
ABSTRACT
Hepatitis B virus (HBV) immune escape and Pol/RT mutations account for HBV immunoprophylactic, therapeutic, and diagnostic failure globally. Little is known about circulating HBV immune escape and Pol/RT mutants in Nigeria. This study focused on narrowing the knowledge gap of the pattern and prevalence of the HBV mutants across clinical cohorts of infected patients in southwestern Nigeria. Ninety-five enrollees were purposively recruited across clinical cohorts of HBV-infected patients with HBsAg or anti-HBc positive serological outcome and occult HBV infection. Total DNA was extracted from patients’ sera. HBV S and Pol gene-specific nested PCR amplification was carried out. The amplicons were further sequenced for serotypic, genotypic, phylogenetic, and mutational analysis. HBV S and Pol genes were amplified in 60 (63.2%) and 19 (20%) of HBV isolates, respectively. All the sixty HBV S gene and 14 of 19 Pol gene sequences were exploitable. The ayw4 serotype was predominant (95%) while ayw1 serotype was identified in 5% of isolates. Genotype E predominates in 95% of sequences, while genotype A, sub-genotype A3 was observed in 5%. Prevalence of HBV IEMs in the “a” determinant region was 29%. Commonest HBV IEM was S113T followed by G145A and D144E. The Pol/RT mutations rtV214A and rtI163V among others were identified in this study. This study provided data on the occurrence of existing and new HBV IEMs and Pol gene mutations in Nigeria.
Introduction
The World Health Organisation’s (WHO) global health-sector strategy aims for the elimination of viral hepatitis as a major public health threat by the year 2030 [Citation1]. The strategic direction number two and priority action entails interventions for impact ranging from prevention, early diagnosis and treatment of viral hepatitis [Citation1]. Hepatitis B virus (HBV) drug resistance mutations (DRM), vaccine, and diagnostic escape mutations may impair the current treatment and prevention strategy against HBV infection and delay the achievement of the goal [Citation2].
Hepatitis B virus (HBV) infection has varied clinical manifestations determined by factors such as level of HBV replication, patient’s age at infection, and infection with HBV mutant strains among others [Citation3]. Clinical entities that define HBV infection include acute HBV infection – this is defined by HBV infection with detectable sero-markers of acute infection such as HBsAg, and anti-HBc IgM within the first 6 months of infection. A large proportion of this clinical cohort of patients is asymptomatic, while some patients may present with fever, arthralgias, rash, right upper quadrant pain, and in extreme cases, fulminant liver failure [Citation4]. Chronic HBV infection is characterized by the persistence of HBsAg for more than 6 months [Citation5]. Nigeria and some sub-Saharan African countries are hyperendemic for HBV infection [Citation6]. The overall HBsAg seroprevalence of HBV infection in Nigeria was estimated to be about 12.2% based on an HBV national survey conducted in 2016 [Citation7]. Chronic HBV infection is asymptomatic in the majority of individuals [Citation8]; however, about 15–40% will progress to develop complications of chronic HBV infection ranging from liver cirrhosis to hepatocellular carcinoma (HCC) [Citation8]. Also, an increasingly important clinical entity known as occult hepatitis B infection (OBI) is being identified. It explains a phenomenon whereby HBV DNA material is detectable in the liver or the serum of an infected patient, with a negative HBsAg serological outcome [Citation9].
The Hepatitis B virus belongs to the genus Orthohepadnavirus and the family Hepadnaviridae [Citation10]. The HBV genome consists of an incomplete double-stranded DNA of approximately 3.2kb. It consists of four overlapping open reading frames (ORFs) which code for the HBV Polymerase enzyme, the S antigen, Core proteins (PreC/C), and the X protein [Citation11]. HBV replicates asymmetrically through an RNA intermediate using the viral genome-encoded reverse transcriptase [Citation12]. [Citation13] The HBV has a variable genome with 10 genotypes based on at least 8% divergence across the complete genome sequence [Citation14] and sub-genotypes based on 4–8% intra-genotypic divergence [Citation15]. HBV genotype displays distinct geographical distribution worldwide and genotypes A, D, and E are the commonest in Africa. HBV genotype E is the commonest genotype in West Africa [Citation6]
The HBV S gene ORF codes for the PreS1, PreS2, and the Surface antigen. The HBV S gene encoding the small HBsAg contains clusters of B-cell epitopes known as the Major Hydrophilic Region (MHR) which spans amino-acid position 99–169 [Citation16]. Within the MHR lies the antigenic determinant region (ADR), which is highly conserved across all HBV genotypes, and spans amino-acid position 124–147 of the Major Hydrophilic Region (MHR). It contains two major and one minor loop with a cysteine disulphide bond extending from the outer surface of the virus. HBsAg mutations associated with the natural course of infection target the first loop (amino acids 107 to 138) [Citation16]. The second hydrophilic loop (aa 139–147) is the main target for neutralizing antibodies from vaccines and immunoglobulins [Citation16,Citation17]. This confers protective immunity against all types of HBV genotypes by hepatitis B vaccines. Variations in amino-acid residues within this region may result in the evolution of viruses with altered conformational structures in the antigenic region, non-recognisable by neutralizing antibodies from vaccination, host natural immune system, or antibody-mediated diagnostic modalities targeting the antigenic region [Citation16]. HBV variants with a polymerase gene mutation may evolve under drug selective pressure with associated concomitant mutations in the antigenic region of the overlapping S gene, which is capable of altering the immune reactivity of the S antigen [Citation16,Citation17].
Mutations in HBV arise due to the unique replicative strategy involving a non-proofreading reverse transcriptase with resultant greater mutability than other DNA viruses [Citation16–18]. The rate of mutation in HBV ranges between 1.4–3.2 × 10−5 base substitutions/site/year [Citation19]. The evolution of HBV mutations involves selective pressure from endogenous factors like host immune pressure or exogenous factors, such as HBV vaccines, HBV immunoglobulins, and treatment with antivirals. The presence of such HBV immune escape variants is associated with immunoglobulin, vaccine failure, and diagnostic failure, and has been known to persist in hepatocytes. These are the potential causes of HBV reactivation following immunosuppression [Citation18,Citation19]. The earliest report of the HBV immune escape mutant was from a vaccinated Italian child who had a vaccine breakthrough HBV infection due to a mutant strain characterized to be G145R [Citation20]. Subsequently, other vaccine escape mutants had been identified involving amino-acid substitutions at positions 120,123,124,126,129,131,133,141,144 of the antigenic determinant [Citation21,Citation22].
In Nigeria, Faleye et al (2015) reported evidence of HBV immune escape mutants among pregnant women and asymptomatic individuals in southwestern Nigeria, and all belong to HBV genotype E [Citation23]. Adesina et al (2021) reported Q129H mutant variants among apparently healthy individuals in southwestern Nigeria [Citation24]. Among the repertoire of circulating immune escape mutants globally, only a few had been reported in Nigeria and none evaluated the occurrence of the immune escape mutants across various clinical cohorts of hepatitis B virus infection. Good knowledge of the frequencies of existing and new HBV IEMs, and clinical attributes of the mutant strains will promote the efficiency of diagnostic procedures, universal HBV immunization programmes and optimize HBV therapeutic guidelines. This study aims to evaluate the circulating HBV immune escape and Pol/RT mutations across clinical cohorts of patients with acute HBV infection, chronic HBV infection, chronic HBV infection with liver cirrhosis, hepatocellular carcinoma, and occult hepatitis B infection in southwestern Nigeria.
Materials and methods
Study location and sample collection
This work is a multi-centred, cross-sectional, hospital-based study carried out in the southwestern region of Nigeria. This study is part of a larger project on HBV serological and virological characterisation across various clinical cohorts in Nigeria. Outpatients attending Gastroenterology clinics (referred from the General Outpatient Department, Blood banks, and antenatal clinics based on incidental findings of positive HBsAg serological profiles) and inpatients being managed for complications of chronic HBV infection including liver cirrhosis and hepatocellular carcinoma were recruited through purposive sampling technique at the Ladoke Akintola University Teaching Hospital, Osogbo, Osun State, Nigeria, Federal Teaching Hospital, Ido- Ekiti and Ekiti State University Teaching Hospital, Ado-Ekiti in Ekiti State, Nigeria. Ethical approval was obtained from the Institutional Review Boards (IRB) of all the tertiary hospitals listed above. The protocol numbers of the ethical clearance certificates issued by the IRBs of the tertiary hospitals were LTH/EC/2020/08/467, ERC/2020/07/06/386B and EKSUTH/A67/2020/12/006, respectively. The study design conformed to the 2013 declaration of Helsinki. All patients with positive HBsAg or anti-HBc serological outcomes who consented were recruited into the study. Clinico-social data were collected from each respondent at the time of sample collection using self-administered structured questionnaires. For the larger study, the sample size was determined based on the prevalence of HBV infection in Nigeria. A total of 95 plasma samples were collected from clinical cohorts of patients ranging from acute HBV infection, n = 5 (the diagnosis of acute HBV infection was made based on positive anti-HBc IgM qualitative assay, clinical history, symptomatology, and patients were followed up for at least 8 months with evidence of anti-HBc IgM seroconversion), chronic HBV infection, n = 59, chronic HBV infection with liver cirrhosis, n = 6, and chronic HBV with hepatocellular carcinoma, n = 18, based on clinical information obtained between April 2019 and December 2020. Known patients with occult hepatitis B infection (OBI), n = 7 were purposively recruited from ongoing larger HBV studies. The patients with occult HBV infection (OBI) were defined by negative HBsAg, detectable HBV DNA in the serum with/without anti-HBc positive serological outcome. The HBV drug (nucleoside analogues) naïve and drug-experienced patients were recruited irrespective of their medication status from the clinical cohort of patients with HBV infection. Recruitment criteria for patients with chronic HBV infection with hepatocellular carcinoma include elevated levels of alpha-fetoprotein above the diagnostic limit (1000 ng/ml), presence of typical focal liver lesions detected by triphasic abdominal computed tomography and histology of biopsied liver mass while patients with chronic HBV-induced liver cirrhosis were recruited based on suggestive histological findings on liver biopsy, abdominal computed tomography, liver function test asides positive HBV serological markers. Patients with hepatocellular carcinoma were recruited irrespective of their treatment modalities. Five millilitres (5 mls) of blood were collected from each study participant by trained phlebotomists into appropriately labelled EDTA bottles. Blood samples were separated by centrifugation at 1500 g for 15 min. Plasma samples were aliquoted in pairs into labelled sterile cryovials, transported on ice and stored at −200C at the reference laboratory, African Centre of Excellence for Genomics of Infectious Diseases, Redeemers University, Nigeria for further serological and molecular analysis
HBV serological studies
Sandwich enzyme-linked immunosorbent assay (ELISA) was used for the detection of HBsAg, HBeAg, anti-HBs, anti-HBc IgG, and anti-HBc IgM from the serum of patients using Melsin Diagnostic kits, China, following the manufacturer’s instruction. The Emax endpoint microplate reader (Molecular Devices, California, USA) was used to determine the optical density and the result was interpreted according to the manufacturer’s instructions. The analytical endpoint sensitivity (lower detection limit) of the ELISA kit used was 5 mIU/ml. The clinical specificity and sensitivity of the Melsin ELISA kits used in Nigeria were 99.88% and 100% respectively. The batch numbers of the kits used range from MID-0002 to MID-00014.
