in Eastern Africa. Prev Vet Med. 2015;122(4):406–416.
- Swallow BM. Impacts of trypanosomiasis on African agriculture. PAAT Technical and Scientific Series (FAO). 1999.
- Franco JR, Cecchi G, Paone M, et al. The elimination of human African trypanosomiasis: achievements in relation to WHO road map targets for 2020. PLoS Negl Trop Dis. 2022;16(1):e0010047.
- Auty H, Morrison LJ, Torr SJ, et al. Transmission dynamics of rhodesian sleeping sickness at the interface of wildlife and livestock areas. Trends Parasitol. 2016;32(8):608–621.
- Mugnier MR, Cross GA, Papavasiliou FN. The in vivo dynamics of antigenic variation in Trypanosoma brucei. Science. 2015;347(6229):1470–1473.
- Jamonneau V, Truc P, Grebaut P, et al. Trypanosoma brucei gambiense Group 2: the Unusual Suspect. Trends Parasitol. 2019;35(12):983–995.
- Weir W, Capewell P, Foth B, et al. Population genomics reveals the origin and asexual evolution of human infective trypanosomes. Elife. 2016;5:e11473.
- Koffi M, De Meeus T, Bucheton B, et al. Population genetics of Trypanosoma brucei gambiense, the agent of sleeping sickness in Western Africa. Proc Natl Acad Sci U S A. 2009;106(1):209–214.
- Morrison LJ, Tait A, McCormack G, et al. Trypanosoma brucei gambiense Type 1 populations from human patients are clonal and display geographical genetic differentiation. Infect Genet Evol. 2008;8(6):847–854.
- Balmer O, Beadell JS, Gibson W, et al. Phylogeography and taxonomy of Trypanosoma brucei. PLoS Negl Trop Dis. 2011;5(2):e961.
- Capewell P, Cooper A, Duffy CW, et al. Human and animal Trypanosomes in Côte d’Ivoire form a single breeding population. PLoS One. 2013b;8(7):e67852.
- Cooper A, Tait A, Sweeney L, et al. Genetic analysis of the human infective trypanosome Trypanosoma brucei gambiense: chromosomal segregation, crossing over, and the construction of a genetic map. Genome Biol. 2008;9(6):R103.
- Duffy CW, MacLean L, Sweeney L, et al. Population genetics of Trypanosoma brucei rhodesiense: clonality and diversity within and between foci. PLoS Negl Trop Dis. 2013;7(11):e2526.
- Gibson W, Garside L, Bailey M. Trisomy and chromosome size changes in hybrid trypanosomes from a genetic cross between Trypanosoma brucei rhodesiense and T. b. brucei. Mol Biochem Parasitol. 1992;51(2):189–199.
- Kennedy PG. Clinical features, diagnosis, and treatment of human African trypanosomiasis (sleeping sickness). Lancet Neurol. 2013;12(2):186–194.
- Sudarshi D, Lawrence S, Pickrell WO, et al. Human African trypanosomiasis presenting at least 29 years after infection—what can this teach us about the pathogenesis and control of this neglected tropical disease? PLoS Negl Trop Dis. 2014;8(12):e3349.
- Hoare CA. The trypanosomes of mammals. A zoological monograph. Oxford, UK: Blackwell Scientific Publications; 1972.
- Kennedy PGE, Rodgers J. Clinical and neuropathogenetic aspects of human African Trypanosomiasis. Front Immunol. 2019;10:39.
- Sudarshi D, Brown M. Human African trypanosomiasis in non-endemic countries. Clin Med. 2015;15(1):70–73.
- Jamonneau V, Ilboudo H, Kabore J, et al. Untreated human infections by Trypanosoma brucei gambiense are not 100% fatal. PLoS Negl Trop Dis. 2012;6(6):e1691.
- Ilboudo H, Bras-Goncalves R, Camara M, et al. Unravelling human trypanotolerance: iL8 is associated with infection control whereas IL10 and TNFα are associated with subsequent disease development. PLoS Pathog. 2014;10(11):e1004469.
- Kabore JW, Camara O, Ilboudo H, et al. Macrophage migrating inhibitory factor expression is associated with Trypanosoma brucei gambiense infection and is controlled by trans-acting expression quantitative trait loci in the Guinean population. Infect Genet Evol. 2019;71:108–115.
- Cooper A, Ilboudo H, Alibu VP, et al. APOL1 renal risk variants have contrasting resistance and susceptibility associations with African trypanosomiasis. Elife. 2017;6:6.
- Kabore JW, Ilboudo H, Noyes H, et al. Candidate gene polymorphisms study between human African trypanosomiasis clinical phenotypes in Guinea. PLoS Negl Trop Dis. 2017;11(8):e0005833.
- Kabore J, Koffi M, Bucheton B, et al. First evidence that parasite infecting apparent aparasitemic serological suspects in human African trypanosomiasis are Trypanosoma brucei gambiense and are similar to those found in patients. Infect Genet Evol. 2011;11(6):1250–1255.
- Kabore J, Camara O, Koffi M, et al. Differences in pathogenicity and virulence of Trypanosoma brucei gambiense field isolates in experimentally infected Balb/C mice. Infect Genet Evol. 2018;63:269–276.
- Muller LSM, Cosentino RO, Forstner KU, et al. Genome organization and DNA accessibility control antigenic variation in trypanosomes. Nature. 2018;563(7729):121–125.
- Xong HV, Vanhamme L, Chamekh M, et al. A VSG Expression Site–Associated Gene Confers Resistance to Human Serum in Trypanosoma rhodesiense. Cell. 1998;95(6):839–846.
- MacLeod A, Tait A, Turner CM. The population genetics of Trypanosoma brucei and the origin of human infectivity. Philos Trans R Soc Lond B Biol Sci. 2001;356(1411):1035–1044.
- Sternberg JM, Maclean L. A spectrum of disease in human African trypanosomiasis: the host and parasite genetics of virulence. Parasitology. 2010;137(14):2007–2015.
- MacLean LM, Odiit M, Chisi JE, et al. Focus–specific clinical profiles in human African trypanosomiasis caused by Trypanosoma brucei rhodesiense. PLoS Negl Trop Dis. 2010;4(12):e906.
- MacLean L, Odiit M, MacLeod A, et al. Spatially and genetically distinct African trypanosome virulence variants defined by host interferon-γ response. J Infect Dis. 2007;196(11):1620–1628.
- Smith DH, Bailey JW. Human African trypanosomiasis in south-eastern Uganda: clinical diversity and isoenzyme profiles. Ann Trop Med Parasitol. 1997;91(7):851–856.
- Goodhead I, Capewell P, Bailey JW, et al. Whole-genome sequencing of Trypanosoma brucei reveals introgression between subspecies that is associated with virulence. MBio. 2013;4(4). DOI:10.1128/mBio.00197-13
- Morrison LJ, McLellan S, Sweeney L, et al. Role for parasite genetic diversity in differential host responses to Trypanosoma brucei infection. Infect Immun. 2010;78(3):1096–1108.
- Morrison LJ, Tait A, McLellan S, et al. A major genetic locus in Trypanosoma brucei is a determinant of host pathology. PLoS Negl Trop Dis. 2009b;3:e557.
- De Muylder G, Daulouede S, Lecordier L, et al. A Trypanosoma brucei kinesin chain promotes parasite growth by triggering host arginase activity. PLoS Pathog. 2013;9(10):e1003731.
