
ABSTRACT
Cysteiniphilum is a newly discovered genus in 2017 and is phylogenetically closely related to highly pathogenic Francisella tularensis. Recently, it has become an emerging pathogen in humans. However, the complete genome sequence of genus Cysteiniphilum is lacking, and the genomic characteristics of genetic diversity, evolutionary dynamics, and pathogenicity have not been characterized. In this study, the complete genome of the first reported clinical isolate QT6929 of genus Cysteiniphilum was sequenced, and comparative genomics analyses to Francisella genus were conducted to unveil the genomic landscape and diversity of the genus Cysteiniphilum. Our results showed that the complete genome of QT6929 consists of one 2.61 Mb chromosome and a 76,819 bp plasmid. The calculated average nucleotide identity and DNA–DNA hybridization values revealed that two clinical isolates QT6929 and JM-1 should be reclassified as two novel species in genus Cysteiniphilum. Pan-genome analysis revealed genomic diversity within the genus Cysteiniphilum and an open pan-genome state. Genomic plasticity analysis exhibited abundant mobile genetic elements including genome islands, insertion sequences, prophages, and plasmids on Cysteiniphilum genomes, which facilitated the broad exchange of genetic material between Cysteiniphilum and other genera like Francisella and Legionella. Several potential virulence genes associated with lipopolysaccharide/lipooligosaccharide, capsule, and haem biosynthesis specific to clinical isolates were predicted and might contribute to their pathogenicity in humans. Incomplete Francisella pathogenicity island was identified in most Cysteiniphilum genomes. Overall, our study provides an updated phylogenomic relationship of members of the genus Cysteiniphilum and comprehensive genomic insights into this rare emerging pathogen.
Introduction
Cysteiniphilum, a genus of Gram-negative bacteria, belongs to the family Fastidiosibacteraceae along with the genera Caedibacter, Facilibium, Fangia, and Fastidiosibacter [Citation1]. This genus was first reported in 2017 and so far consisted of three species, Cysteiniphilum halobium, Cysteiniphilum litorale, and Cysteiniphilum marinum [Citation1–3]. All of these species were initially isolated from water samples from coastal areas in China. Recently, we reported for the first time that C. litorale QT6929 caused skin and soft tissue infections in immunocompetent patients in Wenzhou, China [Citation4]. Subsequently, a case with similar symptoms caused by C. litorale JM-1 infection occurred in Guangdong, China. The patients of the two cases all had the experience of being stabbed by river prawns. This raises our concern that the genus Cysteiniphilum might act as a novel potential pathogen and could infect humans in a specific way.
Infectious diseases are the leading cause of mortality globally, accounting for about a quarter to a third of all deaths [Citation5]. Despite the fact that there are millions of microbial species on Earth, only a small number of them significantly contribute to human disease [Citation6]. Francisella tularensis is one of the most infectious and pathogenic bacteria known [Citation7]. It is important to note that the genus Francisella currently lacks any closely related pathogens or commensals of the human flora [Citation7,Citation8]. The genus Cysteiniphilum is closely related to the Francisella tularensis based on 16S rRNA gene [Citation2]. This raises further concerns about the true pathogenic potential of this genus. To understand the mechanisms of pathogenic bacterial infection and pathogenesis, research and analysis at the genome level are essential. Pan-genome analysis is a powerful method to explore genomic heterogeneity and diversity of bacterial species [Citation8]. The pan-genome plays an important role in the analysis of important drug resistance and virulence genes by analyzing gene diversity within species and has been widely used to mine virulence factors related to pathogenic bacteria [Citation9]. Therefore, it is necessary to resolve the virulence-related genomic features of genus Cysteiniphilum through genomics and pan-genome analysis.
To date, there are only 12 available draft genome sequences of genus Cysteiniphilum in GenBank. Here, we achieved the complete genomes sequence of the first clinical isolate QT6929 of Cysteiniphilum genus, which also represented the first complete sequence of Cysteiniphilum genus and even the family Fastidiosibacteraceae. In addition, we performed a comprehensive comparative genomic analysis and pan-genome analysis of Cysteiniphilum and Francisella genomes. Our results provided important information about the genomic diversity among species of this genus and important insights into their pathogenic potential.
Materials and methods
Bacterial strain
QT6929 was isolated from a 19-year-old female in a tertiary teaching hospital (Wenzhou, China) in November 2018. The patient’s finger was stabbed by an estuarine shrimp, resulting in skin ulceration and accompanying abscess. After culturing the wound exudate on Columbia blood agar plate (BIO-KONT) at 35 ℃ and 5% CO2 atmosphere for 2 days, a Gram-negative coccus was obtained and named QT6929.
Whole-Genome sequencing and bioinformatic analysis
QT6929 was cultured in Luria-Bertani (LB) broth at 37 ℃ for 16 h. The genomic DNA was extracted using an AxyPrep bacterial genomic DNA miniprep kit (Axygen Scientific, Union City, CA, USA) according to the manufacturer’s instructions. The whole genome of QT6929 was sequenced using Illumina NovaSeq and Oxford Nanopore Technologies (ONT) platforms. Hybrid assembly of the Illumina and Nanopore sequences was performed using Unicycler software (v0.5.0). The genome sequences of QT6929 were annotated using Bakta [Citation10] and then corrected by BLAST searches against the UniProtKB/Swiss-Prot, RefSeq. The clusters of orthologous groups (COG) classification of the predicted genes were achieved by eggNOG-mapper v2 [Citation11]. The genome-based taxonomy classification of QT6929 was conducted using Type Strain Genome Server (TYGS) tools [Citation12]. Chromosome and plasmid maps were generated using CGView [Citation13].