DNA extraction
Total DNA was extracted from 200ul of HBsAg and anti-HBc positive plasma samples (n = 95) using the Qiagen DNEasy kit (Qiagen Germany) according to the manufacturer’s instructions. Eluted DNA (50ul) was stored at −80°C until analysed.
Amplification of the HBV S gene
The HBV S gene amplification was carried out using a nested PCR procedure targeting a 408 bp fragment, the partial “S” gene region as described earlier [Citation25,Citation26]. The position of the codons in the S gene covered by the PCR ranges from 22 to 170. The first-round PCR primers used were HBV_S1F (5”-CTAGGACCCCTGCTCGTGTT-3‘) and HBV_S1R (5’-CGAACCACTGAACAAATGGCACT-3‘), while the second-round primers were HBV_SNF (5’-GTTGACAAGAATCCTCACAATACC-3‘) and HBV_SNR (5’-GAGGCCCACTCCCATA-3”). First- and second-round PCR reaction conditions were similar except that extracted DNA was used as the template for the first-round PCR, while the first-round PCR products were used as a template for the second-round PCR amplification. The PCR amplification was carried out using a 50ul reaction containing 10ul of Redload Taq (Solis Biodyne, U.S.A), 2 µl of each of the primer stock (made in 24uM concentrations), 5 µl of DNA template and RNase-free water was added to make up reaction volume to 50ul. PCR procedure was carried out using Eppendorf Thermal cycler (Eppendorf, United Kingdom) as follows: 94°C for 3 min, followed by 45 cycles (denaturation, 94°C for 30 s, annealing, 55°C for 60 s and elongation, 70°C for 40 s with a ramp of 40% from 55°C to 70°C). The reaction was further elongated at 72°C for 7 min and held at 4°C until the reaction was terminated. PCR products were resolved on 2% agarose gel stained with ethidium bromide and viewed using a UV transilluminator.
Amplification of polymerase gene
A 742-bp DNA fragment spanning the polymerase region was amplified using HBV DNA polymerase forward and reverse primers as described earlier [Citation27]. The position of the codons in the Pol gene RT domain covered by the PCR ranges from 54 to 271. The Pol gene RT domain-specific primers used were Pol F −5“TCGTGGTGGACTTCTCTCAATT 3” and reverse 5“CGTTGACAGACTTTCCAATCAAT 3” [Citation27]. PCR amplification was carried out using a 50ul reaction containing 10ul of Redload Taq (Solis Biodyne, U.S.A.). Two microlitres of each of the primer stock (made in 25uM concentrations), 5 µl of DNA template and RNase-free water were added to make up to 50ul reaction volume. Thermal cycling was carried out using the Eppendorf Thermal cycler (Eppendorf, United Kingdom) as follows: 95°C for 5 min, and then 45 cycles of 95°C for 45 s, 56°C for 45 s, and 72°C for 45 s. A final elongation was set at 72°C for 10 min and held at 4°C. PCR products were resolved on 2% agarose gel, stained with ethidium bromide and viewed using a transilluminator [Citation27].
Amplicon sequencing
Polymerase chain reaction products positive for HBV partial S gene and Pol gene fragments with the expected molecular band sizes (408 bp and 742 bp respectively, codon positions 22–170 and 54–271 respectively) after viewing with an ultraviolet transilluminator were subjected to polymerase-chain reaction product purification and Bigdye chemistry sequencing at the African Centre of Excellence for Genomics of Infectious Diseases, Redeemers University, Nigeria. PCR second-round primers were used for each sequencing run.
Phylogenetic and serotypic analysis of HBV partial S gene and Pol gene sequences
The forward and reverse sequences per isolate were stitched into contigs for the HBV S and Pol genes. Each contig was subjected to a Blastn search on the National Centre for Biotechnology Information (NCBI) page. GenBank sequences selected from different countries globally with high percentage similarity scores were retrieved from the GenBank database. Reference sequences of HBV genotypes A-J were also downloaded from the HBV database (http://hbvdb.ibcp.fr/HBVdb/). The phylogram was constructed using the Maximum Likelihood method of the MEGA X software with the Kimura-2 parameter model and 1000 bootstrap replicates [Citation28,Citation29]. The Newick version of the phylogram constructed in Mega X was imported into the Interactive Tree of Life (iTOL) for annotation and presentation [Citation30]. HBV partial S gene sequences were aligned using the CLUSTAL W program in MEGA X software with default settings. Serotypic analysis of HBV isolates was carried out using amino-acid at positions 122, 127, 134, 159, and 160 of the HBV S gene sequences [Citation31,Citation32]. The exploitable forward and reverse HBV Pol gene sequences per isolate were merged into contigs and subjected to BLASTn search. Highly similar sequences were downloaded from the GenBank for Pol gene mutational analysis studies
DNA sequence accession numbers
The HBV S gene and Pol/RT domain sequences generated in this study were deposited into the NCBI GenBank under accession numbers OP420514 to OP420522, OP428653 to OP428701
Statistical analysis
The self-administered structured questionnaires were checked for accuracy and completeness, and data were double-entered to minimize data entry errors and then merged subsequently. Categorical data and median of continuous variables were compared by Chi-square and Mann–Whitney tests using SPSS version 25.0 (IBM, USA). For all statistical analyses, a p-value less than 0.05 was considered significant at a 95% confidence interval (CI).
Result
Socio-demographic characteristics of study participants
In this study, a total of ninety-five (95) participants who appropriately filled out the self-administered structured questionnaires and donated 5mls of blood were enrolled. Among these, 57(60%) were males while 38(40%) were females (). The mean age of study participants was 38.4 ± 11.7 years (age range: 21–66 years). The age groups 21–30 years and 31–40 years had the highest prevalence of 30.5% (n = 29) each (). The participants were recruited from clinical cohorts of patients with acute HBV infection, 5(5.3%), chronic HBV infection, 59(62.1%), chronic HBV infection with liver cirrhosis, 6(6.3%), chronic HBV infection with hepatocellular carcinoma, 18(18.9%). Occult hepatitis B infection (apparently healthy subjects with no clinical signs or symptoms, negative HBsAg with detectable HBV DNA in serum, 5(5.3%) and patients with hepatocellular carcinoma with occult HBV infection, 2(2.1%) (). It is pertinent to state here that this study is part of a larger HBV study in Nigeria, therefore seven known occult hepatitis B infected patients were recruited as earlier reported (two apparently HBV-negative hepatocellular carcinoma patients and five apparently healthy patients with occult HBV infection) were purposively recruited from the larger study. All the occult hepatitis B infected patients were HBsAg negative, anti-HBc positive with detectable HBV DNA in the serum. Study participants with HBV infection (chronic HBV infection and HBV-induced liver cirrhosis cohorts) were recruited from a pool of HBV drug (Tenofovir) experienced and drug naïve patients as well as participants with HBV-induced hepatocellular carcinoma irrespective of treatment modality. Among the study participants with HBV-induced hepatocellular carcinoma, 4(4.2%) were on Sorafenib chemotherapy while among patients with chronic HBV infection and HBV-induced liver cirrhosis, 11 (11.6%) were on Tenofovir.
Figure 1. Distribution of study participants across clinical cohorts of HBV infection.
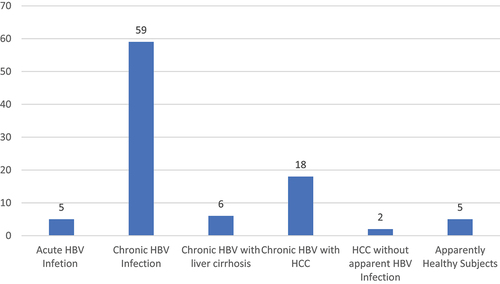
Table 1. Socio-demographic characteristics and HBV serological profiles of study participants.