- Auty H, Torr SJ, Michoel T, et al. Cattle Trypanosomosis: the diversity of trypanosomes and implications for disease epidemiology and control. Rev Sci Tech. 2015;34(2):587–598.
- Steketee PC, Dickie EA, Iremonger J, et al. Divergent metabolism between Trypanosoma congolense and Trypanosoma brucei results in differential sensitivity to metabolic inhibition. PLoS Pathog. 2021;17(7):e1009734.
- Jackson AP, Allison HC, Barry JD, et al. A cell-surface phylome for African trypanosomes. PLoS Negl Trop Dis. 2013;7(3):e2121.
- Jackson AP, Berry A, Aslett M, et al. Antigenic diversity is generated by distinct evolutionary mechanisms in African trypanosome species. Proc Natl Acad Sci U S A. 2012;109(9):3416–3421.
- McCulloch R, Cobbold CA, Figueiredo L, et al. Emerging challenges in understanding trypanosome antigenic variation. Emerg Top Life Sci. 2017;1(6):585–592.
- Silva Pereira S, Casas-Sanchez A, Haines LR, et al. Variant antigen repertoires in Trypanosoma congolense populations and experimental infections can be profiled from deep sequence data using universal protein motifs. Genome Res. 2018;28(9):1383–1394.
- Silva Pereira S, de Almeida Castilho Neto KJG, Duffy CW, et al. Variant antigen diversity in Trypanosoma vivax is not driven by recombination. Nat Commun. 2020;11(1):844.
- Taylor K, Authie E. Pathogenesis of Animal Trypanosomiasis. In: Maudlin PHH MA Miles, editors. The trypanosomiases. Vol. I. London: CAB International; 2004.
- Connor RJ, Van den Bossche P. African animal trypanosomoses. In: Coetzer JAW RC Tustin, editors. Infectious diseases of livestock. Vol. 1. Cape Town: Oxford University Press Southern Africa; 2004. pp. 251–296.
- Grace D, Himstedt H, Sidibe I, et al. Comparing FAMACHA© eye color chart and Hemoglobin Color Scale tests for detecting anemia and improving treatment of bovine trypanosomosis in West Africa. Vet Parasitol. 2007;147(1–2):26–39.
- Anene BM, Chime AB, Anika SM. The production performance of imported Friesian cattle under heavy Trypanosoma challenge in a rain forest zone of Nigeria. Br Vet J. 1991;147(3):275–282.
- Gardiner PR. Recent studies of the biology of Trypanosoma vivax. Adv Parasitol. 1989;28:229–317.
- Taye M, Belihu K, Bekana M, et al. Assessment of impacts of tsetse and trypanosomosis control measures on cattle herd composition and performance in southern region, Ethiopia. Trop Anim Health Prod. 2012;44(7):1759–1763.
- Faye D, Sulon J, Kane Y, et al. Effects of an experimental Trypanosoma congolense infection on the reproductive performance of West African Dwarf goats. Theriogenology. 2004;62(8):1438–1451.
- Okech G, Dolan RB, Stevenson P, et al. The effect of trypanosomosis on pregnancy in trypanotolerant Orma Boran cattle. Theriogenology. 1996;46(3):441–447.
- Batista JS, Riet-Correa F, Teixeira MM, et al. Trypanosomiasis by Trypanosoma vivax in cattle in the Brazilian semiarid: description of an outbreak and lesions in the nervous system. Vet Parasitol. 2007;143(2):174–181.
- Galiza GJ, Garcia HA, Assis AC, et al. High mortality and lesions of the central nervous system in trypanosomosis by Trypanosoma vivax in Brazilian hair sheep. Vet Parasitol. 2011;182(2–4):359–363.
- Faye D, Pereira de Almeida PJ, Goossens B, et al. Prevalence and incidence of trypanosomosis in horses and donkeys in the Gambia. Vet Parasitol. 2001;101(2):101–114.
- Pinchbeck GL, Morrison LJ, Tait A, et al. Trypanosomosis in the Gambia: prevalence in working horses and donkeys detected by whole genome amplification and PCR, and evidence for interactions between trypanosome species. BMC Vet Res. 2008;4(1):7.
- Raftery AG, Jallow S, Coultous RM, et al. Variation in disease phenotype is marked in equine trypanosomiasis. Parasit Vectors. 2020;13(1):148.
- Rodrigues A, Fighera R, Souza T, et al. Neuropathology of naturally occurring Trypanosoma evansi infection of horses. Vet Pathol; 2009.
- Savage VL, Christley R, Pinchbeck G, et al. Co-infection with Trypanosoma congolense and Trypanosoma brucei is a significant risk factor for cerebral trypanosomosis in the equid population of the Gambia. Prev Vet Med. 2021;197:105507.
- Garcia HA, Ramirez OJ, Rodrigues CM, et al. Trypanosoma vivax in water buffalo of the Venezuelan Llanos: an unusual outbreak of wasting disease in an endemic area of typically asymptomatic infections. Vet Parasitol. 2016;230:49–55.
- Hutchinson R, Gibson W. Rediscovery of Trypanosoma (Pycnomonas) suis, a tsetse-transmitted trypanosome closely related to T. brucei. Infect Genet Evol. 2015;36:381–388.
- McNamara JJ, Mohammed G, Gibson WC. Trypanosoma (Nannomonas) godfreyi sp. nov. from tsetse flies in the Gambia: biological and biochemical characterization. Parasitology. 1994;109(4):497–509.
- Peel E, Chardome M. Trypanosoma suis, Ochmann 1905, monomorphic trypanosome pathogenic to mammals, occurring in the salivary glands of Glossina brevipalpis Newst., Mosso, (Urundi). Ann Soc Belg Med Trop (1920). 1954;34:277–295.
- Stevens JR, Brisse S. Systematics of Trypanosomes of Medical and Veterinary Importance. In: Maudlin PHH MA Miles, editors. The Trypanosomiases. Vol. I. Cambridge, UK: CABI Publishing; 2004. pp. 1–24.
- Auty H, Anderson NE, Picozzi K, et al. Trypanosome diversity in wildlife species from the serengeti and Luangwa Valley ecosystems. PLoS Negl Trop Dis. 2012a;6(10):e1828.
- Rodrigues CM, Garcia HA, Rodrigues AC, et al. New insights from Gorongosa National Park and Niassa National Reserve of Mozambique increasing the genetic diversity of Trypanosoma vivax and Trypanosoma vivax-like in tsetse flies, wild ungulates and livestock from East Africa. Parasit Vect. 2017;10(1):337.
- Taylor KA. Immune responses of cattle to African trypanosomes: protective or pathogenic? Int J Parasitol. 1998;28(2):219–240.
- Gibson W. Resolution of the species problem in African trypanosomes. Int J Parasitol. 2007;37(8–9):829–838.
- Holzmuller P, Herder S, Cuny G, et al. From clonal to sexual: a step in T. congolense evolution? Trends Parasitol. 2010;26(2):56–60.
- Morrison LJ, Tweedie A, Black A, et al. Discovery of mating in the major African livestock pathogen Trypanosoma congolense. PLoS One. 2009c;4(5):e5564.
- Tihon E, Imamura H, Dujardin JC, et al. Discovery and genomic analyses of hybridization between divergent lineages of Trypanosoma congolense, causative agent of Animal African Trypanosomiasis. Mol Ecol. 2017;26(23):6524–6538.
- Bengaly Z, Sidibe I, Ganaba R, et al. Comparative pathogenicity of three genetically distinct types of Trypanosoma congolense in cattle: clinical observations and haematological changes. Vet Parasitol. 2002b;108(1):1–19.