Comparative genomics and pan-genome analysis
All 20 publicly available whole-genome sequences (May 2022) belonging to the family Fastidiosibacteraceae were downloaded from the NCBI assembly database (Table S1). The average nucleotide identity (ANI) and in silico DNA–DNA hybridization (DDH) among these genomes and QT6929 were calculated using JspeciesWS [Citation14] and genome-to-genome distance calculator 3.0 (GGDC) [Citation15], respectively. For comparative genomic analysis with Francisella, a total of 1063 assemblies of Francisella genus were downloaded from the NCBI assembly database (Nov 2022). The quality of assembly was assessed by CheckM v1.2.2, resulting in 1031 high-quality assemblies for downstream analysis (Table S1) [Citation16]. A total of 165 representative genomes were selected from these highly related Francisella genomes (mash-distance <0.001). Pan-genome analysis of 13 Cysteiniphilum spp. genomes and 165 Francisella spp. representative genomes individual or combined was performed using PPanGGOLin with default parameters [Citation17]. The orthogroups were defined as orthologous genes sharing >60% identity and >80% coverage. Regions of genomic plasticity (RGPs) were detected using panRGP with default parameters [Citation18]. The taxonomic profiling of RGPs sequences was analyzed using SprayNPray [Citation19]. The insertion sequences (IS), prophage regions, and plasmid sequences were predicted using ISEScan, PHASTER, and Platon, respectively [Citation20–22].
Phylogenetic analysis
For 13 Cysteiniphilum spp. genomes, genome-based phylogenetic tree was generated by TYGS. To investigate the phylogenetic rrelationship of Cysteiniphilum and Francisella, the paired mash distance of 13 Cysteiniphilum spp. genomes and 165 representative Francisella spp. genomes were calculated, and a neighbor-joining tree was constructed using mathtree v1.2.0 [Citation23,Citation24]. In addition, a highly resolved maximum likelihood tree based on the aligned bacterial core genes was obtained using UBCG2 (Up-to-date bacterial core genes) [Citation25]. All these phylogenetic trees were visualized using iTOL [Citation26].
Identification of the virulence factors, antimicrobial resistance genes, and Francisella pathogenicity island
To fully investigate the potential resistance and virulence factors of Cysteiniphilum and Francisella genus, a total of 14,544 orthogroups were clustered using 13 Cysteiniphilum spp. genomes and 1031 Francisella spp. genomes. To annotate the virulence factors, the orthogroups were aligned against the Virulence Factors Database (VFDB) full dataset (setB) using BLASTp with an E-value cutoff of <10−6, identity >50%, and coverage >60% [Citation27]. The identification of antimicrobial resistance genes was performed using AMRFinderPlus [Citation28]. The gene clusters of Francisella pathogenicity island (FPI) among Cysteiniphilum and Francisella genomes were identified using cblaster v1.3.16 with default parameters [Citation29]. The FPI cluster of the F. novicida strain U112 (accession number NZ_CP009633) was used as input query.
Results
Genomic features of strain QT6929
The complete QT6929 genome consisted of a 2.61 Mb chromosome with GC content of 38.92% and one circular plasmid (designated pQT6929) of 76,819 bp in length with a GC content of 36.42% (). The general genomic features were given in . In total, 2,324 protein coding sequences (CDS) were predicted, with 2,252 in the chromosome and 72 in the plasmid. The functional annotations of CDS were listed in Table S2. Among them, 1,498 genes could be annotated in the COG database (), which were mapped to the categories of metabolism (primarily amino acid transporter and metabolism, 11.28%), cellular processes and signaling (primarily cell wall/membrane/envelope biogenesis, 7.41%), and information processing and storage (translation, ribosomal structure, and biogenesis, 13.22%). The CDS associated with conjugal transfer, plasmid maintenance, and mobile elements were identified in the plasmid.
Figure 1. Genome visualization and annotation of strain QT6929. (A) Chromosome; (B) Plasmid. The element colour of each circle is indicated in the legend; (C) Clusters of Orthologous Groups (COG) analysis of strain QT6929 genome.
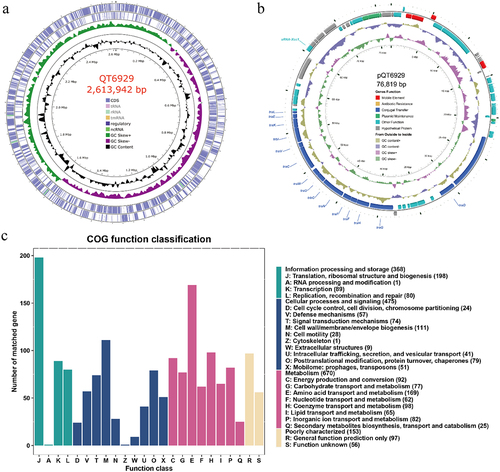
Table 1. General features of the QT6929 genome.
Phylogenetic analysis of strain QT6929
In our previous study, QT6929 was initially classified as C. litorale by the 16S rRNA gene sequence. Here, we confirmed the taxonomy with the TYGS tools based on a state-of-the-art genome-based taxonomy. The results showed that QT6929 was most closely related to C. littorale DSM101832 from genus Cysteiniphilum. In addition, TYGS also suggested that QT6929 belongs to a novel species. To better identify QT6929 at the species level, we calculated the ANI and in silico DDH values of QT6929 for other genomes from Fastidiosibacteraceae family (). ANI value of 95–96% and DDH value of 70% were used as a boundary for species delineation [Citation30]. The results showed that QT6929 had the highest ANI value of approximately 95% to three C. littorale genomes and one Cysteiniphilum genome JM-1 from clinical sample. The results of DDH heatmap also showed that QT6929 had the highest DDH value of approximately 65% to three C. littorale genomes followed by JM-1. In addition, we found that the ANI and DDH values of JM-1 to QT6929 were higher than that of three C. littorale and below the boundary for species delineation. Taken together, these results suggested that QT6929 and JM-1 represented a novel species within the Cysteiniphilum genus based on the combined ANI and DDH. We assigned the species name of QT6929 to Cysteiniphilum wenzhou based on the first discovered region.