HBV serologic profiles of study participants
The HBV HBsAg, HBeAg, anti-HBs, anti-HBc IgM, and anti-HBc IgG serological markers were assayed in all study participants. A significant proportion of participants was positive for HBsAg, 88 of 95 (). This was dominant in the age groups 21–30 years (30.5%, n = 29) and 31–40 years, 30.5% (n = 29), followed by the 41–50 years age group with a prevalence of 21.1% (n = 20) (). Interestingly, one (1.1%) study participant had positive anti-HBs with concurrent HBsAg positivity and positive anti-HBc IgG serological outcome, while all others were negative for anti-HBs. A high proportion of study participants were HBeAg negative (75.8%) (). Nineteen study participants (20.0%) had detectable anti-HBc IgM suggesting acute infection; however, 14 out of these 19 study participants had already been diagnosed HBV positive for more than 6 months, also had other serological features of chronic HBV infection coupled with the fact that they had already developed long-term complications of chronic HBV infection ranging from liver cirrhosis to hepatocellular carcinoma (). The remaining five patients with anti-HBc IgM were asymptomatic and followed up for at least 8 months with evidence of anti-HBc IgM seroconversion. The proportion of patients with positive anti-HBc IgM increases with age. Furthermore, on patients’ clinical cohort-specific analysis, chronic HBV-infected patients accounted for the bulk of patients recruited into the study (62.1%) while acute HBV-infected patients were the least (5.3%). Of note, is that the proportion of patients that were positive for anti-HBc IgM, a presumptive serological marker of acute HBV infection, was interestingly, relatively high among patients with chronic HBV with hepatocellular carcinoma, n = 12, 66.7% (). Also, the proportion of participants with HBeAg negative serological outcome was highest among occult HBV-infected participants, n = 6, 85.7% ().
S gene and Pol gene RT domain-specific nested polymerase chain reaction
Total DNA was extracted from plasma samples of all 95 study participants irrespective of their serological outcome. Of the 95 patients’ samples tested by HBV S region-specific nested PCR, 60 (63.2%) of the HBV isolates yielded the expected 408 bp amplicon [Citation26] while 19 (20%) of HBV isolates tested by Pol gene-specific nested PCR yielded the expected 742 bp amplicons [Citation27]. Out of the 19 Pol gene-specific amplicons, the sequence data of five samples were not exploitable due to the presence of multiple peaks. Sequence data of the remaining exploitable 14 sequences were used for Pol gene mutational analysis. All the 60 HBV S gene-specific amplicons yielded exploitable sequences for HBV serotypic, genotypic, phylogenetic, and mutational analyses.
Serotypic, genotypic, phylogenetic, and mutational analysis of HBV isolates
The HBV serotypes of the 60 S gene sequences were identified using the inferred amino acid residues at positions S122, S127, S134, S159, and S160 (Supplementary ) [Citation31,Citation32]. Fifty-seven (57) of the HBV isolates (all genotype E) belong to the serotype ayw4 while the remaining 3 isolates (all genotype A, sub-genotype A3) were serotype ayw1 (Supplementary , Supplementary ). The phylogram of HBV isolates in supplementary below showed that 57 of the 60 HBV S gene sequences generated from this study clustered around the genotype E while 3 HBV S gene sequences clustered around genotype A, sub-genotype A3 specifically. Two out of the three HBV genotype A sequences clustered around the Cameroonian strains while the remaining HBV isolate clustered around the Haitian strain (). We used the NCBI genotyping tool to confirm the HBV genotypes and sub-genotypes.
Figure 2. Rectangular phylogenetic tree depicts the rare genotype A3 sequences obtained from this study. Tree Scale: 0.05 -

Evolutionary analysis by maximum likelihood method
The evolutionary history was inferred by using the Maximum likelihood method [Citation27]. The tree with the highest log likelihood (−750.86) is shown in above. The percentage of trees in which the associated taxa clustered together is shown next to branches. Initial tree(s) for the heuristic search were obtained automatically by applying neighbor-join and BioNJ algorithms to a matrix of pairwise distances estimated using the Tamura-nei model, and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The proportion of sites where at least 1 unambiguous base is present in at least 1 sequence for each descendant clade is shown next to each internal node in the tree. This analysis involved 150 nucleotide sequences. All positions containing gaps and missing data were eliminated (complete deletion option). There were a total of 136 positions in the final dataset. Evolutionary analyses were conducted in MEGA X [Citation28]. The phylogenetic tree was imported in Newick format into the Interactive Tree of Life (iTOL) software for phylogenetic tree editing and viewing [Citation30]. The outlier sequence was obtained from the Chimpanzee HBV isolate. Alphabet and number of each label typifies the HBV Genotype and the sub-genotype separated by a space. The next series of alphabets and number is the genbank accession number which is followed by the country of origin of these sequences when available. HBV reference genotypes and study-derived sequences were collapsed and represented by a black triangle, the size of the triangle corresponds to the number of genomes making up the reference and study derived genotypes. The labels in red depict the sequences derived from the study which belong to the rare genotypes A3 from Nigeria.
The figure above depicts alignment of HBV S gene reference and study-derived amino acid sequences showing amino-acid substitutions across various segments. The HBV S gene immunodominant antigenic determinant region (ADR) which spans amino-acid position 124–147 is outlined in black box above with HBV immune escape mutants identified in selected HBV S gene sequences from the study.
The prevalence of HBV S gene amino acid substitutions among HBV isolates studied was 80% (n = 48/60, where 48 HBV sequences had S gene amino-acid substitutions and a total of 60 HBV sequences were studied). The HBV immune escape mutants identified in this study which span the major hydrophilic region (MHR) (amino acid residues position 99–169) of the HBV S gene include Y100C, M103K, L109V, I110L, L109P, G112R, S113T, S114T, T123I, T126I, Q129R, G130R, T131N, F134I, S140T, S140L, S143T, D144E, G145R, G145A, G159A, F161Y, and R169P. () (Supplementary , Supplementary ). Prevalence of HBV IEMs in the restricted, immunodominant “a” determinant region (ADR) between amino-acid positions 124–147 was 29% (n = 9/31, 9 connotes numbers of HBV S gene mutations between amino-acid positions 124–147 while 31 stands for the total number of HBV S gene mutations). Some HBV S gene amino-acid mutations occurring upstream (amino-acid residue position 22–98) of the Major Hydrophilic Region (MHR) were also observed in this study (, supplementary , Supplementary ). Supplementary shows the various HBV S gene amino-acid substitutions and immune escape mutants observed among HBV isolates studied. The HBV S gene region analysed in this study includes an area upstream of the Major Hydrophilic Region (MHR) spanning amino-acid position 22–98, the upstream region of the MHR is sub-divided into the first transmembrane helix (aa 1–20), the internal loop of the HBsAg (aa 27–79), second transmembrane helix (aa 80–98) [Citation33]. The MHR spans amino-acid region 99–169. Within the MHR lies the antigenic determinant region (ADR) which is an immunodominant region with clusters of B cell epitopes spanning amino-acid position 124–147. The HBV S gene region downstream of the antigenic determinant region occupies amino-acid position 148–169 [Citation16].
Figure 3. HBV S gene amino acid sequence alignment using bioedit software.
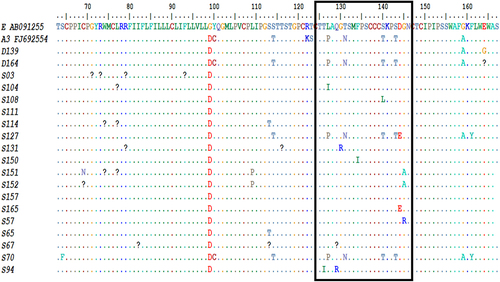
Table 2. Rate of HBV S gene pointmutation and mutation types across clinical cohorts of patients.
In this study, clinical cohorts of HBV-infected patients showed the variable inter-cohort rate of HBV mutation and mutational pattern. Patients’ cohort with occult HBV infection had the highest rate of HBV S gene mutation i.e., number of patients with HBV S gene mutations divided by the total number of HBV S gene sequences studied, n = 5/7, 71.4% (), followed by the clinical cohort of patients with HBV induced hepatocellular carcinoma, n = 11/17, 64.7%, while the least rate of HBV S gene mutation was observed among a clinical cohort of patients with acute HBV infection, n = 2/5, 40% and patient’s cohort with HBV-induced liver cirrhosis, n = 2/5, 40.0% (). The studied patient cohorts also displayed intra-cohort variation in mutation type and frequency. Across clinical cohorts of patients with chronic HBV infection, HBV-induced liver cirrhosis and HBV-induced hepatocellular carcinoma, K24R mutation predominates with a prevalence of n = 12, 12.6%, n = 2, 25% and n = 5, 14.7%, respectively, while among patients with occult HBV infection, S113T Mutation predominates, n = 3, 18.8% (). The highest number of HBV S gene point mutations in this study was observed among the clinical cohort of patients with chronic HBV infection (91 point mutations) followed by the clinical cohort of patients with chronic HBV infection complicated by hepatocellular carcinoma (34 point mutations) () Also, it is interesting to note that the clinical cohort of subjects without apparent HBV infection (occult hepatitis B infection) had a relatively high number of HBV S gene point mutations (13 point mutations) ()
Seven confirmed cases of occult hepatitis B virus infection were purposively recruited from the ongoing larger HBV study, two were identified as hepatocellular carcinoma with negative HBsAg serological profile. One of the patients was a 56 year old farmer, with positive anti-HBc IgG while all other HBV serological parameters were negative. The observed HBV S gene mutations include S113T+ R169A The second patient was a 50 year old male who had all HBV serological parameters negative except the anti-HBc IgG. The only HBV S gene mutation detected in this patient was K24R. Among the apparently healthy subjects, four of five HBV isolates had HBV IEMs. One patient had atypical serologic presentation of HBV evidenced by negative findings of HBsAg, positive HBeAg and anti-HBc IgG, while all other serological parameters were negative. This patient had HBV S gene mutations which include K24R+A45S+F134I+ R169A shown in Table 4 below. One of the apparently healthy enrollees currently do not harbour any HBV S gene immune escape mutation. ()
Table 3. Socio-demographic, serological, and HBV S gene mutational profiles of enrollees with occult HBV infection.