- Bengaly Z, Sidibe I, Boly H, et al. Comparative pathogenicity of three genetically distinct Trypanosoma congolense-types in inbred Balb/c mice. Vet Parasitol. 2002a;105(2):111–118.
- Masumu J, Marcotty T, Geysen D, et al. Comparison of the virulence of Trypanosoma congolense strains isolated from cattle in a trypanosomiasis endemic area of eastern Zambia. Int J Parasitol. 2006;36(4):497–501.
- Chiweshe SM, Steketee PC, Jayaraman S, et al. Parasite specific 7SL-derived small RNA is an effective target for diagnosis of active trypanosomiasis infection. PLoS Negl Trop Dis. 2019;13(2):e0007189.
- Contreras Garcia M, Walshe E, Steketee PC, et al. Comparative sensitivity and specificity of the 7SL sRNA diagnostic test for animal trypanosomiasis. Front Vet Sci. 2022;9:868912.
- Mamoudou A, Zoli A, Tanenbe C, et al. 2006. Evaluation sur le terrain et sur souris de la resistance des trypanosomes des bovins du plateau de l’Adamaoua au Cameroun a l’acaturate de diminazene et au chlorure d’isometamidium. Revue Elev Med Vet Pays Trop. 59:11–16.
- Fasogbon AI, Knowles G, Gardiner PR. A comparison of the isoenzymes of Trypanosoma (Duttonella) vivax isolates from East and West Africa. Int J Parasitol. 1990;20(3):389–394.
- Nakayima J, Nakao R, Alhassan A, et al. Genetic diversity among Trypanosoma (Duttonella) vivax strains from Zambia and Ghana, based on cathepsin L-like gene. Parasite. 2013;20:24.
- Cortez AP, Rodrigues AC, Garcia HA, et al. Cathepsin L-like genes of Trypanosoma vivax from Africa and South America – characterization, relationships and diagnostic implications. Mol Cell Probes. 2009;23(1):44–51.
- Cortez AP, Ventura RM, Rodrigues AC, et al. The taxonomic and phylogenetic relationships of Trypanosoma vivax from South America and Africa. Parasitology. 2006;133(02):159–169.
- Garcia HA, Rodrigues AC, Rodrigues CM, et al. Microsatellite analysis supports clonal propagation and reduced divergence of Trypanosoma vivax from asymptomatic to fatally infected livestock in South America compared to West Africa. Parasit Vectors. 2014;7(1):210.
- Vos GJ, Gardiner PR. Antigenic relatedness of stocks and clones of Trypanosoma vivax from east and west Africa. Parasitology. 1990;100(1):101–106.
- Duffy CW, Morrison LJ, Black A, et al. Trypanosoma vivax displays a clonal population structure. Int J Parasitol. 2009;39(13):1475–1483.
- Barry JD. Antigenic variation during Trypanosoma vivax infections of different host species. Parasitology. 1986;92(1):51–65.
- Uzoigwe NR. Self-cure in zebu calves experimentally infected with Trypanosoma vivax. Vet Parasitol. 1986;22(1–2):141–146.
- Jackson AP, Goyard S, Xia D, et al. Global gene expression profiling through the complete life cycle of Trypanosoma vivax. PLoS Negl Trop Dis. 2015;9(8):e0003975.
- Dirie MF, Wallbanks KR, Molyneux DH, et al. Haemorrhagic syndrome associated with T. vivax infections of cattle in Somalia. Acta Trop. 1988 45(3):291–292.
- Gardiner PR, Assoku RK, Whitelaw DD, et al. Haemorrhagic lesions resulting from Trypanosoma vivax infection in Ayrshire cattle. Vet Parasitol. 1989;31(3–4):187–197.
- Magona JW, Walubengo J, Odimin JT. Acute haemorrhagic syndrome of bovine trypanosomosis in Uganda. Acta Trop. 2008;107(2):186–191.
- Wellde BT, Chumo DA, Adoyo M, et al. Sindrome hemorragico En Bovinos Asociado Con La Infeccion PorTrypanosoma vivax. Trop Anim Health Prod. 1983;15(2):95–102.
- Assoku RK, Gardiner PR. Detection of antibodies to platelets and erythrocytes during infection with haemorrhage-causing Trypanosoma vivax in Ayrshire cattle. Vet Parasitol. 1989;31(3–4):199–216.
- D’Archivio S, Medina M, Cosson A, et al. Genetic engineering of Trypanosoma (Dutonella) vivax and in vitro differentiation under axenic conditions. PLoS Negl Trop Dis. 2011;5(12):e1461.
- Morrison LJ, Vezza L, Rowan T, et al. Animal African trypanosomiasis: time to increase focus on clinically relevant parasite and host species. Trends Parasitol. 2016;32(8):599–607.
- Radwanska M, Vereecke N, Deleeuw V, et al. Salivarian trypanosomosis: a review of parasites involved, their global distribution and their interaction with the innate and adaptive mammalian host immune system. Front Immunol. 2018;9:2253.
- Sileghem MJ, Flynn JN, Darji A, et al. African Trypanosomiasis. In: Kierszenbaum F, editor. Parasitic infections and the immune system. London: Academic Press; 1994. (pp. 1-51).
- Stijlemans B, Guilliams M, Raes G, et al. African trypanosomosis: from immune escape and immunopathology to immune intervention. Vet Parasitol. 2007;148(1):3–13.
- Horn D. Antigenic variation in African trypanosomes. Mol Biochem Parasitol. 2014;195(2):123–129.
- Morrison LJ, Marcello L, McCulloch R. Antigenic variation in the African trypanosome: molecular mechanisms and phenotypic complexity. Cell Microbiol. 2009a;11(12):1724–1734.
- Silva Pereira S, Jackson AP, Figueiredo LM. Evolution of the variant surface glycoprotein family in African trypanosomes. Trends Parasitol. 2022b;38(1):23–36.
- Engstler M, Pfohl T, Herminghaus S, et al. Hydrodynamic flow-mediated protein sorting on the cell surface of trypanosomes. Cell. 2007;131(3):505–515.
- Bargul JL, Jung J, McOdimba FA, et al. Species-specific adaptations of trypanosome morphology and motility to the mammalian host. PLoS Pathog. 2016;12(2):e1005448.
- Schwede A, Macleod OJ, MacGregor P, et al. How does the VSG coat of bloodstream form African trypanosomes interact with external proteins? PLoS Pathog. 2015;11(12):e1005259.
- Bartossek T, Jones NG, Schafer C, et al. Structural basis for the shielding function of the dynamic trypanosome variant surface glycoprotein coat. Nat Microbiol. 2017;2(11):1523–1532.
- Lanca AS, de Sousa KP, Atouguia J, et al. Trypanosoma brucei: immunisation with plasmid DNA encoding invariant surface glycoprotein gene is able to induce partial protection in experimental African trypanosomiasis. Exp Parasitol. 2011;127(1):18–24.
- Magez S, Caljon G, Tran T, et al. Current status of vaccination against African trypanosomiasis. Parasitology. 2010;137(14):2017–2027.
- Baral TN, Magez S, Stijlemans B, et al. Experimental therapy of African trypanosomiasis with a nanobody-conjugated human trypanolytic factor. Nat Med. 2006;12(5):580–584.
- Autheman D, Crosnier C, Clare S, et al. An invariant Trypanosoma vivax vaccine antigen induces protective immunity. Nature. 2021;595(7865):96–100.