Figure 2. Average nucleotide identity (ANI), in silico DNA-DNA hybridization (DDH) and phylogenetic analysis of strain QT6929. (A) Heatmap based on ANIb values between each pair of genome sequences from strain QT6929 and strains within genus Cysteiniphilum. Eleven groups were formed based on a cut-off score of >95%; (B) Heatmap based on DDH values. Twelve groups were formed based on a cut-off score of >70%; (C) Maximum likelihood phylogenomic tree of QT6929 and family Fastidiosibacteraceae genomes. The information of genus, species and genome size was indicated using a coloured box.
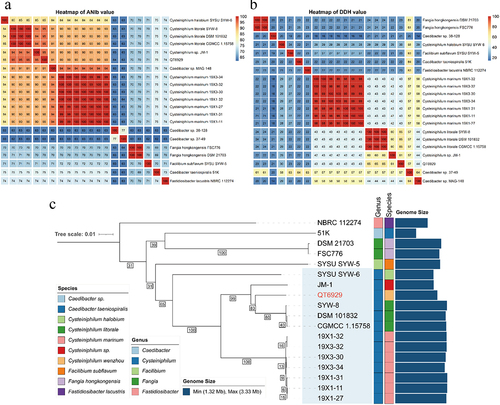
Next, a genome-based phylogenetic tree of the Fastidiosibacteraceae family was constructed based on the currently available genome (). The genome of the type strain F. tularensis was used as the outgroup, and three genomes (MAG-148, 38–128, and 37–49) were excluded because of possibly incomplete assembly. Consistent with the above analysis, we found that QT6929 had a closer phylogenetic relationship to C. littorale strains than JM-1 and C. halobium SYW-6 was the outermost branch among these Cysteiniphilum genomes. Genome sizes of Fastidiosibacteraceae family vary widely, ranging from 1.32 to 3.33 Mb (). In the genus Cysteiniphilum, the genome size of QT6929 (2.69 Mb) was much closer to that of JM-1 (2.46 Mb) and C. halobium SYW-6 (2.44 Mb). C. halobium SYW-6 and Caedibacter taeniospiralis 51K had the smallest genomes in the Cysteiniphilum genus and Fastidiosibacteraceae family, respectively.
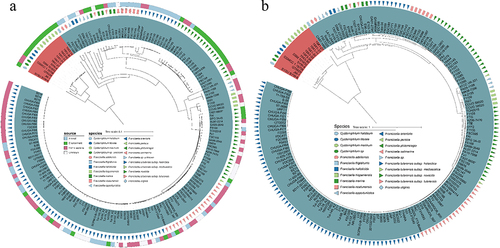
Since the genus Cysteiniphilum is close to the pathogenic Francisella genus as per 16S rRNA gene sequence, we further analyzed the whole genomic distance between the two genera. We selected 165 representative strains from the 1031 genomes of Francisella genus available in NCBI to construct a mash-distance-based phylogenetic tree together with Cysteiniphilum strains (). These representative Francisella spp. strains cover almost the complete Francisella genus including 13 species and 3 F. tularensis subspecies. The results showed that Cysteiniphilum strains were most closely related to F. frigiditurris CA97–1460 strain and most distantly related to those F. tularensis subspecies strains. In addition, we also constructed a highly resolved maximum likelihood tree based on the 81 universal bacterial core genes. The overall topology of the core-gene tree was highly consistent with the mash tree, and these Cysteiniphilum spp. strains were still closest to F. frigiditurris CA97–1460.
Figure 3. Phylogenetic analysis of Cysteiniphilum and Francisella genus. (A) Mash-distance based neighbour-joining tree of 13 Cysteiniphilum and 165 representative Francisella genomes. (B) Maximum likelihood phylogenetic tree based on 81 up-to-date bacterial core genes.
Pan-genome analysis of genus cysteiniphilum and Francisella
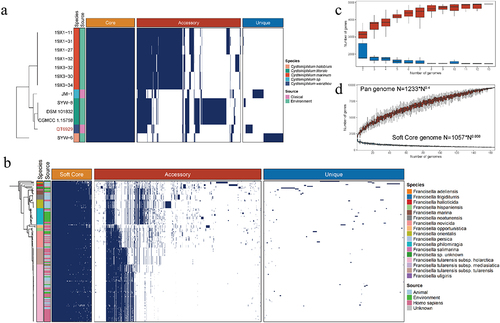
To investigate the genomic diversity of Cysteiniphilum and Francisella genus, pan-genome analysis was performed. Thirteen Cysteiniphilum genomes consisted of 4,913 non-redundant orthogroups (defined as gene families) that were classified into three classes: core, accessory, and unique genes (). About 1,443 orthogroups (25.18%) shared by all strains formed the core genome, while the remaining 3,470 orthogroups formed a variable genome. Genome of 165 Francisella spp. consisted of 9,604 orthogroups, of which only 753 (7.84%) belong to the soft core (present in at least 95% of the Francisella genomes). The pan-genome accumulation curve of Francisella genus fit Heaps’ law pan-genome model with exponent γ = 0.4 (), indicating an open pan-genome. The exponent γ of soft-core-genome accumulation curve of Francisella was close to zero, indicating that its core-genomes are in a stable state. The accumulation curve of Cysteiniphilum could not be fit by Heaps’ law. However, as more genomes were evaluated, the number of pan-genomes of Cysteiniphilum likewise rose without plateauing (), indicating that Cysteiniphilum also possesses an open pan-genome.