The overall prevalence of HBV sequences with Pol gene RT domain amino-acid substitutions among exploitable HBV Pol gene sequences studied was 100% (n = 14/14). All the 14 HBV Pol gene RT domain sequences studied had one or more amino acid substitutions. A total of 52 different Pol gene RT domain amino acid substitutions were observed at different sites shown in below, supplementary below. The I121N mutation had the highest frequency of all the observed mutations, occurring in 10 of the 14 HBV isolates, this was followed by the Y126H mutation observed in 8 of 14 HBV isolates () and D263E observed in 7 of 14 HBV isolates studied (). Specific putative reverse transcriptase mutation found in this study include the rtI163V mutation () implicated in Adefovir and Lamivudine drug resistance and rtV214A mutation () associated with Adefovir, Lamivudine, and Tenofovir resistance.
Figure 4. HBV Pol/RT mutational analysis using bioedit software. image depicts HBV polymerase gene sequence at amino- acid position 115–223 with the amino-acid substitutions observed in the sequences.
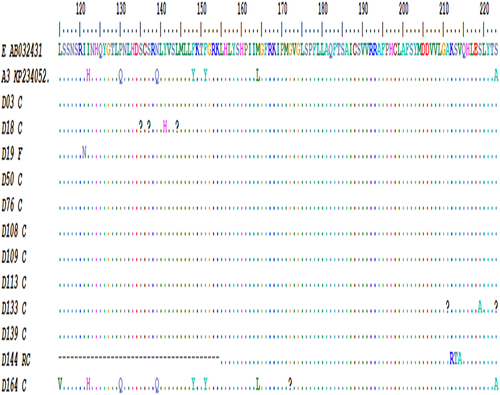
Figure 5. Distribution of Pol gene RT domain amino-acid substitutions among 14 HBV isolates analysed.
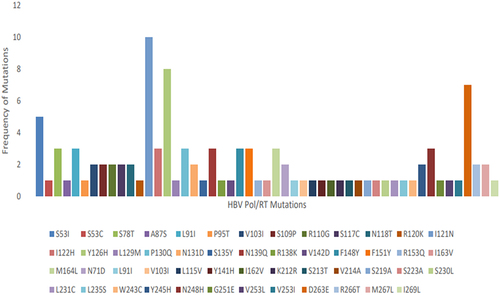
Each colour connotes specific amino-acid substitution observed in the study. The legend shows various colour-coded squares representing specific mutations starting from amino acid position 53–269.
Discussion
Hepatitis B virus (HBV) infection is a leading cause of end-stage liver diseases, liver cirrhosis and hepatocellular carcinoma-related morbidity and mortality in sub-Saharan African and south-east Asian countries [Citation34]. The HBV reverse transcriptase has a low-replicative fidelity with the resultant evolution of HBV mutants across all the genes of the virus [Citation16–19]. HBV immune escape mutants (IEM) and drug resistance mutants (DRM) had been associated with HBV vaccine, immunoglobulin, therapeutic and diagnostic failure globally [Citation18,Citation19]. Very few studies in Nigeria reported the circulating HBV immune escape mutants [Citation23,Citation24], and those studies were limited to the population of healthy, asymptomatic individuals and pregnant women. None evaluated the clinical characterization of the HBV mutants, and this is important to improve the efficiency and effectiveness of HBV diagnosis, immunoprophylactic programmes, therapeutic modalities, and guidelines. Therefore, this study focused on identifying the pattern and prevalence of HBV mutants across clinical cohorts of patients with acute HBV infection, chronic HBV infection, chronic HBV with liver cirrhosis, HBV-induced hepatocellular carcinoma, and occult hepatitis B virus infection. The study hypothesized the presence of HBV IEMs across various clinical cohorts of patients, intra and inter-cohort variability in the mutation type, mutation rate, and frequency across various cohorts of HBV-infected patients in southwestern Nigeria. We recruited 95 study participants with an overall male gender preponderance (Males, 60%, females, 40%) among the enrollees. However, age-specific gender analysis showed a preponderance of the female gender among the most populous age group 21–30 years. This is important because, this is the most sexually active, childbearing, and productive age group, infection with HBV IEMs of the productive women of this age group may increase the population of infants and neonates that may be infected perinatally with HBV vaccine and diagnostic escape mutants. On clinical cohort stratification of gender, the female gender also predominates among cohorts of patients with occult hepatitis B infection (71.4%). This has serious implications in light of the findings from earlier studies [Citation35,Citation36] and our study on the presence of specific HBV S gene mutations associated with occult hepatitis B infection. These mutations had been established to be very stable and vertically transmissible from infected mothers to children perinatally [Citation37–39]. Therefore, these infected women with occult HBV infection are potential reservoirs for maintaining inter- and intragenerational HBV vaccine and diagnostic escape mutants in this geographical space.
On HBV serological profiling of the total study population (n = 95), we observed about 93% were positive for HBsAg, while 7% were negative, the HBsAg negative population was attributable to the occult HBV infected patients. Interestingly, one study participant had concurrent HBsAg and anti-HBs serological outcome. This constitutes an atypical serological profile of HBV infection. Different reasons had been advanced for this observation ranging from the presence of an HBV S gene immune escape mutant with immunodominant epitopes incapable of eliciting neutralizing antibody response [Citation40,Citation41], HBV re-infection with a different strain of HBV that targets different HBsAg epitopes [Citation42], elicitation of heterologous subtype-specific antibodies targeting HbsAg subtypes different from the wild HBsAg [Citation43]. We also observed a relatively high proportion of participants with anti-HBcIgM presenting with clinical features in keeping with chronic HBV infection and its sequelae. In fact, patients with chronic HBV infection with hepatocellular carcinoma relatively had a high prevalence of anti-HBcIgM, n = 12, 66.7% (). Reasons proffered for the observation above may be that the presence of low-titre of anti-HBcIgM in chronic HBV infection may be attributable to co-existing presence of rheumatoid factor or IgG aggregates [Citation44]. Anti-HBcIgM antibodies may persist at low titres in patients with chronic HBV infection, and increase significantly during periods of acute exacerbation [Citation45]. About a quarter of the study participants recruited had positive HBeAg serological outcomes (n = 23, 24.2%). This seroprevalence of HBeAg positive outcome falls within the range of seroprevalence findings from other studies conducted locally here in Nigeria [Citation46–48]. Among the study participants with negative HBeAg serological outcome, the clinical cohort of patients with occult HBV infection had the highest proportion (85.7%). This is not entirely strange because it is known that HBV epigenetic alterations involving methylation and acetylation of the HBV genome had been associated with deficient production of HBsAg and concurrent reduced expression of HBV precore/core gene [Citation49,Citation50]. Also, the process of HBV genome integration in the host nucleus in the course of the HBV replication cycle disrupts the HBV genome architecture with resultant loss of HBsAg, inadequate virion production, reduced HBV Precore/Core gene expression and invariably HBeAg synthesis [Citation51].
The three forms of hepatitis B virus surface antigen (HBsAg), the large, medium, and small uniquely possess 226 amino-acid residues at the carboxyl end [Citation52]. The major hydrophilic region (MHR) of the HBsAg is found within the core of these amino acid residues, it occupies the amino-acid position 99–169 and it is made up of B cell-rich epitope clusters [Citation52]. Specifically, the immunodominant clusters of B cell epitopes within the MHR between amino-acid residues 124–147 known as the antigenic “a” determinant region (ADR) are unique sites aimed by neutralising antibodies. Amino acid substitutions at the “a” determinant region may therefore alter the antigenicity and the immunogenic conformation of HBsAg with the resultant evolution of immune escape mutants of HBV [Citation52]. These mutant strains may exhibit reduced response to HBV vaccines, immunoglobulins and evade detection by antibody-based commercial assays [Citation52]. Few studies had identified some of the circulating IEMs in Nigeria but there exists a dearth of information on the evaluation of the clinical attributes of these HBV mutants across clinical cohorts of HBV infection in Nigeria.