- Black SJ, Guirnalda P, Frenkel D, et al. Induction and regulation of Trypanosoma brucei VSG-specific antibody responses. Parasitology. 2010;137(14):2041–2049.
- Nguyen HTT, Guevarra RB, Magez S, et al. Single-cell transcriptome profiling and the use of AID deficient mice reveal that B cell activation combined with antibody class switch recombination and somatic hypermutation do not benefit the control of experimental trypanosomosis. PLoS Pathog. 2021;17(11):e1010026.
- Baral TN, De Baetselier P, Brombacher F, et al. Control of Trypanosoma evansi infection is IgM mediated and does not require a type I inflammatory response. J Infect Dis. 2007;195(10):1513–1520.
- Barroso R, Morrison WI, Morrison LJ. Molecular dissection of the antibody response: opportunities and needs for application in cattle. Front Immunol. 2020;11:1175.
- Wang F, Ekiert DC, Ahmad I, et al. Reshaping antibody diversity. Cell. 2013;153(6):1379–1393.
- Radwanska M, Guirnalda P, De Trez C, et al. Trypanosomiasis-induced B cell apoptosis results in loss of protective anti-parasite antibody responses and abolishment of vaccine-induced memory responses. PLoS Pathog. 2008;4(5):e1000078.
- Moon S, Janssens I, Kim KH, et al. Detrimental effect of Trypanosoma brucei brucei infection on memory B cells and host ability to recall protective B-cell responses. J Infect Dis. 2022;226(3):528–540.
- Obishakin E, de Trez C, Magez S. Chronic Trypanosoma congolense infections in mice cause a sustained disruption of the B-cell homeostasis in the bone marrow and spleen. Parasite Immunol. 2014;36(5):187–198.
- Blom-Potar MC, Chamond N, Cosson A, et al. Trypanosoma vivax infections: pushing ahead with mouse models for the study of Nagana. II. Immunobiological dysfunctions. PLoS Negl Trop Dis. 2010;4(8):4.
- Chamond N, Cosson A, Blom-Potar MC, et al. Trypanosoma vivax infections: pushing ahead with mouse models for the study of Nagana. I. Parasitological, hematological and pathological parameters. PLoS Negl Trop Dis. 2010;4(8):e792.
- Morrison WI, Murray M, Akol GWO. Immune responses of cattle to African trypanosomes. In: Tizard I, editor. Immunology and pathogenesis of trypanosomiasis. New York: CRC Press Inc; 1985. (pp.103-131).
- Lejon V, Mumba Ngoyi D, Kestens L, et al. Gambiense human african trypanosomiasis and immunological memory: effect on phenotypic lymphocyte profiles and humoral immunity. PLoS Pathog. 2014;10(3):e1003947.
- Frenkel D, Zhang F, Guirnalda P, et al. Trypanosoma brucei co-opts NK cells to kill Splenic B2 B Cells. PLoS Pathog. 2016;12(7):e1005733.
- Holland WG, My LN, Dung TV, et al. The influence of T. evansi infection on the immuno-responsiveness of experimentally infected water buffaloes. Vet Parasitol. 2001;102(3):225–234.
- Ilemobade AA, Adegboye DS, Onoviran O, et al. Immunodepressive effects of trypanosomal infection in cattle immunized against contagious bovine pleuropneumonia. Parasite Immunol. 1982;4(4):273–282.
- Mwangi DM, Munyua WK, Nyaga PN. Immunodepression En Tripanosomiasis Caprina: Efecto De La Infeccion Aguda ConTrypanosoma congolense Sobre La Respuesta De Anticuerpos A La Vacuna De Esporos De Antrax. Trop Anim Health Prod. 1990;22(2):95–100.
- Rurangirwa FR, Musoke AJ, Nantulya VM, et al. Immune depression in bovine trypanosomiasis: effects of acute and chronic Trypanosoma congolense and chronic Trypanosoma vivax infections on antibody response to Brucella abortus vaccine. Parasite Immunol. 1983;5(3):267–276.
- Sharpe RT, Langley AM, Mowat GN, et al. Immunosuppression in bovine trypanosomiasis: response of cattle infected with Trypanosoma congolense to foot-and-mouth disease vaccination and subsequent live virus challenge. Res Vet Sci. 1982;32(3):289–293.
- Camara M, Soumah AM, Ilboudo H, et al. Extravascular Dermal Trypanosomes in Suspected and Confirmed Cases of gambiense Human African Trypanosomiasis. Clin Infect Dis. 2021;73(1):12–20.
- Capewell P, Cren-Travaille C, Marchesi F, et al. The skin is a significant but overlooked anatomical reservoir for vector-borne African trypanosomes. Elife. 2016;5. DOI:10.7554/eLife.17716
- Trindade S, Rijo-Ferreira F, Carvalho T, et al. Trypanosoma brucei Parasites Occupy and Functionally Adapt to the Adipose Tissue in Mice. Cell Host Microbe. 2016;19(6):837–848.
- De Niz M, Bras D, Ouarne M, et al. Organotypic endothelial adhesion molecules are key for Trypanosoma brucei tropism and virulence. Cell Rep. 2021;36(12):109741.
- D’Archivio S, Cosson A, Medina M, et al. Non-invasive in vivo study of the Trypanosoma vivax infectious process consolidates the brain commitment in late infections. PLoS Negl Trop Dis. 2013;7(1):e1976.
- Silva Pereira S, De Niz M, Serre K, et al. Immunopathology and Trypanosoma congolense parasite sequestration cause acute cerebral trypanosomiasis. Elife. 2022a;11:11.
- Hemphill A, Ross CA. Flagellum-mediated adhesion of Trypanosoma congolense to bovine aorta endothelial cells. Parasitol Res. 1995;81(5):412–420.
- Losos GJ, Paris J, Wilson AJ, et al. Distribution of Trypanosoma congolense in tissues of cattle. Trans R Soc Trop Med Hyg. 1973;67(2):278.
- Mills JN, Valli VE, Boo KS, et al. The quantitation of Trypanosoma congolense in calves. III. A quantitative comparison of trypanosomes in jugular vein and microvasculature and tests of dispersing agents. Tropenmed Parasitol. 1980 31(3):299–312.
- Costa RVC, Abreu APM, Thome SMG, et al. Parasitological and clinical-pathological findings in twelve outbreaks of acute trypanosomiasis in dairy cattle in Rio de Janeiro state, Brazil. Vet Parasitol Reg Stud Rep. 2020;22:100466.
- Hajduk SL, Moore DR, Vasudevacharya J, et al. Lysis of Trypanosoma brucei by a toxic subspecies of human high density lipoprotein. J Biol Chem. 1989;264(9):5210–5217.
- Rifkin MR. Trypanosoma brucei: some properties of the cytotoxic reaction induced by normal human serum. Exp Parasitol. 1978;46(2):189–206.
- Rifkin MR. Trypanosoma brucei: cytotoxicity of host high-density lipoprotein is not mediated by apolipoprotein A-I. Exp Parasitol. 1991;72(2):216–218.
- Hager KM, Pierce MA, Moore DR, et al. Endocytosis of a cytotoxic human high density lipoprotein results in disruption of acidic intracellular vesicles and subsequent killing of African trypanosomes. J Cell Biol. 1994;126(1):155–167.
- Pays E, Vanhollebeke B, Uzureau P, et al. The molecular arms race between African trypanosomes and humans. Nature Rev Microbiol. 2014;12(8):575–584.