Figure 4. Pan-genome analysis of Cysteiniphilum and Francisella genus individually. (A) Distributions of core gene (present in all genomes), accessory gene (present in 5%~95% of the genomes) and strain-specific gene (present in one genomes) in Cysteiniphilum spp. genomes. (B) Distributions of soft-core gene (present in at least 95% of genomes), accessory gene and strain-specific gene in Francisella spp. genomes. (C) Gene accumulation curve of core genes (blue) and pan genome (red) of Cysteiniphilum spp. genomes. The upper and lower edges of the boxes indicate the first quartile (25th percentile of the data) and third quartile (75th percentile), respectively, of 1,00 random different input orders of the genomes. (D) Gene accumulation curve of soft-core genes (blue) and pan genome (red) of Francisella spp. genomes.
To illustrate the genetic relatedness of Cysteiniphilum and Francisella genus, we also analyzed the core-pan genomes of them together (). The combined sets of both Cysteiniphilum and Francisella genomes consisted of 13,997 orthogroups, of which 327 orthogroups were shared by the soft-core set of Cysteiniphilum and Francisella genomes and 98 orthogroups were shared by the accessory-set of them. We also observed 50 and 33 orthogroups of the accessory genomes of Francisella, forming part of the soft-core and unique genomes of Cysteiniphilum, respectively.
Figure 5. Union analysis of pan-genome of Cysteiniphilum and Francisella genus. (A) Horizontal bars indicate the number of soft-core, accessory and unique orthogroups of Cysteiniphilum and Francisella genus. Vertical bars represent the number of these six sets of orthogroups and their shared orthogroups. (B) Distribution of COG function category for each set of orthogroups in (A).
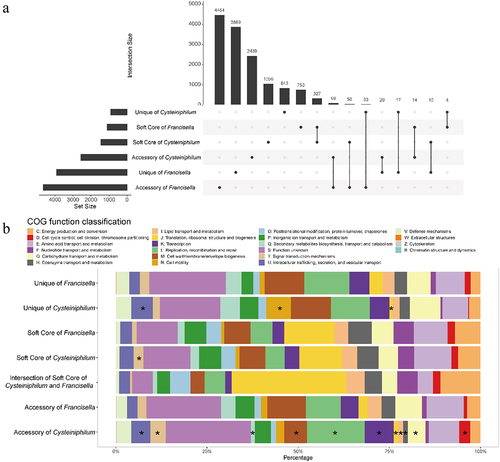
To further understand the pan-genome function enrichment of the Cysteiniphilum and Francisella, we performed a COG analysis to categorize their functions (). Totals of 51.28% (7178/13997) orthogroups can be assigned to COG functional categories, and 1507 of them belonged to “function unknown” category. In addition to the “T: signal transduction mechanisms” category, there is no significant difference in the distribution of each COG category between the soft-cores of Cysteiniphilum and Francisella. The intersection of the soft-core set of Cysteiniphilum and Francisella had the highest proportion of “J: Translation, ribosomal structure and biogenesis” category.
Genomic plasticity analysis of genus Cysteiniphilum
To explore the genomic plasticity of Cysteiniphilum genomes, we predicted the regions of genomic plasticity using a pangenome-based method. The RGPs are gene clusters located in highly variable genomic regions, most of which are derived from HGTs [Citation18]. The number of RGPs of species of Cysteiniphilum ranged from 33 to 85, with C. marinum being the most and C. halobium the least (). The size of these RGPs ranged from approximately 3 kbp to 13.53 kbp (Table S3). The amount of RGPs was significantly associated with the genome size (P < 0.05, Pearson test). Next, we analyzed the origin of the closest homologs to proteins encoded on RPGs at genus level, and the top six enriched genus for each strain were shown (). The result showed that the origin of proteins encoded on RPGs mainly enriched in 12 genera and 3 unclassified species, of which the genera of fastidiosibacteraceae accounted for the vast majority. Homologs from the genus Francisella were enriched in three C. litorale strains, C. halobium SYW-6, Cysteiniphilum spp. QT6929 and JM-1. Homologs from the genus Vibrio and Candidatus Pelagibacter were enriched in five out of seven C. marinum strains. Compared to other species of Cysteiniphilum, we found that a large proportion (35.53%, 108/304) of homologous of QT6929‘s RGPs were categorized into “Others” (up to 61 genera). Moreover, the homologs from the genus Legionella were identified on QT6929.
Figure 6. Distribution of RGPs and MGEs in Cysteiniphilum genomes. From left to right represent strain species, genome size, number of RGPs, genus levels source of the closest homologs of proteins encoded on RGPs, number of IS, number of phages and total length of predicted plasmid sequences.