In this study, the overall prevalence of HBV S gene point mutations among HBV S gene sequences studied was 80% (n = 48/60). We identified HBV immune escape mutants (IEM) across clinical cohorts of patients ranging from acute HBV infection, 5 (5.2%), chronic HBV infection, 59 (62.1%), chronic HBV infection with liver cirrhosis, 6 (6.3%), HBV infection with hepatocellular carcinoma, 18 (18.9%). We also purposively selected seven confirmed cases of occult hepatitis B virus infection, 5, (5.3%) apparently healthy subjects, two patients (2.1%) with apparent HBV-negative hepatocellular carcinoma, among patients presenting at the gastroenterology clinics of selected tertiary healthcare facilities in southwestern Nigeria. The HBV IEMs identified in this study spanning the major hydrophilic region (MHR) (amino acid residue position 99–169) of the HBV S gene include Y100C, M103K, L109V, I110L, L109P, G112R, S113T, S114T, T123I, T126I, L127P, L127I, Q129R, G130R, T131N, F134I, S140T, S140L, S143T, D144E, G145R, G145A, G159A, F161Y, R169P, and R169A. Prevalence of HBV IEMs in the restricted, immunodominant “a” determinant region (ADR) between amino-acid positions 124–147 was 29% (n = 9/31, where 9 connotes the number of HBV S gene mutations occurring in the antigenic determinant region and 31 stands for the total number of HBV S gene mutations). The prevalence of HBV IEMs in this study was slightly higher than that reported by Araujo et al, 2020 (10.7%, Brazil) [Citation53], Di Lello et al, (2019) (7.5%–10.7%, Argentina) [Citation54], Yan et al, (2017) (9.01%, China) [Citation55], while it was similar to that reported by Adesina et al, in Nigeria (2021) 18% [Citation24]. This suggests a high circulating rate of HBV IEMs in southwestern Nigeria with implications for HBV diagnosis, vaccine effectiveness, and prognosis. From this study, asides the repertoire of HBV IEMs already established to be circulating in Nigeria by Faleye et al [Citation23] and Adesina et al [Citation24], we identified new putative HBV IEMs in Nigeria to the best of our knowledge. These include Y100C, M103K, L109P, I110L, L109P, G112R, S113T, S114T, T123I, T126I, G130R, T131N, F134I, S140L, D144E, G145A, and G159A across various clinical cohorts of HBV infection in southwestern Nigeria.
The commonest HBV IEM observed in this study was the S113T (11.6%), followed by the G145A, D144E and T131N with 3.3% prevalence each. The highest frequency of mutations was found in HBV isolate S152, followed by S151 and S157, respectively. All the three HBV isolates belong to genotype E. Interestingly, the HBV isolate with the highest frequency of HBV S gene mutations belongs to the occult hepatitis B infection cohort. The observation from this study is at variance with earlier studies that reported genotype A-related predominance of immune escape mutants. It is pertinent to note that HBV Genotype A had been reported globally to have higher rates of HBV IEMs [Citation56–58]. One of the reasons advanced for this observation may be that the current HBV recombinant vaccine in use globally is synthesized from the HBV genotype A, sub-genotype A2 [Citation56,Citation57]. Therefore, under the influence of vaccine selective pressure at the vaccine-specific antigenic determinant region of amino-acid positions 139–145, HBV vaccine escape mutants may evolve, specifically involving genotype A. However, the findings from this study on the genotype E predominance of HBV IEMs may not be representative as only three HBV genotype A isolates were studied in comparison with 57 HBV genotype E isolates.
Across the population of HBV-infected patients studied, the HBV S gene mutation rate was highest among the clinical category of patients with occult HBV infection (71.4%) and HBV-induced hepatocellular carcinoma (64.7%) while the acute HBV-infected clinical cohort had the least mutation rate (40%). It is instructive to note that among patients with HBV-induced HCC and chronic HBV clinical cohort, mutations upstream of the MHR had the highest mutational frequencies (K24R, G44E). This pattern was also observed among occult HBV-infected patients. These mutations belong to the class1 HLA-A2 restricted cytotoxic lymphocyte (CTL) epitope between amino-acid residues 29–53 [Citation59,Citation60]. Patients with hepatocellular carcinoma and chronic hepatitis have been established to have higher frequencies of these mutations [Citation59,Citation60]. This is also validated in our study. These mutations had been proposed as immune escape mutants that evolved under the T cell-mediated immune response [Citation35]. Also, it is known that the prevalence and frequency of HBV IEMs increase with time and age in an infected individual [Citation61,Citation62] which may be explained by the increased evolution of escape mutants under persistent host immune pressure, immunoprophylactic, and therapeutic pressure. This invariably explains the finding in our study on why patients with long-standing HBV infection and its sequelae; chronic HBV, HBV with HCC, and even occult HBV infection had the highest mutational rate as well as absolute numbers of observed mutations (). Similarly, for the same reason, it has been established there is an increased accumulation of HBV S gene mutations in HBV-associated end-stage liver diseases [Citation59].
The earliest reported HBV immune escape mutant ever discovered was the G145R HBV mutant involving Glycine replacement with Arginine at amino-acid position 145, an established and stable vaccine escape mutant [Citation35]. This HBV mutant was also observed in this study, interestingly, in a 52-year-old unvaccinated male patient with HBV-induced hepatocellular carcinoma. The serological findings in this patient include positive HBsAg and anti-HBcIgM, negative anti-HBs, HBeAg, and anti-HBe, serological outcomes. This lends credence to the work by Oon and Chen that attributed G145R HBV mutant to the causation of liver cancer among Singaporeans [Citation61]. This mutant strain is very stable, transmissible, and infectious as it possesses an efficient attachment to Heparan Sulphate and demonstrable infectivity in vitro [Citation63]. The protein docking method and other computational biology approaches employed in the study of the G145R mutant revealed suboptimal immunogenic activities of the HBsAg as a result of the change in the HBsAg loop structural conformation [Citation64]. Characteristically, most mutations present at the antigenic determinant region can change the hydrophilic, electrical, and acidity conformation of the ADR loops thus changing the loop structure and antigenicity of HBsAg [Citation35,Citation59]. The presence of this G145R mutant, a vaccine escape mutant in an unvaccinated individual raises some pertinent questions about the evolution of this HBV mutant becoming the dominant strain in the absence of immunoprophylactic pressure in an infected host. This appears to be a bit divergent from an earlier report by Lazarevic et al [Citation35] on the role of HBV vaccine in the evolution and dominance of this HBV mutant, in an attempt to proffer an answer to this observation, we can safely infer that this patient was infected ab initio with a vaccine escape mutant – G145R. Also, it is known that unvaccinated patients may harbour this mutation as a minor population, which on immunoprophylactic pressure may become the major population of mutants in an infected host [Citation35]. However, the question is, can a minor population of viral escape mutants assume a major population in the absence of immunoprophylactic pressure in an infected host? If not, then, the earlier reason advanced remains the most valid option. Traditionally, the G145R vaccine escape mutant is understandably observed in the majority of patients with positive serological anti-HBs outcome [Citation35,Citation65,Citation66]; however, this index patient had a negative anti-HBs serological profile and therefore establishes that G145R mutants may also be found in patients with negative anti-HBs serological profile. Other specific HBV vaccine escape mutants identified in this study include Y100C, T126I, Q129R, D144E, and G145A [Citation67].
The established and common HBV diagnostic escape mutants associated with occult hepatitis B infection present in this study include Y100C, T123I, T126I, P127L, Q129R, G145R, and G145A [Citation67]. It is instructive to note that most of the patients who had the HBV diagnostic escape mutants had no phenotypic expression of the mutations as most of these patients showed positive HBsAg serological outcomes. Among the clinical cohort of patients with occult HBV infection, two were identified as hepatocellular carcinoma without apparent HBV infection. One of the patients had a combination of the HBV S gene mutations – S113T+ R169A while the identified HBV S gene mutation in the second patient was K24R only. Among the apparently healthy subjects, four of five HBV isolates had HBV IEMs. One patient had an atypical serologic presentation of HBV evidenced by negative findings of HBsAg, positive HBeAg and anti-HBc IgG. This patient had HBV S gene mutation which includes K24R+A45S+F134I+ R169A. The second patient, a 53-year-old woman with the HBV S gene K24R+G44E+A45S+L109P+G145A mutational combination. All the serological markers were negative except the anti-HBc IgG. Also, from this study, we observed that phenotypic expression of the HBV diagnostic escape mutants among isolates with occult hepatitis B infection is combinatorial in most instances.
Occult HBV infection (OBI) has also been associated with an increased risk of transmitting HBV through blood transfusion, haemodialysis, and transplantation [Citation68]. It has also been implicated in cryptogenic liver diseases, development of hepatocellular carcinoma, and progressive chronic HBV infection [Citation68]. Among various mechanisms implicated in the development of occult HBV infection, presence of HBV S gene diagnostic escape mutant had been identified [Citation68]. One of such important mutation identified in this study is the G145A mutation. The G145A mutation had been traditionally associated with OBI with demonstrable capacity invitro and invivo to significantly reduce extracellular and intracellular HBsAg expression. This effect was more pronounced when G145A mutation co-exists with M103I, K122R, and G145A [Citation68]. One of the patients from this study had detectable HBsAg despite harbouring this mutation, with other co-existent mutations which includes L109P, K24R, and A45S. This implies there is complex interaction between this mutation (G145A) and its expression phenotypically. An explanation for the observation above may be that co-existent mutations may mask the phenotypic effect of the phenotypic of the G145A with resultant evolution of a revertant strain which phenotypically mimics the wild variant [Citation69]. Another known HBV diagnostic escape mutant documented to be associated with OBI identified in our study is the T131N. This mutation is characterised by an additional N-linked glycosylation site at amino acid position 131 [Citation35]. N-linked glycosylation is essential for survival and maintenance of biological processes of enveloped viruses [Citation70]. The HBsAg has many additional N-linked glycosylation sites such as those found in position 146, 115, 123, 113, and 131. This additional glycosylation of the HBsAg reduces HBsAg antigenicity and B cell epitope configuration with resultant development of occult HBV infection. The additional N-linked glycosylation sites hide the HBsAg B-cell epitope with non-recognition by humoral immune system [Citation71,Citation72].
