
ABSTRACT
Influenza viruses, including four major types (A, B, C, and D), can cause mild-to-severe and lethal diseases in humans and animals. Influenza viruses evolve rapidly through antigenic drift (mutation) and shift (reassortment of the segmented viral genome). New variants, strains, and subtypes have emerged frequently, causing epidemic, zoonotic, and pandemic infections, despite currently available vaccines and antiviral drugs. In recent years, avian influenza viruses, such as H5 and H7 subtypes, have caused hundreds to thousands of zoonotic infections in humans with high case fatality rates. The likelihood of these animal influenza viruses acquiring airborne transmission in humans through viral evolution poses great concern for the next pandemic. Severe influenza viral disease is caused by both direct viral cytopathic effects and exacerbated host immune response against high viral loads. Studies have identified various mutations in viral genes that increase viral replication and transmission, alter tissue tropism or species specificity, and evade antivirals or pre-existing immunity. Significant progress has also been made in identifying and characterizing the host components that mediate antiviral responses, pro-viral functions, or immunopathogenesis following influenza viral infections. This review summarizes the current knowledge on viral determinants of influenza virulence and pathogenicity, protective and immunopathogenic aspects of host innate and adaptive immune responses, and antiviral and pro-viral roles of host factors and cellular signalling pathways. Understanding the molecular mechanisms of viral virulence factors and virus-host interactions is critical for the development of preventive and therapeutic measures against influenza diseases.
1. Introduction
Influenza viruses (flu) have a global distribution and are both human and animal pathogens. Human influenza viruses cause annual epidemics (seasonal), which are highly contagious respiratory infections that can lead to severe illness and life-threatening complications in high-risk groups [Citation1]. Occasionally, a new human influenza virus strain would arise from an animal origin and spread rapidly among human populations that have no pre-existing immunity, causing excessive mortality and morbidity globally, known as a pandemic. There have been four influenza pandemics in modern times, the 1918–19 “Spanish” flu, the 1957 “Asia” flu, the 1968 “Hong Kong” flu, and the 2009 “Swine flu.” The 1918–19 “Spanish” flu was the most severe pandemic in recent history, which was estimated to have caused ~ 500 million infections and 50–100 million deaths worldwide [Citation2]. Humans can also be sporadically infected by animal influenza viruses (zoonotic), most often avian and swine flu; however, they have yet to establish sustained infection in humans. In particular, avian H5 and H7 influenza A viruses have caused hundreds to thousands of infections in humans, with a high case fatality rate (30–50%) (reviewed in [Citation3]). The likelihood of these avian influenza viruses gaining efficient human-to-human transmissions to cause the next pandemic poses a significant public health risk.
Current control measures for influenza infections include vaccines and antiviral drugs. However, influenza viruses have evolved rapidly to escape vaccine immunity and develop drug resistance. New viral variants exhibit reduced binding of vaccine-elicited neutralizing antibodies, causing vaccine mismatches or reduced vaccine efficacy. Mutations resistant to US Food and Drug Administration (FDA)-approved antiviral drugs have been detected [Citation4]. Some become predominant in circulating strains, making the class of antivirals, such as M2 inhibitors, ineffective. Broadly protective vaccines and novel therapeutics are urgently needed to combat ever-changing influenza viruses.
Significant progress has been made in the understanding of the molecular mechanisms of influenza viral replication, transmission, and disease pathogenesis. This review summarizes the current knowledge of viral genetic markers and host responses affecting influenza virus pathogenicity and virulence, which will help in the development of better prevention and treatment options.
2. Mechanism of influenza virus replication and pathogenesis
2.1. Influenza virus genome and genes
Influenza viruses belong to the Orthomyxoviridae family and include four major types A, B, C, and D. Influenza A virus (IAV) can infect a wide range of avian and mammalian species, including humans, birds, ducks, chickens, turkeys, pigs, horses, and dogs [Citation5]. Influenza B virus (IBV) infects humans and seals, whereas influenza C virus (ICV) infects humans and pigs [Citation5]. IAV, IBV, and ICV are human pathogens. The influenza D virus (IDV), discovered in 2011, infects pigs and cattle, with no human infections reported to date [Citation6]. This review focuses primarily on IAV as it causes most human and animal flu infections.
IAV are enveloped RNA viruses with eight single-stranded negative-sense RNA segments (). Viral envelope proteins include two major glycoproteins, haemagglutinin (HA) and neuraminidase (NA), and the transmembrane protein M2. Beneath the viral membrane is the matrix lattice formed by the M1 protein. Inside viral particles are eight viral RNA (vRNA) segments in the form of the viral ribonucleoprotein complex (vRNP), of which each vRNA is encapsidated by nucleoproteins (NPs) and associated with the PB1/PB2/PA polymerase complex ().
Figure 1. Illustration of influenza virion components, genomic organization, and viral genes. (a) Influenza virus is an enveloped RNA virus, which has 3 envelope proteins (HA, NA, and M2) on the viral membrane, an M1-formed matrix layer, and eight vRnps. (b) Each vRNP consists of one vRNA segment wrapped with NP and associated with polymerase complex PB2/PB1/PA. (c) Each vRNA segment encodes 1–3 genes, through alternative splicing (NS2/NEP and M2) and frameshifting (such as PA-X and PB1-F2). Accessory proteins expressed through frameshifting are shown as filled dark green bars/boxes.