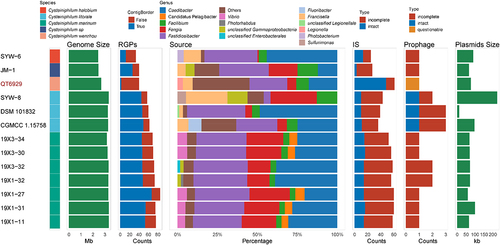
We further analyzed mobile genetic elements (MGEs) that promote genomic plasticity, including insertion sequence (IS), prophage, and plasmids. A total of 20 IS families were identified in genus Cysteiniphilum, of which members of the ISNCY family were the most common, followed by IS3 family (Table S4). The ISs counts of Cysteiniphilum strains ranged from 25 to 61, with QT6929 having the most. A total of 20 prophage regions were found in Cysteiniphilum genomes with an average length of 20.6 kb. These prophage sequences can be clustered into nine clades with a 99% sequence identity threshold (Table S5). All three intact prophages were on the C. littorale strains. Plasmid sequences were predicted in all Cysteiniphilum genomes with an average length of 82.3 kb (). In addition, we found that the circular plasmid, pQT6929 shared the highest similarity (94.63% identity and 71% coverage) with the plasmid sequence of C. halobium SYW-6 (Figure S1).
Compare the pathogenic factors of genus Cysteiniphilum and Francisella
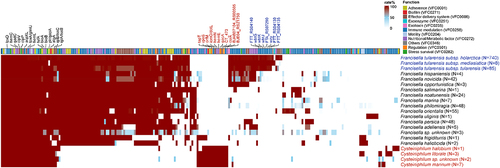
To illustrate the pathogenic potential of Cysteiniphilum strains, we scanned the virulence-related factors on 13 Cysteiniphilum genomes and 1088 Francisella genomes (). A total of 73 genes were associated with the production of known or putative virulence factors in Cysteiniphilum, mainly involving immune modulation (n = 27), nutritional/metabolic (n = 12), adherence (n = 9), effector delivery system (n = 9), stress survival (n = 5), and so on (Table S6). Cysteiniphilum carried fewer virulence factors than Francisella genus, with the undefined Cysteiniphilum spp. carrying the highest number of virulence genes. Totals of 26 virulence factors were conserved on both Cysteiniphilum and Francisella genomes, mainly related to the synthesis of haem (hemC, hemL), biotin (bioA, bioB), capsule (galE), lipopolysaccharide (kdsA, lpxD, and glmU), etc. (Table S6). Sixteen virulence factors were specific in Cysteiniphilum, like alginate synthesis-related gene algC, type IV secretion system (T4SS) components (CBU_2076, CT_473, and icmO), and so on. Among these Francisella spp., F. tularensis subspecies owned the highest counts of virulence genes. Their additional virulence factors were involved in the synthesis of capsule (FTA_RS07290, FTT_RS04130, FTT_RS04135 and wzy) and LPS (wbtG, wbtJ and wbtK), which are related to host immunity.
Figure 7. The genotypic profiles of potential virulence factors on 13 Cysteiniphilum genomes and 1031 Francisella genomes. Color coding is based on the prevalence rate of genes in each species.
Analysis of antimicrobial resistance gene in Cysteiniphilum and Francisella spp
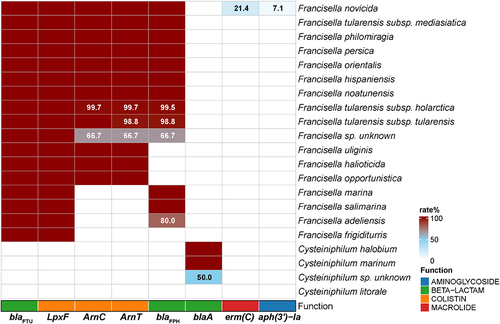
To investigate the antimicrobial resistance of Cysteiniphilum and Francisella, we explored their enzyme producing ARGs. The results showed that eight ARGs were identified on Cysteiniphilum and Francisella genomes, conferring resistance to β-lactam, colistin, aminoglycoside, and macrolide (). The β-lactamase-encoding gene blaFTU and lipid A 4’-phosphatase-encoding gene lpxF were carried by almost all of the Francisella species. Another class A type β-lactamase was found in C. halobium, C. marinum, and Cysteiniphilum spp. strain JM-1, which exhibit low similarity with blaFTU (34.74% identity and 95% coverage). Of note, we also identified the methyltransferase-encoding gene erm(C) and phosphotransferase-encoding gene aph(3’)-Ia in some F. novicida strains.
Figure 8. The genotypic profiles of antimicrobial resistance genes on 13 Cysteiniphilum genomes and 1031 Francisella genomes. Color coding is based on the prevalence rate of genes in each species.
Analysis of Francisella pathogenicity island
FPI was a cluster of genes encoding non-canonical T6SS, which was critical for its pathogenicity. Thus, we examined the existence of FPI on the Cysteiniphilum and Francisella genomes. The results showed that intact or incomplete FPI clusters were present in most of these genomes except for C. littorale and F. halioticida strains (). For Cysteiniphilum strains, we only found IglABC and IglG genes, which exhibited approximately 32% to 61% identities for the reference sequence. In terms of Francisella, most of these FPI genes were conserved except for pdpD, orf9, and pdpC, which were lost in more than half of the Francisella species ().
Figure 9. Analysis of Francisella pathogenicity island on Cysteiniphilum and Francisella genomes. (A) Prevalence rate of 17 FPI genes among Cysteiniphilum and Francisella spp. (B) Comparative analysis of FPI clusters in Cysteiniphilum and Francisella. Colored arrows indicate homologs to the corresponding gene in the same colour in the reference sequence.

Discussion
Cysteiniphilum spp. may pose a threat to human health as an emerging potential pathogen, but its genomic characterization remains unknown. In this study, we achieve the complect genome sequence of the first reported clinical Cysteiniphilum strain QT6929 and conducted a comprehensive comparative genomic analysis of Cysteiniphilum spp. and its phylogenetic neighbor Francisella spp.