The HBV Pol gene codes for the DNA Polymerase. An essential domain of the Pol gene, the reverse transcriptase (RT) enzyme domain serves as the nucleos(t)ide analogue (NA) drug target. The nucleos(t)ide analogue selective pressure on the RT domain may result in the evolution of HBV drug resistance mutants. The categories of RT drug resistance mutations to nucleos(t)ide analogues include: 1. The primary RT drug resistance mutations affecting amino-acid residues at positions 169,181,184, 202, 204, 236, 250 confers reduced HBV susceptibility to nucleos(t)ide analogues [Citation68,Citation69]. 2. The secondary (compensatory) RT drug resistance mutation restores or improves the functional capacity of the reverse transcriptase enzyme [Citation70,Citation71]. 3. The putative RT nucleoside analogue antiviral resistance mutation may arise in a setting of long-term usage of nucleos(t)ide analogue agents [Citation68]. 4. The pre-treatment RT drug resistance mutation [Citation67,Citation68]. Categories 3 and 4 drug resistance mutations are poorly characterized with inadequate phenotypic data and clinical implications of these mutations.
In this study, the prevalence of RT domain amino-acid substitution among HBV isolates was 100% (n = 14/14)). A total of 52 RT domain amino-acid substitutions spread across different sites were observed. The rtI121N mutation had the highest frequency, occurring in 10 of 14 HBV isolates studied, followed by the Y126H amino-acid substitutions observed in 8 HBV Pol gene/RT domain sequences out of 14 HBV sequences studied. The specific putative reverse transcriptase mutation observed in the study includes the rtV214A associated with Adefovir, Lamivudine, and Tenofovir resistance [Citation72]. It is instructive to note that this specific mutation was identified in a 50-year-old drug naïve patient with HBV-induced hepatocellular carcinoma suggesting that the mutation can arise pre-treatment with nucleos(t)ide analogues disproving earlier reports that it is associated with long-term NA therapy. We also identified rtI163V mutation implicated in drug resistance associated with Adefovir and Lamivudine long-term usage [Citation73]. The rtI163V had also been associated with Entecavir drug resistance [Citation74]. Only 4 (4.2%) of patients were on nucleoside analogue (Tenofovir), while the rest were not on any antiviral medication. The bulk of mutations observed in this drug naïve cohort of patients belong to the pre-treatment category of reverse transcriptase drug resistance.
Of the 60 HBV sequences analysed in this study, expectedly, a large proportion, 57 (95%) were found to belong to genotype E and three sequences (5%) belong to genotype A, specifically, sub-genotype A3. Nigeria belongs to the genotype E crescent [Citation75], and available inferences using the Bayesian inference method suggest that HBV genotype E is indigenous to West Africa, and originated from Nigeria [Citation76]. It is interesting to note that one of the HBV genotype A3 sequences (S127) clustered around the Haitian strain while another clustered around the Cameroonian strain (S70). On further investigation of the patient with isolate S127, he is a male teacher, whose state of origin is from one of the states in the south-south geopolitical zone of Nigeria but currently resides in Ekiti State in Nigeria. Neither patient himself nor his three generations earlier to the best of his knowledge had ever visited Haiti. These findings lend credence to the report by Andernach et al in 2009 on the role of phylogenetics and phylogeographical analysis of HBV in transatlantic slave trade migration studies in Haiti [Citation77]. A large proportion of the Haitian population are descendants of the African slaves [Citation78] who migrated through the transatlantic slave route about two centuries earlier from the Bight of Benin region in the Atlantic Ocean [Citation78]. Vertical and horizontal transmission of HBV infection among African slaves from their countries of origin enhances intra and intergenerational transfer of HBV. The circulating strains of HBV in Haiti (specifically Genotype A1 and A3) may therefore cluster around that of the country of origin of the slaves in Africa, especially those residing in countries around the Bight of Benin in the Atlantic Ocean [Citation79]. Coincidentally, this patient of interest is from the southern region of Nigeria close to the Bight of Benin in the Atlantic Ocean. Unlike Genotype E which available pieces of evidence suggested was introduced into the human population as late as towards the end of the 19th century when the slave trade had ended and therefore has very little genetic diversity and is largely confined to the African countries [Citation76,Citation80,Citation81]
It is pertinent to note that the serotypes identified in this study using the amino acids at position 122, 127, 136, 159, and 160 from the interpretable 60 HBV sequences revealed serotype ayw4 as the predominant serotype (95%) while 5% of the sequences belong to serotype ayw1. (). All the HBV sequences serotyped ayw4 belong to genotype E while the serotype ayw1 belongs to genotype A3. The prevalent serotype in Nigeria is the ayw4 and it follows the pattern of the genotype E. The serotype ayw1 is rare in Nigeria so also is the genotype A [Citation76]. Earlier studies in Nigeria showed the predominance of ayw4 serotype among genotype E sequences from southwestern Nigeria [Citation23] while Forbi et al in 2010 established the predominance of serotype ayw4 in two remote villages in the north-central region of Nigeria with very few HBV isolates classified as serotypes ayw1 and ayw2 [Citation76].
Conclusion
This study added to the repertoire of circulating hepatitis B virus (HBV) immune escape mutants (IEM) and Pol/RT domain mutants across different clinical cohorts of HBV infected patients in southwestern Nigeria. It provided data on the frequencies of existing and new HBV IEMs, clinical attributes of the mutant strains to promote the efficiency of diagnostic procedures, universal HBV immunization programmes and optimise HBV therapeutic guidelines. We therefore recommend larger studies on surveillance of HBV IEMs and DRMs spread across all the geopolitical zones of Nigeria.
Study limitation
A limitation of this study was that additional laboratory procedures such as the HBV viral load, liver function tests which would have further validated some of our findings could not be carried out due to financial constraints. Also, the yield of the exploitable Pol gene sequences were relatively low, therefore the observed circulating HBV drug resistance mutants were few.
Author contributions
Osasona G. Oluwadamilola (O.G.O) – conceptualisation, project management, sample collection, software, formal analysis, laboratory investigation, original draft preparation; Oguntoye Oluwatosin Oluwagbenga (O.O.O) – Manuscript review, editing, sample collection; Abdulkareem O.Lukman (A.O.L.) – manuscript review, editing, sample collection; Arowosaye Abiola Opeyemi (A.A.O.) – Sample collection, manuscript review, editting; Adewumi Moses Olubusuyi (A.M.O.)- Manuscript review and editing; Christian Happi (C.H.)- Conceptualisation, manuscript review and editing; Folarin Onikepe (F.O.) – Supervision, conceptualisation,manuscript review and editing.
Supplemental Material
Download MS Word (3.9 MB)Acknowledgements
The authors thank Dr Ijarotimi, Dr Abiola and the entire staff of the Gastroenterology unit of the Ladoke Akintola University Teaching Hospital, Osogbo, Nigeria for guidance and assistance in the course of the field in the unit. We also appreciate Dr Yusuf Musa and Dr Ariyo Olumuyiwa of the Federal Teaching Hospital, Ido-Ekiti, Dr Ijeoma Ifeorah for their kind gestures and support in the set-up of this study. We sincerely appreciate Mr Ekerette Udoh (E.U) for his support with data analysis. The authors would like to appreciate the African Centre of Excellence for Genomics of Infectious Diseases, Redeemers University, Ede for making laboratory facilities available to conduct this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplemental data
Supplemental data for this article can be accessed online at https://doi.org/10.1080/21505594.2023.2218076.
Data Availability statement
The hepatitis B virus Polymerase and S gene sequences obtained from this study had been deposited in the Genbank sequence database under accession numbers OP420514 to OP420522, OP428653 to OP428701 https://mail.google.com/mail/u/0/#search/gb-admin%40ncbi.nlm.nih.gov/FMfcgzGqQSSScVjrPsPRrPvRSnpZmLjr.
Additional information
Funding
References
- World Health Organisation Global Health Sector Strategies on respectively, HIV, Viral hepatitis and sexually transmitted infections for the period 2022-2023, 2016-2021. Accessed on 24 November, 2021.
- Mokaya J, McNaughton AL, Hadley MJ, et al. A systematic review of hepatitis B virus (HBV) drug and vaccine escape mutations in Africa. A call for urgent action. PLoS Negl Trop Dis. 2018;12(8):e0006629.