- Pays E, Vanhollebeke B, Vanhamme L, et al. The trypanolytic factor of human serum. Nature Rev Microbiol. 2006;4(6):477–486.
- Perez-Morga D, Vanhollebeke B, Paturiaux-Hanocq F, et al. Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science. 2005;309(5733):469–472.
- Vanwalleghem G, Fontaine F, Lecordier L, et al. Coupling of lysosomal and mitochondrial membrane permeabilization in trypanolysis by APOL1. Nat Commun. 2015;6(1):8078.
- De Greef C, Hamers R. The serum resistance-associated (SRA) gene of Trypanosoma brucei rhodesiense encodes a variant surface glycoprotein-like protein. Mol Biochem Parasitol. 1994;68(2):277–284.
- Genovese G, Friedman DJ, Ross MD, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. 2010;329(5993):841–845.
- Kamoto K, Noyes H, Nambala P, et al. Association of APOL1 renal disease risk alleles with Trypanosoma brucei rhodesiense infection outcomes in the northern part of Malawi. PLoS Negl Trop Dis. 2019;13(8):e0007603.
- Capewell P, Clucas C, DeJesus E, et al. The TgsGP gene is essential for resistance to human serum in Trypanosoma brucei gambiense. PLoS Pathog. 2013a;9(10):e1003686.
- Cooper A, Capewell P, Clucas C, et al. A primate APOL1 variant that kills Trypanosoma brucei gambiense. PLoS Negl Trop Dis. 2016;10(8):e0004903.
- Uzureau P, Uzureau S, Lecordier L, et al. Mechanism of Trypanosoma brucei gambiense resistance to human serum. Nature. 2013;501(7467):430–434.
- Alsford S, Currier RB, Guerra-Assuncao JA, et al. Cathepsin-L can resist lysis by human serum in Trypanosoma brucei brucei. PLoS Pathog. 2014;10(5):e1004130.
- Mabille D, Caljon G. Inflammation following trypanosome infection and persistence in the skin. Curr Opin Immunol. 2020;66:65–73.
- Kumar R, Gupta S, Bhutia WD, et al. Atypical human trypanosomosis: potentially emerging disease with lack of understanding. Zoonoses Public Health. 2022;69(4):259–276.
- Vanhollebeke B, Truc P, Poelvoorde P, et al. Human Trypanosoma evansi infection linked to a lack of apolipoprotein L-I. N Engl J Med. 2006;355(26):2752–2756.
- Van Vinh Chau N, Buu Chau L, Desquesnes M, et al. A clinical and epidemiological investigation of the first reported human infection with the zoonotic parasite Trypanosoma evansi in Southeast Asia. Clinl Infect Dis. 2016;62(8):1002–1008.
- McConville MJ, Naderer T. Metabolic pathways required for the intracellular survival of Leishmania. Annu Rev Microbiol. 2011;65(1):543–561.
- Moreira D, Rodrigues V, Abengozar M, et al. Leishmania infantum modulates host macrophage mitochondrial metabolism by hijacking the SIRT1-AMPK axis. PLoS Pathog. 2015;11(3):e1004684.
- Shah-Simpson S, Lentini G, Dumoulin PC, et al. Modulation of host central carbon metabolism and in situ glucose uptake by intracellular Trypanosoma cruzi amastigotes. PLoS Pathog. 2017;13(11):e1006747.
- Grebaut P, Chuchana P, Brizard JP, et al. Identification of total and differentially expressed excreted–secreted proteins from Trypanosoma congolense strains exhibiting different virulence and pathogenicity. Int J Parasitol. 2009;39(10):1137–1150.
- Holzmuller P, Biron DG, Courtois P, et al. Virulence and pathogenicity patterns of Trypanosoma brucei gambiense field isolates in experimentally infected mouse: differences in host immune response modulation by secretome and proteomics. Microbes Infect. 2008;10(1):79–86.
- Tizard I, Nielsen KH, Seed JR, et al. Biologically active products from African Trypanosomes. Microbiol Rev. 1978;42(4):664–681.
- McGettrick AF, Corcoran SE, Barry PJ, et al. Trypanosoma brucei metabolite indolepyruvate decreases HIF-1α and glycolysis in macrophages as a mechanism of innate immune evasion. Proc Natl Acad Sci U S A. 2016;113(48):E7778–7787.
- Gobert AP, Daulouede S, Lepoivre M, et al. L-Arginine availability modulates local nitric oxide production and parasite killing in experimental trypanosomiasis. Infect Immun. 2000;68(8):4653–4657.
- Coller SP, Mansfield JM, Paulnock DM. Glycosylinositolphosphate soluble variant surface glycoprotein inhibits IFN-γ-induced nitric oxide production via reduction in STAT1 phosphorylation in African Trypanosomiasis. J Immunol. 2003;171(3):1466–1472.
- Magez S, Stijlemans B, Radwanska M, et al. The glycosyl-inositol-phosphate and dimyristoylglycerol moieties of the glycosylphosphatidylinositol anchor of the trypanosome variant-specific surface glycoprotein are distinct macrophage-activating factors. J Immunol. 1998 160(4):1949–1956.
- Hambrey PN, Mellors A, Tizard IR. The phospholipases of pathogenic and non-pathogenic Trypanosoma species. Mol Biochem Parasitol. 1981;2(3–4):177–186.
- Hambrey PN, Tizard IR, Mellors A Accumulation of phospholipase A1 in tissue fluid of rabbits infected with Trypanosoma brucei. Tropenmed Parasitol. 1980 31(4):439–443.
- Samad A, Licht B, Stalmach ME, et al. Metabolism of phospholipids and lysophospholipids by Trypanosoma brucei. Mol Biochem Parasitol. 1988;29(2–3):159–169.
- Wang Y, Utzinger J, Saric J, et al. Global metabolic responses of mice to Trypanosoma brucei brucei infection. Proc Nat Acad Sci. 2008;105(16):6127–6132.
- El Sawalhy A, Seed JR, Hall JE, et al. Increased excretion of aromatic amino acid catabolites in animals infected with Trypanosoma brucei evansi. J Parasitol. 1998;84(3):469–473.
- Hall JE, Seed JR, Sechelski JB. Multiple alpha-keto aciduria in Microtus montanus chronically infected with Trypanosoma brucei gambiense. Comp Biochem Physiol B. 1985;82(1):73–78.
- Newport GR, Page CR, Ashman PU, et al. Alteration of free serum amino acids in voles infected with Trypanosoma brucei gambiense. J Parasitol. 1977;63(1):15–24.
- Seed JR, Hall JE, Sechelski J. Phenylalanine metabolism in Microtus montanus chronically infected with Trypanosoma brucei gambiense. Comp Biochem Physiol Part B. 1982;71(2):209–215.
- El Sawalhy A, Seed JR, el Attar H, et al. Catabolism of tryptophan by Trypanosoma evansi. J Eukaryot Microbiol. 1995;42(6):684–690.
- Hall JE, Seed JR. Increased urinary excretion of aromatic amino acid catabolites by Microtus montanus chronically infected with Trypanosoma brucei gambiense. Comp Biochem Physiol B. 1984;77(4):755–760.
- Marciano D, Llorente C, Maugeri DA, et al. Biochemical characterization of stage-specific isoforms of aspartate aminotransferases from Trypanosoma cruzi and Trypanosoma brucei. Mol Biochem Parasitol. 2008;161(1):12–20.
- Cockram PE, Dickie EA, Barrett MP, et al. Halogenated tryptophan derivatives disrupt essential transamination mechanisms in bloodstream form Trypanosoma brucei. PLoS Negl Trop Dis. 2020;14(12):e0008928.