First, we rebuild the taxonomic relationship of Cysteiniphilum spp. based on genome sequences. With the increasing use of WGS, sequence data began to be used as the basis for taxonomic assignments and revealed misidentifications and misclassifications of bacteria using single-gene 16S rRNA phylogeny [Citation31]. The two clinical strains QT6929 and JM-1 were initially assigned to C. littorale based on 16S rRNA gene sequence. Although the ANI values of these strains with C. littorale were around 95%, which was in the transition zone, the DDH value of them with C. littorale was far below the boundary for species delineation. Moreover, compared to C. littorale, the genome sizes of QT6929 and JM-1 are significantly reduced and were closer to C. halobium SYW-6. Differences in genome size might be related to their ecological niches, especially when bacteria evolve to live in symbiosis, losing a large number of useless genes and causing extreme genome reductions [Citation32]. The difference in genome size between the two clinical isolates and environmental source C. litorale strains might indicate their adaptation to new ecological niches, such as mammals [Citation8]. Taken together, it is reasonable to reclassify the two isolates into two distinct novel species and QT6929 represented an evolutionary lineage that exists between JM-1 and C. littorale strains.
As a genetic neighbor of Francisella, we confirm that Cysteiniphilum was closest to F. frigiditurris among various Francisella spp. based on the mash distance and 81 universal core gene sets. The Francisella genus consisted of several recognized species associated with a variety of clinical and environmental sources, including highly virulent human and animal pathogens, opportunistic human pathogens, fish pathogens, tick endosymbionts, and seemingly free-living organisms inhabiting brackish water [Citation33,Citation34]. The F. frigiditurris strain was first isolated from a cooling tower in California and its pathogenicity to human remain unknown [Citation34]. Through union pan-genome analysis, we found that the core genomes of Cysteiniphilum and Francisella maintained a certain degree of similarity, while the accessory genomes were more divergent. This suggests that the two genera might maintain some continuation in their growth patterns but evolved different species characteristics through adaptation to their respective ecological niches [Citation35].
The RGPs of varying sizes were widespread on the Cysteiniphilum genomes. These RGPs are mainly derived from horizontal gene transfer, which is a major mechanism for the formation of bacterial species gene repertoires and an important basis for maintaining species diversity and gene novelty [Citation36,Citation37]. The distinct counts of RGPs among different species are mainly attributed to their genome size differences. These large fragments of RGPs often represented genomic islands, while the small ones might be associated with transposons, integrons, or result from incomplete assembly. Importantly, we found that QT6929 had the highest amount of IS, which might enhance its exchange of genetic material to other strains and promoted its broader genome plasticity. The analysis results of the possible origin of RGPs revealed that in addition to these sibling genera, a rich and diverse type of bacterial genera contributed to their genetic material. For C. marinum strains, RGPs derived from Vibrio and Candidatus Pelagibacter were identified which imply its preference for marine habitats in the past or present. For other species of Cysteiniphilum, Francisella sourced RGPs indicated their adaptation to terrestrial habitats. Importantly, we noticed that RGPs from Legionella appeared in Cysteiniphilum spp. strain QT6929. The genus Legionella includes Legionella pneumophila was a facultative intracellular pathogen like Francisella that causes Legionnaires’ disease and Pontiac fever [Citation38]. This suggests that QT6929 might have a distinct evolutionary process from other species of Cysteiniphilum and unique microhabitats.
In view of the fact that some species of Cysteiniphilum were pathogenic to humans, we further conducted an extensive genomic evaluation of their virulence factor profile. Up to 10 categories of virulence factors were identified, among which LPS/LOS, capsule, haem and biotin-related factors were conserved on all Cysteiniphilum genomes. These genes might form the general property of species of the genus Cysteiniphilum. For the two clinical isolates of Cysteiniphilum spp., three additional putative virulence factors were identified associated with LPS/LOS and capsule. LPS and LOS are essential components of the outer membrane of Gram-negative bacteria, which regulate the response of the host immune system and play crucial roles in bacteria-host interactions [Citation39]. Notably, F. tularensis subspecies, the most virulent of the Francisella species, also possessed additional virulence factors for capsule and LPS synthesis compared with other Francisella spp. There are considerable diversities in LPS structures, and certain pathogens evade host immune responses by producing the less immunogenic LPS/LOS [Citation40,Citation41]. Whether additionally acquired LPS/LOS-related genes in the clinical isolates altered the structure and immunogenicity of LPS needs to be further explored. Overall, the acquisition of these genes might confer them different virulence characteristics and even stronger host pathogenicity. Further work is required to explore the effect of these genes on the pathogenicity of clinical isolates.
The understanding of resistome of Cysteiniphilum spp. was still insufficient in the current work. Previous studies showed that most of these Cysteiniphilum spp. were resistant to cephalosporins, positive for β-lactamase, and sensitive to chloramphenicol, ciprofloxacin, doxycycline, gentamicin, levofloxacin, and tetracycline [Citation1–4]. However, we only found a class A β-encoding gene blaA in C. halobium, C. marinum, and Cysteiniphilum spp. strain JM-1. Searches with more lenient thresholds or more sensitive methods also failed. A more complete database might be helpful to identify their potential β-encoding gene in the future. Surprisingly, we found erm(C) and aph(3”)-Ia genes in some F. novicida strains, which had not been reported before. By analyzing the genetic environment of erm(C) and aph(3”)-Ia, we found that the ARGs, in most sequences, were located in the FPI gene cluster (Figure S2). Whether the source of these ARGs were naturally obtained or derived from laboratory remain unkown.