- Han YF, Zhao J, Ma LY, et al. Factors predicting occurrence and prognosis of hepatitis B related hepatocellular carcinoma. World J Gastroenterol. 2011 Oct 14;17(38):4258–18. DOI:10.3748/wjg.v17.i38.4258
- Burns GS, Thompson AJ. Viral hepatitis B: clinical and epidemiological characteristics. Cold Spring Harb Perspect Med. 2014 Oct 30;4(12):a024935. PMID: 25359547; PMCID: PMC4292086. DOI:10.1101/cshperspect.a024935
- Noordeen F. Hepatitis B virus infection: An insight into infection outcomes and recent treatment options. VirusDis. 2015 Jun;26(1–2):1–8. Epub 2015 Apr 5. PMID: 26436115; PMCID: PMC4585049. DOI:10.1007/s13337-015-0247-y
- Kramvis A, Kew MC. Epidemiology of hepatitis B virus in Africa, its genotypes and clinical associations of genotypes. Hepatol Res. 2007 Jul;37(s1):S9–S19. PMID: 17627641. DOI:10.1111/j.1872-034X.2007.00098.x
- Olayinka AT, Oyemakinde A, Balogun MS, et al. Seroprevalence of hepatitis b infection in Nigeria: A national survey. Am J Trop Med Hyg. 2016 Oct 5;95(4):902–907. Epub 2016 Aug 15. PMID: 27527630; PMCID: PMC5062798. DOI:10.4269/ajtmh.15-0874
- Krajden M, McNabb G, Petric M. The laboratory diagnosis of hepatitis B virus. Can J Infect Dis Med Microbiol. 2005 Mar;16(2):65–72. PMID: 18159530; PMCID: PMC2095015. DOI:10.1155/2005/450574
- Kwak MS, Kim YJ. Occult hepatitis B virus infection. WJH. 6(12): PMID: 25544873; PMCID: PMC4269905:860–869. 2014 Dec 27. DOI:10.4254/wjh.v6.i12.860
- Schaefer S. Hepatitis B virus taxonomy and hepatitis B virus genotypes world J. Gastroenterol. 2007. Jan 7;13(1):14–21. DOI:10.3748/wjg.v13.i1.14
- Ganem D, Schneider R. Hepadnaviridae : the viruses and their replication. In: Howley P, editor knipe Dm. Fields Virology.Philadelphia: Lippincott-Raven; 2001. pp. 2923–2970.
- Utsumi T, Yano Y, Hotta H. Molecular epidemiology of hepatitis B virus in Asia. WJMG. 2014;4(2):19–26.
- Sunbul M. Hepatitis B virus genotypes: global distribution and clinical importance. World J Med Genetics. 2014;4(2):19–26.
- Norder H, Courance AM, Coursaget P, et al. Genetic Diversity of hepatitis b virus strains derived worldwide: Genotypes, subgenotypes, and HBsAg subtypes. Intervirology. 2004;47(6):289–309. DOI:10.1159/000080872
- Kramvis A. Genotypes and genetic variability of hepatitis B virus. Intervirology. 2014;57(3–4):141–150. DOI:10.1159/000360947. Epub 2014 Jul 15. PMID: 25034481.
- Cuestas ML, Mathet VL, Ruiz V, et al. Unusual naturally occurring humoral and cellular mutated epitopes of hepatitis B virus in a chronically infected argentine patient with anti-HBs antibodies. J Clin Microbiol. 2006 Jun;44(6):2191–2198. PMID: 16757620; PMCID: PMC1489447. DOI:10.1128/JCM.00057-06
- Romanò L, Paladini S, Galli C, et al. Hepatitis B vaccination. Hepatitis B Vaccination Hum Vaccin Immunother. 2015;11(1):53–57. DOI:10.4161/hv.34306. Epub 2014 Nov 1. PMID: 25483515; PMCID: PMC4514213.
- Yano Y, Azuma T, Hayashi Y. Variations and mutations in the hepatitis B virus genome and their associations with clinical characteristics. WJH. 2015;7(3):583–592.
- Salpini R, Colagrossi L, Bellocchi MC, et al. Hepatitis B surface antigen genetic elements critical for immune escape correlate with hepatitis B virus reactivation upon immunosuppression. Hepatology. 2015;61:823–833. CrossRef. DOI:10.1002/hep.27604
- Zanetti AR, Tanzi E, Manzillo G, et al. Hepatitis B variant in Europe. Lancet. 1988;2(8620):1132–1133. DOI:10.1016/S0140-6736(88)90541-7.
- Weber B. Genetic variability of the S gene of hepatitis B virus: Clinical and diagnostic impact. J Clin Virol. 2005;32(2):102–112.
- Kreutz C. Molecular, immunological and clinical properties of mutated hepatitis B virus. J Cell Mol Med. 2022;6(1):113–143.
- Faleye TO, Adewumi OM, Ifeorah IM, et al. Detection and circulation of hepatitis B virus immune escape mutants among asymptomatic community dwellers in Ibadan, southwestern Nigeria. Int J Infect Dis. 2015;39:102–109. Epub 2015 Aug 14. PMID: 26283552. DOI:10.1016/j.ijid.2015.08.008
- Adesina OA, Akanbi OA, Opaleye OO, et al. Detection of Q129H immune escape mutation in apparently healthy hepatitis B virus carriers in southwestern Nigeria. Viruses. 2021;1273(7):29. DOI:10.3390/v13071273. PMID: 34210073; PMCID: PMC8310067.
- Forbi JC, Vaughan G, Purdy MA, et al. Epidemic history and evolutionary dynamics of hepatitis b virus infection in two remote communities in rural Nigeria. PLoS ONE. 2011;5(7):e11615. DOI:10.1371/journal.pone.0011615
- Faleye TOC, Adewumi MO, Ifeorah IM, et al. Detection of hepatitis B virus isolates with mutations associated with immune escape mutants among pregnant women in Ibadan, southwestern Nigeria. SpringerPlus. 2015;4(1):43. DOI:10.1186/s40064-015-0813-1
- Koyaweda GW, Ongus JR, Machuka E, et al. Detection of circulating hepatitis B virus immune escape and polymerase mutants among HBV-positive patients attending institut pasteur de Bangui, central African republic. Int J Infect Dis. 2020; Jan;90:138–144. DOI:10.1016/j.ijid.2019.10.039. Epub 2019 Nov 1. PMID: 31682960; PMCID: PMC6912157.
- Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10(3):512–526.
- Kumar S, Stecher G, Li M, et al. Molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–1549.
- Letunic I, Bork P. Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 2 July 2019;47(W1):W256–W259. DOI:10.1093/nar/gkz239
- Forbi JC, Ben-Ayed Y, Xia GL, et al. Disparate distribution of hepatitis B virus genotypes in four sub-Saharan African countries. J Clin Virol. 2013 Sep;58(1):59–66. Epub 2013 Jul 17. PMID: 23871163; PMCID: PMC4591023. DOI:10.1016/j.jcv.2013.06.028
- Velkov S, Protzer U, Michler T. Global occurrence of clinically relevant hepatitis B Virus variants as found by analysis of publicly available sequencing data. Viruses. 12(11): PMID: 33238650; PMCID: PMC7700573:1344. 2020 Nov 23. DOI:10.3390/v12111344
- Schädler S, Hildt E. HBV life cycle: entry and morphogenesis. Viruses. 2009 Sep;1(2):185–209. Epub 2009 Sep 1. PMID: 21994545; PMCID: PMC3185491. DOI:10.3390/v1020185
- Kedar Mukthinuthalapati VVP, Sewram V, Ndlovu N, et al. Hepatocellular carcinoma in Sub-Saharan Africa. JCO Glob Oncol. 2021 May;7(7):756–766. PMID: 34043413; PMCID: PMC8457845. DOI:10.1200/GO.20.00425
- Lazarevic I, Banko A, Miljanovic D, et al. Immune-escape hepatitis B virus mutations associated with viral reactivation upon Immunosuppression. Viruses. 11(9): PMID: 31450544; PMCID: PMC6784188:778. 2019 Aug 24. DOI:10.3390/v11090778
- Elkady A, Iijima S, Aboulfotuh S, et al. Characteristics of escape mutations from occult hepatitis B virus infected patients with hematological malignancies in South Egypt. WJH. 9(9): PMID: 28396718; PMCID: PMC5368625:477–486. 2017 Mar 28. DOI:10.4254/wjh.v9.i9.477
- Locarnini S. Molecular virology of hepatitis B virus. Semin Liver Dis. 2004;24:3–10. DOI:10.1055/s-2004-828672. [PubMed Google Scholar].
- Mathet VL, Feld M, Espínola L, et al. 69.Hepatitis B virus S gene mutants in a patient with chronic active hepatitis with circulating anti-HBs antibodies. J Med Virol. 2003;69(1):18–26. PubMed] [Google Scholar. DOI:10.1002/jmv.10267
- Thakur V, Kazim SN, Guptan RC, et al. 70.Transmission of G145R mutant of HBV to an unrelated contact. J Med Virol. 2005;76(1):40–46. PubMed] [Google Scholar. DOI:10.1002/jmv.20321
- Carman WF, Zanetti AR, Karayiannis P, et al. Vaccine induced escape mutants of hepatitis B virus. Lancet. 1990;336:325–329.
- Zhang JM, Xu Y, Wang XY, et al. Co-existence of hepatitis B surface antigen (HBsAg) and heterologous subtype specific antibodies to HBsAg among patients with chronic hepatitis B virus infection. Clin Infect Dis. 2007;44(9):1161–1169.
- Aydin N, Kirdar S, Uzun N, et al. Atypical serological profiles in hepatitis B infections: investigations of S gene mutations in cases with concurrently positive for HBsAg and anti-HBs. Mikrobiyoloji bulteni. 2016;50(4):533–543.
- Ponde RA. Atypical Serological Profiles in Hepatitis B virus infection. Er J Clin Microbiol Inf Dis. 2013;32(4):461–476.
- Tedder RS, Wilson Croome R. IgM antibody response to the hepatitis B core antigen in acute and chronic hepatitis B. J Hyg. 1981;86(2):163–173.