- Diskin C, Corcoran SE, Tyrrell VJ, et al. The trypanosome-derived metabolite Indole-3-pyruvate inhibits prostaglandin production in macrophages by targeting COX2. J Immunol. 2021;207(10):2551–2560.
- Fitzgerald HK, O’Rourke SA, Desmond E, et al. The Trypanosoma brucei-derived ketoacids, indole pyruvate and hydroxyphenylpyruvate, induce HO-1 expression and suppress inflammatory responses in human dendritic cells. Antioxidants (Basel). 2022;11(1):11.
- Lamour SD, Alibu VP, Holmes E, et al. Metabolic profiling of central nervous system disease in Trypanosoma brucei rhodesiense infection. J Infect Dis. 2017;216(10):1273–1280.
- Rodgers J, Stone TW, Barrett MP, et al. Kynurenine pathway inhibition reduces central nervous system inflammation in a model of human African trypanosomiasis. Brain. 2009;132(5):1259–1267.
- Creek DJ, Mazet M, Achcar F, et al. Probing the metabolic network in bloodstream-form Trypanosoma brucei using untargeted metabolomics with stable isotope labelled glucose. PLoS Pathog. 2015;11(3):e1004689.
- Smith TK, Bringaud F, Nolan DP, et al. Metabolic reprogramming during the Trypanosoma brucei life cycle. F1000Res. 2017;6:6.
- Greig N, Wyllie S, Patterson S, et al. A comparative study of methylglyoxal metabolism in trypanosomatids. FEBS J. 2009;276(2):376–386.
- Alesandro PAD, Sherman IW. Changes in lactic dehydrogenase levels of Trypanosoma lewisi associated with appearance of ablastic immunity. Exp Parasitol. 1964;15(5):430–438.
- Darling TN, Balber AE, Blum JJ. A comparative study of D-lactate, L-lactate and glycerol formation by four species of Leishmania and by Trypanosoma lewisi and Trypanosoma brucei gambiense. Mol Biochem Parasitol. 1988;30(3):253–257.
- Herbert WJ, Mucklow MG, Lennox B. The cause of death in acute murine trypanosomiasis. Trans R Soc Trop Med Hyg. 1975;69:4.
- Hoppe JO, Chapman CW. Role of glucose in acute parasitemic death of the rat infected with Trypanosoma equiperdum. J Parasitol. 1947;33(6):509–516.
- Voorheis HP. The effect of T. brucei (S-42) on host carbohydrate metabolism: liver production and peripheral utilization of glucose. Trans R Soc Trop Med Hyg. 1969;63(1):122–123.
- Levy DJ, Goundry A, Laires RSS, et al. Role of the inhibitor of serine peptidase 2 (ISP2) of Trypanosoma brucei rhodesiense in parasite virulence and modulation of the inflammatory responses of the host. PLoS Negl Trop Dis. 2021;15(6):e0009526.
- O’Brien TC, Mackey ZB, Fetter RD, et al. A parasite cysteine protease is key to host protein degradation and iron acquisition. J Biol Chem. 2008;283(43):28934–28943.
- Saric J, Li JV, Swann JR, et al. Integrated cytokine and metabolic analysis of pathological responses to parasite exposure in rodents. J Proteome Res. 2010;9(5):2255–2264.
- Misek DE, Saltiel AR. An inositol phosphate glycan derived from a Trypanosoma brucei glycosyl-phosphatidylinositol mimics some of the metabolic actions of insulin. J Biol Chem. 1992;267(23):16266–16273.
- Kovarova J, Nagar R, Faria J, et al. Gluconeogenesis using glycerol as a substrate in bloodstream-form Trypanosoma brucei. PLoS Pathog. 2018;14(12):e1007475.
- Huet G, Lemesre JL, Grard G, et al. Serum lipid and lipoprotein abnormalities in human African trypanosomiasis. Trans R Soc Trop Med Hyg. 1990;84(6):792–794.
- Lamour SD, Gomez-Romero M, Vorkas PA, et al. Discovery of infection associated metabolic markers in human African trypanosomiasis. PLoS Negl Trop Dis. 2015;9(10):e0004200.
- Nakamura Y. Alterations of serum lipid, lipoprotein and inflammatory cytokine profiles of rabbits infected with Trypanosoma brucei brucei. Vet Parasitol. 1998;80(2):117–125.
- Assoku RK, Tizard IR, Nielsen KH. Free fatty acids, complement activation, and polyclonal B-cell stimulation as factors in the immunopathogenesis of African trypanosomiasis. Lancet. 1977;310(8045):956–959.
- Chamond N, Cosson A, Coatnoan N, et al. Proline racemases are conserved mitogens: characterization of a Trypanosoma vivax proline racemase. Mol Biochem Parasitol. 2009;165(2):170–179.
- Boada-Sucre AA, Rossi Spadafora MS, Tavares-Marques LM, et al. Trypanosoma vivax adhesion to red blood cells in experimentally infected sheep. Patholog Res Int. 2016;2016:4503214.
- Hemphill A, Frame I, Ross CA. The interaction of Trypanosoma congolense with endothelial cells. Parasitology. 1994;109(5):631–641.
- Buratai LB, Nok AJ, Ibrahim S, et al. Characterization of sialidase from bloodstream forms of Trypanosoma vivax. Cell Biochem Funct. 2006;24(1):71–77.
- Coustou V, Plazolles N, Guegan F, et al. Sialidases play a key role in infection and anaemia in Trypanosoma congolense animal trypanosomiasis. Cell Microbiol. 2012;14(3):431–445.
- Guegan F, Plazolles N, Baltz T, et al. Erythrophagocytosis of desialylated red blood cells is responsible for anaemia during Trypanosoma vivax infection. Cell Microbiol. 2013;15(8):1285–1303.
- Nok AJ, Balogun EO. A bloodstream Trypanosoma congolense sialidase could be involved in anemia during experimental trypanosomiasis. J Biochem. 2003;133(6):725–730.
- Murray M, Clifford DJ, Gettinby G, et al. Susceptibility to African trypanosomiasis of N’Dama and Zebu cattle in an area of Glossina morsitans submorsitans challenge. Vet Rec. 1981 109(23):503–510.
- Ramirez-Barrios R, Reyna-Bello A, Parra O, et al. Trypanosoma vivax infection in sheep: different patterns of virulence and pathogenicity associated with differentially expressed proteomes. Vet Parasitol. 2019;276:100014.
- Puppel K, Kuczynska B. Metabolic profiles of cow’s blood; a review. J Sci Food Agric. 2016;96(13):4321–4328.
- Cozzi G, Ravarotto L, Gottardo F, et al. Short communication: reference values for blood parameters in Holstein dairy cows: effects of parity, stage of lactation, and season of production. J Dairy Sci. 2011;94(8):3895–3901.
- Mair B, Drillich M, Klein-Jobstl D, et al. Glucose concentration in capillary blood of dairy cows obtained by a minimally invasive lancet technique and determined with three different hand-held devices. BMC Vet Res. 2016;12(1):34.
- Nafikov RA, Beitz DC. Carbohydrate and lipid metabolism in farm animals. J Nutr. 2007;137(3):702–705.
- Black SJ, Jack RM, Morrison WI Host-parasite interactions which influence the virulence of Trypanosoma (Trypanozoon) brucei brucei organisms. Acta Trop. 1983 40(1):11–18.