The FPI, a non-canonical type Ⅵ secretion system (T6SS) presented in most of the Francisella genomes, was critical for its phagosome escaping, intracellular replication, and pathogenicity in animals [Citation42]. The intact FPI was an island of ~30-kb encoding 18 genes, 14 of which have been shown to be essential for growth in macrophages [Citation43]. We found that Cysteiniphilum strains lost most of the FPI genes during evolution, which might be an evolutionary choice for its adaptation to the local environment. Francisella employs ClpB ATPase, encoded outside of the FPI, to disassemble its contracted T6SS sheath [Citation44]. Interestingly, we identified the clpV gene encoding the ClpB homologs in all these Cysteiniphilum genomes. The roles of the ubiquitous and conserved IglABCG and clpV genes in Cysteiniphilum remained unclear. Although the pathogenicity of other species of genus Cysteiniphilum except of QT6929 and JM-1 was not yet clear, study of this genus might help to reveal the formation and evolution of the virulence of Francisella.
In conclusion, our study contributes to the first complete genome of the genus Cysteiniphilum. More importantly, we perform a systematic comparative genomic analysis of Cysteiniphilum and Francisella genus, which allows us to obtain a landscape of the population diversity, evolution, and pathogenicity of this potential pathogen. Combining ANI and DDH, we reclassified the clinical isolates QT6929 and JM-1 into two novel species that are closely related but distinct from C. litorale. The pan-genome analysis revealed a genomic diversity within the genus. Several potential virulence factors associated with LPS/LOS, capsule, and haem biosynthesis specific to clinical isolates might contribute to their pathogenicity in humans, requiring further experimental confirmation. Incomplete FPI-like structures were also identified in most of the Cysteiniphilum genomes. Our study enriches the information of the Cysteiniphilum genomes and contributes to provide a key genetic framework for assessing and understanding the molecular events of Cysteiniphilum pathogenesis as well.
Ethics approval
This study uses strains obtained from a teaching hospital at Wenzhou Medical University. It did not require the study to be reviewed or approved by an ethics committee because individual patient data was not involved, and only anonymous clinical residual samples during routine hospital laboratory procedures were used in this study.
Supplemental Material
Download Zip (813.1 KB)Acknowledgements
The authors would like to acknowledge all study participants and individuals who contributed to this study.
Disclosure statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Data availability statement
The complete genomic sequences of QT6929 have been deposited in GenBank at https://www.ncbi.nlm.nih.gov/nucleotide/, under the accession numbers CP103983 (chromosome) and CP103984 (plasmid).
Supplemental data
Supplemental data for this article can be accessed online at https://doi.org/10.1080/21505594.2023.2214416.
Additional information
Funding
References
- Luo HM, Feng JH, Li LH, et al. Cysteiniphilum marinum sp. nov., isolated from coastal seawater. Antonie Van Leeuwenhoek. 2021 Jul;114(7):1079–13.
- Liu L, Salam N, Jiao JY, et al. Cysteiniphilum litorale gen. nov., sp. nov., isolated from coastal seawater. Int J Syst Evol Microbiol. 2017 Jul;67(7):2178–2183.
- Xiao M, Zheng ML, Salam N, et al. Facilibium subflavum gen. nov., sp. nov. and Cysteiniphilum halobium sp. nov., new members of the family Fastidiosibacteraceae isolated from coastal seawater. Int J Syst Evol Microbiol. 2019 Dec;69(12):3757–3764.
- Xu CQ, Zhang XC, Wu Q, et al. Skin and soft tissue infection caused by Cysteiniphilum litorale in an immunocompetent patient: a case report. Indian J Med Microbiol. 2021 Oct-Dec;39(4):545–547.
- Balloux F, van Dorp L. Q&A: what are pathogens, and what have they done to and for us? BMC Biol. 2017 Oct 19;15(1):91.
- Diard M, Hardt WD. Evolution of bacterial virulence. FEMS Microbiol Rev. 2017 Sep 1;41(5):679–697.
- Pechous RD, McCarthy TR, Zahrt TC. Working toward the future: insights into Francisella tularensis pathogenesis and vaccine development. Microbiol Mol Biol Rev. 2009 Dec;73(4):684–711.
- Kumar R, Broms JE, Sjostedt A. Exploring the diversity within the Genus Francisella - an integrated pan-genome and genome-mining approach. Front Microbiol. 2020;11:1928.
- Golicz AA, Bayer PE, Bhalla PL, et al. Pangenomics comes of age: from bacteria to plant and animal applications. Trends Genet. 2020 Feb;36(2):132–145.
- Schwengers O, Jelonek L, Dieckmann MA, et al. Bakta: rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb Genom. 2021 Nov;7(11). DOI:10.1099/mgen.0.000685.
- Cantalapiedra CP, Hernandez-Plaza A, Letunic I, et al. eggNOG-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol Biol Evol. 2021 Dec 9;38(12):5825–5829. DOI:10.1093/molbev/msab293
- Meier-Kolthoff JP, Goker M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat Commun. 2019 May 16;10(1):2182.
- Stothard P, Wishart DS. Circular genome visualization and exploration using CGView. Bioinformatics. 2005 Feb 15;21(4):537–539.
- Richter M, Rossello-Mora R, Oliver Glockner F, et al. JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics. 2016 Mar 15;32(6):929–931. DOI:10.1093/bioinformatics/btv681
- Meier-Kolthoff JP, Carbasse JS, Peinado-Olarte RL, et al. TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022 Jan 7;50(D1):D801–807. DOI:10.1093/nar/gkab902
- Parks DH, Imelfort M, Skennerton CT, et al. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015 Jul;25(7):1043–1055.