- Konerman MA, Lok AS. Epidemiology, Diagnosis and Natural History of Hepatitis B. Lok in zakim and boyer’s hepatology. 7th ed. Amsterdam, Netherlands: Elsevier; 2018.
- Lesi OA, Audu RA, Okwuraiwe AP, et al. Serological and virological markers of nigerian patients with hepatitis B infection. Niger J Clin Pract. 2019 Apr;22(4):534–538. PMID: 30975959. DOI:10.4103/njcp.njcp_273_17
- Forbi JC, Iperepolu OH, Zungwe T, et al. Prevalence of hepatitis B e antigen in chronic HBV carriers in North-central Nigeria. J Health Popul Nutr. 2012 Dec;30(4):377–382. PMID: 23304903; PMCID: PMC3763608. DOI:10.3329/jhpn.v30i4.13289
- Anaedobe CG, Fowotade A, Omoruyi CE, et al. Prevalence, sociodemographic features and risk factors of hepatitis B virus infection among pregnant women in Southwestern Nigeria. Pan Afr Med J. 20 PMID: 26301010; PMCID: PMC4524914 2015 Apr 24: 406. DOI:10.11604/pamj.2015.20.406.6206
- Miller RH, Robinson WS. Integrated hepatitis B virus DNA sequences specifying the major viral core polypeptide are methylated in PLC/PRF/5 cells. Proc Natl Acad Sci U S A. 1983 May;80(9):2534–2538. PMID: 6302693; PMCID: PMC393860. DOI:10.1073/pnas.80.9.2534
- Vivekanandan P, Thomas D, Torbenson M. Hepatitis B viral DNA is methylated in liver tissues. J Viral Hepat. 2008 Feb;15(2):103–107. PMID: 18184192. DOI:10.1111/j.1365-2893.2007.00905.x
- Raimondo G, Burk RD, Lieberman HM, et al. Interrupted replication of hepatitis B virus in liver tissue of HBsAg carriers with hepatocellular carcinoma. Virology. 1988 Sep;166(1):103–112. PMID: 2842938. DOI:10.1016/0042-6822(88)90151-1
- Coleman PF. Detecting hepatitis B surface antigen mutants. Emerg Infect Dis. 2006 Feb;12(2):198–203. PMID: 16494742; PMCID: PMC3293431. DOI:10.3201/eid1203.050038
- Araujo NM, Teles SA, Spitz N. Spitz comprehensive analysis of clinically significant hepatitis B virus mutations in relation to genotype, subgenotype and geographic region. Front Microbiol. 2020;11. DOI:10.3389/fmicb.2020.616023
- Lello D, A F, Ridruejo E, et al. Molecular epidemiology of hepatitis B virus mutants associated with vaccine escape, drug resistance and diagnosis failure. J Viral Hepat. 2019;26(5):552–560.
- Yan B, Lu J, Feng Y, et al. Temporal trend of hepatitis B surface mutations in the post-immunization period: 9 years of surveillance (2005–2013) in eastern China. Sci Rep. 2017;7(1):6669. DOI:10.1038/s41598-017-07085-z
- Ma Q, Wang Y. Comprehensive analysis of the prevalence of hepatitis B virus escape mutations in the major hydrophilic region of surface antigen. J Med Virol. 2012;84(2):198–206.
- Kay A, Zoulim F. Hepatitis B virus genetic variability and evolution. Virus Res. 2007;127(2):164–176.
- Echevarría JM, Avellón A. Hepatitis B virus genetic diversity. J Med Virol. 2006;78(S1):S36–S42. DOI:10.1002/jmv.20605. [PubMed] [CrossRef] [Google Scholar] [Ref list].
- Purdy MA. Hepatitis B virus S gene escape mutants. Asian J Transfus Sci. 2007 Jul;1(2):62–70. PMID: 21938236; PMCID: PMC3168123. DOI:10.4103/0973-6247.33445
- Chen WN, Oon CJ. Mutation “hot spot” in HLA class I-restricted T cell epitope on hepatitis B surface antigen in chronic carriers and hepatocellular carcinoma. Biochem Biophys Res Commun. 1999;262(3):757–761.
- Oon CJ, Chen WN. Current aspects of hepatitis B surface antigen mutants in Singapore. J Viral Hepat. 1998;5(s2):17–23.
- Hsu HY, Chang MH, Liaw SH, et al. Changes of hepatitis B surface antigen variants in carrier children before and after universal vaccination in Taiwan. Hepatology. 1999;30:1312–1317.
- Sureau C, Salisse J. A conformational heparan sulfate binding site essential to infectivity overlaps with the conserved hepatitis B virus a-determinant. Hepatology. 2013;57(3):985–994.
- Rezaee R, Poorebrahim M, Najafi S, et al. Impacts of the G145R mutation on the structure and immunogenic activity of the hepatitis B surface antigen: A computational analysis. Hepat Mon. 2016;16(7):e39097.
- Colson P, Borentain P, Coso D, et al. Hepatitis B virus reactivation in HBsAg-negative patients is associated with emergence of viral strains with mutated HBsAg and reverse transcriptase. Virology. 2015;484:354–463. Google Scholar] [CrossRef. DOI:10.1016/j.virol.2015.06.017
- Inoue J, Kondo Y, Wakui Y, et al. Reactivation of resolved hepatitis B virus infection with immune escape mutations after long-term corticosteroid therapy. Clin J Gastroenterol. 2016;9(2):93–98.
- Yang JX, Liu BM, Li XG, et al. Profile of HBV antiviral resistance mutations with distinct evolutionary pathways against nucleoside/nucleotide analogue treatment among Chinese chronic hepatitis B patients. Antivir Ther. 2010;15(8):1171–1178. PMID: 21149924. DOI:10.3851/IMP1677
- Lazarevic I. Clinical implications of hepatitis B virus mutations: recent advances. WJG. 2814;20(24):7653–7664. [PMC free article] [PubMed] [CrossRef] [Google Scholar] [Ref list]. DOI:10.3748/wjg.v20.i24.7653
- Lok AS, Zoulim F, Locarnini S, et al.Antiviral drug-resistant HBV: standardization of nomenclature and assays and recommendations for management. Hepatology. 2007; Jul;46(1):254–265. PMID: 17596850. DOI:10.1002/hep.21698
- Liu BM, Li T, Xu J, et al. Characterization of potential antiviral resistance mutations in hepatitis B virus reverse transcriptase sequences in treatment-naïve Chinese patients. Antiviral Res. 2010 Mar;85(3):512–519. Epub 2009 Dec 23. PMID: 20034521. DOI:10.1016/j.antiviral.2009.12.006
- Deng L, Tang H. Hepatitis B virus drug resistance to current nucleos(t)ide analogs: Mechanisms and mutation sites. Hepatol Res. 2011;41(11):1017–1024.
- Harris BJ, Holzmayer V, Qureshi H, et al. Hepatitis B genotypes and surface antigen mutants present in Pakistani blood donors. PLoS ONE. 2017 Jun 5;12(6):e0178988. PMID: 28582431; PMCID: PMC5459465. DOI:10.1371/journal.pone.0178988
- Archampong TN, Boyce CL, Lartey M, et al. HBV genotypes and drug resistance mutation in anti-retroviral treatment naïve and treatment experienced HBV-HIV-Co-infected patients. Antivir Ther. 2017;22(1):13–20.
- Arrese E, Basaras M, Blanco S, et al. Evolution of hepatitis B virus during long-term therapy in patients with chronic hepatitis B. Ann Hepatol. 2011 Oct-Dec;10(4):434–440.
- Mokaya J, Vasylyeva TI, Barnes E, et al. Global prevalence and phylogeny of hepatitis B virus (HBV) drug and vaccine resistance mutations. J Viral Hepat. 2021 Aug;28(8):1110–1120. Epub 2021 May 7. PMID: 33893696; PMCID: PMC8581767. DOI:10.1111/jvh.13525
- Andernach IE, Hübschen JM, Muller CP. Hepatitis B virus: the genotype E puzzle. Rev Med Virol. 2009 Jul;19(4):231–240. PMID: 19475565. DOI:10.1002/rmv.618
- Forbi JC, Vaughan G, Purdy MA, et al. Epidemic history and evolutionary dynamics of hepatitis b virus infection in two remote communities in rural Nigeria. PLoS ONE. 2010 Jul 19;5(7):e11615. PMID: 20657838; PMCID: PMC2906510. DOI:10.1371/journal.pone.0011615
- Andernach IE, Nolte C, Pape JW, et al. Slave trade and hepatitis B virus genotypes and subgenotypes in Haiti and Africa. Emerg Infect Dis. 2009 Aug;15(8):1222–1228. PMID: 19751583; PMCID: PMC3467954. DOI:10.3201/eid1508.081642
- Central Intelligence Agency. 2022.[cited August16 2022]. World factbook. https://www.cia.gov/library/publications/the-world-factbook/index.html
- Ampah KA, Pinho-Nascimento CA, Kerber S, et al. Limited genetic diversity of hepatitis b virus in the general population of the offin river valley in Ghana. PLoS ONE. 2016;11(6):e0156864. [PMC free article] [PubMed] [Google Scholar] [Ref list]. DOI:10.1371/journal.pone.0156864
- Lago BV, Mello FC, Ribas FS, et al. Analysis of complete nucleotide sequences of angolan hepatitis B virus isolates reveals the existence of a separate lineage within genotype E. PLoS ONE. 2014;9(3):e92223. [PMC free article] [PubMed] [Google Scholar] [Ref list]. DOI:10.1371/journal.pone.0092223