- Turner CM. The use of experimental artefacts in African trypanosome research. Parasitol Today. 1990;6(1):14–17.
- Auty HK, Picozzi K, Malele I, et al. Using molecular data for epidemiological inference: assessing the prevalence of Trypanosoma brucei rhodesiense in tsetse in Serengeti, Tanzania. PLoS Negl Trop Dis. 2012b;6(1):e1501.
- Dyer NA, Rose C, Ejeh NO, et al. Flying tryps: survival and maturation of trypanosomes in tsetse flies. Trends Parasitol. 2013;29(4):188–196.
- Haines LR. Examining the tsetse teneral phenomenon and permissiveness to trypanosome infection. Front Cell Infect Microbiol. 2013;3:84.
- Matthews KR. Trypanosome signaling—quorum sensing. Annu Rev Microbiol. 2021;75(1):495–514.
- MacGregor P, Savill NJ, Hall D, et al. Transmission stages dominate trypanosome within-host dynamics during chronic infections. Cell Host Microbe. 2011;9(4):310–318.
- Seed JR, Sechelski JB. Mechanism of long slender (Ls) to short stumpy (Ss) transformation in the African Trypanosomes. J Protozool. 1989;36(6):572–577.
- Reuner B, Vassella E, Yutzy B, et al. Cell density triggers slender to stumpy differentiation of Trypanosoma brucei bloodstream forms in culture. Mol Biochem Parasitol. 1997;90(1):269–280.
- Vassella E, Reuner B, Yutzy B, et al. Differentiation of African trypanosomes is controlled by a density sensing mechanism which signals cell cycle arrest via the cAMP pathway. J Cell Sci. 1997;110(21):2661–2671.
- Vickerman K. Developmental cycles and biology of pathogenic trypanosomes. Br Med Bull. 1985;41(2):105–114.
- Jensen RE, Simpson L, Englund PT. What happens when Trypanosoma brucei leaves Africa. Trends Parasitol. 2008;24(10):428–431.
- Schnaufer A, Domingo GJ, Stuart K. Natural and induced dyskinetoplastic trypanosomatids: how to live without mitochondrial DNA. Int J Parasitol. 2002;32(9):1071–1084.
- Rojas F, Silvester E, Young J, et al. Oligopeptide signaling through TbGPR89 drives trypanosome quorum sensing. Cell. 2019;176(1–2):306–317 e316.
- Tettey MD, Rojas F, Matthews KR. Extracellular release of two peptidases dominates generation of the trypanosome quorum-sensing signal. Nat Commun. 2022;13(1):3322.
- Rojas F, Matthews KR. Quorum sensing in African trypanosomes. Curr Opin Microbiol. 2019;52:124–129.
- Frasch AP, Carmona AK, Juliano L, et al. Characterization of the M32 metallocarboxypeptidase of Trypanosoma brucei: differences and similarities with its orthologue in Trypanosoma cruzi. Mol Biochem Parasitol. 2012;184(2):63–70.
- Hemerly JP, Oliveira V, Del Nery E, et al. Subsite specificity (S3, S2, S1‘, S2’ and S3’) of oligopeptidase B from Trypanosoma cruzi and Trypanosoma brucei using fluorescent quenched peptides: comparative study and identification of specific carboxypeptidase activity. Biochem J. 2003;373(3):933–939.
- Magez S, Pinto Torres JE, Obishakin E, et al. Infections with extracellular trypanosomes require control by efficient innate immune mechanisms and can result in the destruction of the mammalian humoral immune system. Front Immunol. 2020;11:382.
- Seed JR, Sechelski J. Growth of pleomorphic Trypanosoma brucei rhodesiense in irradiated inbred mice. J Parasitol. 1988;74(5):781–789.
- Lisack J, Morriswood B, Engstler M. Response to comment on ‘Unexpected plasticity in the life cycle of Trypanosoma brucei’. Elife. 2022;11:11.
- Mony BM, MacGregor P, Ivens A, et al. Genome-wide dissection of the quorum sensing signalling pathway in Trypanosoma brucei. Nature. 2014;505(7485):681–685.
- Guegan F, Rajan KS, Bento F, et al. A long noncoding RNA promotes parasite differentiation in African trypanosomes. Sci Adv. 2022;8(24):eabn2706.
- Mony BM, Matthews KR. Assembling the components of the quorum sensing pathway in African trypanosomes. Mol Microbiol. 2015;96(2):220–232.
- McDonald L, Cayla M, Ivens A, et al. Non-linear hierarchy of the quorum sensing signalling pathway in bloodstream form African trypanosomes. PLoS Pathog. 2018;14(6):e1007145.
- Barquilla A, Saldivia M, Diaz R, et al. Third target of rapamycin complex negatively regulates development of quiescence in Trypanosoma brucei. Proc Nat Acad Sci. 2012;109(36):14399–14404.
- Domenicali Pfister D, Burkard G, Morand S, et al. A Mitogen-activated protein kinase controls differentiation of bloodstream forms of Trypanosoma brucei. Eukaryot Cell. 2006;5(7):1126–1135.
- Saldivia M, Ceballos-Perez G, Bart JM, et al. The AMPKα1 pathway positively regulates the developmental transition from proliferation to quiescence in Trypanosoma brucei. Cell Rep. 2016;17(3):660–670.
- Vassella E, Kramer R, Turner CM, et al. Deletion of a novel protein kinase with PX and FYVE-related domains increases the rate of differentiation of Trypanosoma brucei. Mol Microbiol. 2001;41(1):33–46.
- Cayla M, McDonald L, MacGregor P, et al. An atypical DYRK kinase connects quorum-sensing with posttranscriptional gene regulation in Trypanosoma brucei. Elife. 2020;9. DOI:10.7554/eLife.51620
- Briggs EM, Rojas F, McCulloch R, et al. Single-cell transcriptomic analysis of bloodstream Trypanosoma brucei reconstructs cell cycle progression and developmental quorum sensing. Nat Commun. 2021;12(1):5268.
- Shapiro SZ, Naessens J, Liesegang B, et al. Analysis by flow cytometry of DNA synthesis during the life cycle of African trypanosomes. Acta Trop. 1984 41(4):313–323.
- Silvester E, Young J, Ivens A, et al. Interspecies quorum sensing in co-infections can manipulate trypanosome transmission potential. Nat Microbiol. 2017;2(11):1471–1479.
- Silvester E, Ivens A, Matthews KR. A gene expression comparison of Trypanosoma brucei and Trypanosoma congolense in the bloodstream of the mammalian host reveals species-specific adaptations to density-dependent development. PLoS Negl Trop Dis. 2018;12(10):e0006863.
- Nikolskaia OV, de ALAP, Kim YV, et al. Blood-brain barrier traversal by African trypanosomes requires calcium signaling induced by parasite cysteine protease. J Clin Invest. 2006;116(10):2739–2747.
- Elliott EB, McCarroll D, Hasumi H, et al. Trypanosoma brucei cathepsin-L increases arrhythmogenic sarcoplasmic reticulum-mediated calcium release in rat cardiomyocytes. Cardiovasc Res. 2013;100(2):325–335.
- Morty RE, Pelle R, Vadasz I, et al. Oligopeptidase B from Trypanosoma evansi. A parasite peptidase that inactivates atrial natriuretic factor in the bloodstream of infected hosts. J Biol Chem. 2005;280(12):10925–10937.
- Troeberg, R N Pike, R E Morty, R K Berry, T H Coetzer, J D Lonsdale-Eccles, 1996. Eur J Biochem Jun 238(3):728–36. doi: 10.1111/j.1432-1033.1996.0728w.x.