- Gautreau G, Bazin A, Gachet M, et al. PPanGGOLiN: depicting microbial diversity via a partitioned pangenome graph. PLoS Comput Biol. 2020 Mar;16(3):e1007732.
- Bazin A, Gautreau G, Medigue C, et al. panRGP: a pangenome-based method to predict genomic islands and explore their diversity. Bioinformatics. 2020 Dec 30;36(Suppl_2):i651–658. DOI:10.1093/bioinformatics/btaa792
- Garber AI, Armbruster CR, Lee SE, et al. SprayNPray: user-friendly taxonomic profiling of genome and metagenome contigs. BMC Genomics. 2022 Mar 12;23(1):202. DOI:10.1186/s12864-022-08382-2
- Arndt D, Grant JR, Marcu A, et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016 Jul 8;44(W1):W16–21. DOI:10.1093/nar/gkw387
- Xie Z, Tang H. Isescan: automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics. 2017 Nov 1;33(21):3340–3347.
- Schwengers O, Barth P, Falgenhauer L, et al. Platon: identification and characterization of bacterial plasmid contigs in short-read draft assemblies exploiting protein sequence-based replicon distribution scores. Microb Genom. 2020 Oct;6(10). DOI:10.1099/mgen.0.000398.
- Katz LS, Griswold T, Morrison SS, et al. Mashtree: a rapid comparison of whole genome sequence files. J Open Source Softw. 2019 Dec 10;4(44):1762. DOI:10.21105/joss.01762
- Ondov BD, Treangen TJ, Melsted P, et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016 Jun 20;17(1):132. DOI:10.1186/s13059-016-0997-x
- Kim J, Na SI, Kim D, et al. UBCG2: up-to-date bacterial core genes and pipeline for phylogenomic analysis. J Microbiol. 2021 Jun;59(6):609–615.
- Letunic I, Bork P. Interactive Tree of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021 Jul 2;49(W1):W293–296.
- Liu B, Zheng D, Zhou S, et al. VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 2022 Jan 7;50(D1):D912–917. DOI:10.1093/nar/gkab1107
- Feldgarden M, Brover V, Gonzalez-Escalona N, et al. AMRFinderPlus and the Reference Gene Catalog facilitate examination of the genomic links among antimicrobial resistance, stress response, and virulence. Sci Rep. 2021 Jun 16;11(1):12728. DOI:10.1038/s41598-021-91456-0
- Gilchrist CLM, Booth TJ, van Wersch B, et al. Cblaster: a remote search tool for rapid identification and visualization of homologous gene clusters. Bioinformat Adv. 2021;1(1):vbab016.
- Wambui J, Cernela N, Stevens MJA, et al. Whole genome sequence-based identification of clostridium estertheticum complex strains supports the need for taxonomic reclassification within the species clostridium estertheticum. Front Microbiol. 2021;12:727022.
- Tran PN, Savka MA, Gan HM. In-silico taxonomic classification of 373 genomes reveals species misidentification and new genospecies within the genus pseudomonas. Front Microbiol. 2017;8:1296.
- McCutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol. 2011 Nov 8;10(1):13–26.
- Challacombe JF, Pillai S, Kuske CR. Shared features of cryptic plasmids from environmental and pathogenic Francisella species. PLoS ONE. 2017;12(8):e0183554.
- Challacombe JF, Petersen JM, Gallegos-Graves V, et al. Whole-genome relationships among Francisella bacteria of diverse origins define new species and provide specific regions for detection. Appl Environ Microbiol. 2017 Feb 1;83(3). doi:10.1128/AEM.02589-16
- Mesa V, Monot M, Ferraris L, et al. Core-, pan- and accessory genome analyses of Clostridium neonatale: insights into genetic diversity. Microb Genom. 2022 May;8(5). DOI:10.1099/mgen.0.000813.
- Treangen TJ, Rocha EP, Moran NA. Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genet. 2011 Jan 27;7(1):e1001284.
- Niehus R, Mitri S, Fletcher AG, et al. Migration and horizontal gene transfer divide microbial genomes into multiple niches. Nat Commun. 2015 Nov 23;6:8924. doi:10.1038/ncomms9924
- Goncalves IG, Simoes LC, Simoes M. Legionella pneumophila. Trends Microbiol. 2021 Sep;29(9):860–861.
- Zhang G, Meredith TC, Kahne D. On the essentiality of lipopolysaccharide to Gram-negative bacteria. Curr Opin Microbiol. 2013 Dec;16(6):779–785.
- Montminy SW, Khan N, McGrath S, et al. Virulence factors of Yersinia pestis are overcome by a strong lipopolysaccharide response. Nat Immunol. 2006 Oct;7(10):1066–1073.
- Needham BD, Trent MS. Fortifying the barrier: the impact of lipid a remodelling on bacterial pathogenesis. Nat Rev Microbiol. 2013 Jul;11(7):467–481.
- Clemens DL, Lee BY, Horwitz MA. The Francisella type VI secretion system. Front Cell Infect Microbiol. 2018;8:121.
- Broms JE, Sjostedt A, Lavander M. The role of the Francisellatularensis pathogenicity island in type vi secretion, intracellular survival, and modulation of host cell signaling. Front Microbiol. 2010;1:136.
- Brodmann M, Dreier RF, Broz P, et al. Francisella requires dynamic type VI secretion system and ClpB to deliver effectors for phagosomal escape. Nat Commun. 2017 Jun 16;8:15853. doi:10.1038/ncomms15853