- Bastos IMD, Motta FN, Charneau S, et al. Prolyl oligopeptidase of Trypanosoma brucei hydrolyzes native collagen, peptide hormones and is active in the plasma of infected mice. Microbes Infect. 2010;12(6):457–466.
- Morty RE, Bulau P, Pelle R, et al. Pyroglutamyl peptidase type I from Trypanosoma brucei: a new virulence factor from African trypanosomes that de-blocks regulatory peptides in the plasma of infected hosts. Biochem J. 2006;394(3):635–645.
- Morty RE, Lonsdale-Eccles JD, Mentele R, et al. Trypanosome-derived oligopeptidase B is released into the plasma of infected rodents, where it persists and retains full catalytic activity. Infect Immun. 2001;69(4):2757–2761.
- Atyame Nten CM, Sommerer N, Rofidal V, et al. Excreted/secreted proteins from trypanosome procyclic strains. J Biomed Biotechnol. 2010;2010:212817.
- Bossard G, Cuny G, Geiger A. Secreted proteases of Trypanosoma brucei gambiense: possible targets for sleeping sickness control? Biofactors. 2013;39(4):407–414.
- Brossas JY, Gulin JEN, Bisio MMC, et al. Secretome analysis of Trypanosoma cruzi by proteomics studies. PLoS One. 2017;12(10):e0185504.
- Garzon E, Holzmuller P, Bras-Goncalves R, et al. The Trypanosoma brucei gambiense secretome impairs lipopolysaccharide-induced maturation, cytokine production, and allostimulatory capacity of dendritic cells. Infect Immun. 2013;81(9):3300–3308.
- Geiger A, Hirtz C, Becue T, et al. Exocytosis and protein secretion in Trypanosoma. BMC Microbiol. 2010;10(1):20.
- Bayer-Santos E, Aguilar-Bonavides C, Rodrigues SP, et al. Proteomic analysis of Trypanosoma cruzi secretome: characterization of two populations of extracellular vesicles and soluble proteins. J Proteome Res. 2013;12(2):883–897.
- Coakley G, Maizels RM, Buck AH. Exosomes and Other Extracellular Vesicles: the New Communicators in Parasite Infections. Trends Parasitol. 2015;31(10):477–489.
- Eliaz D, Kannan S, Shaked H, et al. Exosome secretion affects social motility in Trypanosoma brucei. PLoS Pathog. 2017;13(3):e1006245.
- Torrecilhas AC, Soares RP, Schenkman S, et al. Extracellular vesicles in trypanosomatids: host cell communication. Front Cell Infect Microbiol. 2020;10:602502.
- Douanne N, Dong G, Amin A, et al. Leishmania parasites exchange drug-resistance genes through extracellular vesicles. Cell Rep. 2022;40:111121.
- Buratta S, Tancini B, Sagini K, et al. Lysosomal exocytosis, exosome release and secretory autophagy: the autophagic- and endo-lysosomal systems go extracellular. Int J Mol Sci. 2020;21(7):2576.
- Silverman JS, Muratore KA, Bangs JD. Characterization of the late endosomal ESCRT machinery in Trypanosoma brucei. Traffic. 2013;14(10):1078–1090.
- Rotureau B, Van Den Abbeele J. Through the dark continent: african trypanosome development in the tsetse fly. Front Cell Infect Microbiol. 2013;3. DOI:10.3389/fcimb.2013.00053
- Kamdem CN, Tiofack AAZ, Mewamba EM, et al. Molecular identification of different trypanosome species in tsetse flies caught in the wildlife reserve of Santchou in the western region of Cameroon. Parasitol Res. 2020;119(3):805–813.
- Kasozi KI, Zirintunda G, Ssempijja F, et al. Epidemiology of Trypanosomiasis in Wildlife-Implications for Humans at the Wildlife Interface in Africa. Front Vet Sci. 2021;8:621699.
- Signaboubo D, Payne VK, Moussa IMA, et al. Diversity of tsetse flies and trypanosome species circulating in the area of Lake Iro in southeastern Chad. Parasit Vectors. 2021;14(1):293.
- Morrison WI, Wells PW, Moloo SK, et al. Interference in the establishment of superinfections with Trypanosoma congolense in cattle. J Parasitol. 1982;68(5):755–764.
- Balmer O, Stearns SC, Schotzau A, et al. Intraspecific competition between co-infecting parasite strains enhances host survival in African trypanosomes. Ecology. 2009;90(12):3367–3378.
- Woolhouse MEJ, Thumbi SM, Jennings A, et al. Co-infections determine patterns of mortality in a population exposed to parasite infection. Sci Adv. 2015;1(2):e1400026.
- Thumbi SM, Bronsvoort BM, Poole EJ, et al. Parasite co-infections and their impact on survival of indigenous cattle. PLoS One. 2014;9(2):e76324.
- Thumbi SM, de CBBM, Poole EJ, et al. Parasite co-infections show synergistic and antagonistic interactions on growth performance of East African zebu cattle under one year. Parasitology. 2013;140(14):1789–1798.
- Venter F, Matthews KR, Silvester E. Parasite co-infection: an ecological, molecular and experimental perspective. Proc R Soc B. 2022;289(1967):20212155.
- Capewell P, Clucas C, DeJesus E, et al. The TgsGP gene is essential for resistance to human serum in Trypanosoma brucei gambiense. PLOS Pathogens. 2013;9:e1003686. DOI:10.1371/journal.ppat.1003686
- Currier RB, Cooper A, Burrell-Saward H et al, Decoding the network ofTrypanosoma bruceiproteins that determines sensitivity to apolipoprotein-L1. PLOS Pathogens. 2018;14:e1006855. DOI:10.1371/journal.ppat.1006855
- McCarroll, C.S., C.L. Rossor, L.R. Morrison, L.J. Morrison, and C.M. Loughrey. 2015. A Pre-clinical Animal Model of Trypanosoma brucei Infection Demonstrating Cardiac Dysfunction. PLoS Neglected Tropical Diseases. 9:e0003811
- Alsford S, Eckert S, Baker N, et al. High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature. 2012;482(7384):232–236.
- Altmann S, Rico E, Carvalho S, et al. Oligo targeting for profiling drug resistance mutations in the parasitic trypanosomatids. Nucleic Acids Res. 2022;50(14):e79.
- Horn D. Genome-scale RNAi screens in African trypanosomes. Trends Parasitol. 2022a;38(2):160–173.
- Horn D, Buscaglia CA. A profile of research on the parasitic trypanosomatids and the diseases they cause. PLoS Negl Trop Dis. 2022b;16(1):e0010040.
- Awuah-Mensah G, McDonald J, Steketee PC, et al. Reliable, scalable functional genetics in bloodstream-form Trypanosoma congolense in vitro and in vivo. PLoS Pathog. 2021;17(1):e1009224.
- Chantal I, Minet C, Berthier D. In vitro cultivation of Trypanosoma congolense bloodstream forms: state of the art and advances. Vet Parasitol. 2021;299:109567.
- Coustou V, Guegan F, Plazolles N, et al. Complete in vitro life cycle of Trypanosoma congolense: development of genetic tools. PLoS Negl Trop Dis. 2010;4(3):e618.
- Gjini E, Haydon DT, Barry JD, et al. Critical interplay between parasite differentiation, host immunity, and antigenic variation in trypanosome infections. Am Nat. 2010;176(4):424–439.