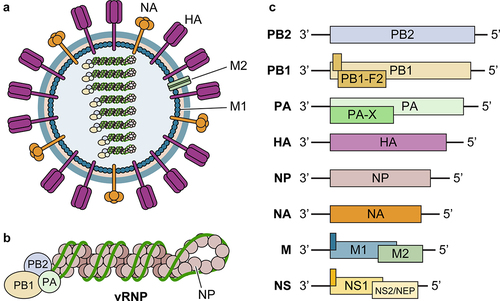
Each vRNA segment encodes one to three proteins via alternative splicing or translation mechanisms (). There are a total of ten essential proteins for viral infection (PB1, PB2, PA, NP, HA, NA, M1, M2, NS1, and NEP/NS2) and several accessory proteins, such as PB1-F2 and PA-X, which mediate virus-host interactions, modulate innate immunity, and affect viral pathogenicity (reviewed in [Citation7]). PB1 (containing an RNA-dependent RNA polymerase domain), PB2 (containing a cap-binding domain), and PA (containing an endonuclease domain) form a heterotrimeric polymerase complex that is essential for viral RNA synthesis. NP encapsidates vRNA and complementary RNA (cRNA) and is also required for viral RNA synthesis as demonstrated by influenza virus minigenome assay [Citation8,Citation9]. HA binds to the host receptor and mediates membrane fusion. NA facilitates the release and spread of progeny virions by cleaving sialic acid on the cell surface. M1 mediates viral particle assembly and budding from the plasma membrane. M2 is an ion channel protein on the viral membrane that is required for viral replication and modulation of cellular homoeostasis. NS1 impairs host antiviral responses via multiple mechanisms as described below in details. NEP/NS2 facilitates the export of vRNAs from the nucleus to the cytoplasm for assembly.
2.2. Influenza virus life cycle
To initiate an infection, the influenza virus first binds to the cell surface receptor, sialic acid residues of glycoproteins or glycolipids, through the receptor-binding site (RBS) in the HA protein. Viruses are internalized into early endosomes and trafficked to late endosomes, where the low pH environment causes conformational changes in HA to expose a fusion peptide that leads to the fusion of viral and endosomal membranes. The M2 proton channels on the viral membrane mediate H+ influx into the viral interior, which lowers the pH to facilitate the release of vRNPs from M1 into the cell cytoplasm in a process called uncoating [Citation1,Citation7].
The released vRNPs are then imported into the nucleus for viral RNA replication and transcription. Viral RNA synthesis is mediated by the heterotrimeric polymerase complex PB1/PB2/PA [Citation10]. The polymerase complex recognizes the terminal promoter sequence of each vRNA segment and generates complementary positive-strand RNA (cRNA), which is then used to produce more copies of vRNA. mRNA is transcribed from vRNA and initiated by cap structures stolen from cellular pre-mRNAs in a cap-snatching mechanism, by which PB2 binds to the 5’ m [Citation7]G cap, and the PA endonuclease cleaves it. The 3’-polyadenylated tail is added to viral mRNAs through a stuttering mechanism. Thus, influenza mRNAs are 5-’ capped and 3-polyadenylated. Some viral transcripts (M and NS) are processed by the host RNA splicing machinery. Both primary and processed viral transcripts are exported to the cytoplasm for translation by the host ribosomes. PB1, PB2, PA, and NP are imported into the nucleus to increase viral RNA synthesis and vRNP formation. Viral membrane proteins (HA, NA, and M2) traffic to the plasma membrane through cellular secretory pathways. Other non-structural proteins, such as NS1, PB1-F2, and PA-X, modulate host cell responses.
The newly synthesized vRNPs are exported from the nucleus with assistance from M1 and NEP/NS2, and transported to the plasma membrane for assembly and budding [Citation11]. M1 interacts with both viral membrane proteins and vRNPs to mediate viral particle formation and is also a major player in virus budding. Eight vRNPs are packaged into viral particles in a “1 + 7” configuration, where one central vRNP is surrounded by 7 vRNPs [Citation12]. The newly formed progeny virions form aggregates at the cell surface because of the binding of HA to sialic acids on viral envelope proteins and on the cell surface. NA neuraminidase activity is thus required to release new virions that can spread to infect new target cells.
Three classes of FDA-approved antiviral drugs target different stages of the viral life cycle [Citation4]. The M2 inhibitors (M2I) – amantadine and rimantadine – block M2 ion channel activity. Zanamivir and oseltamivir are NA inhibitors (NAI) that prevent the release of viral particles from the cell surface and stop the viral spread. Baloxavir marboxil (BXM) is an inhibitor of PA endonuclease activity (PAI) and suppresses viral transcription. Drug-resistant mutations have been identified in each class of antivirals [Citation4]. Resistance to M2Is develops rapidly and requires a single or a limited number of mutations in M2 such as L26F, V27A, and S31N, which are prevalent in circulating influenza viral strains [Citation13]. Thus, the M2Is amantadine and rimantadine are no longer recommended to treat influenza infections. NAI-resistant mutations such as H275Y, E119G, I223R, R292K, and Q136K, as well as PAI-resistant mutations such as I38M or I38T, have been identified from influenza isolates [Citation4,Citation14,Citation15], though their global frequency was still low [Citation16]. Nevertheless, novel classes of antiviral drugs and/or host-based therapeutics should be developed to treat influenza infections.
2.3. Influenza virus pathogenesis
Human influenza viruses are transmitted through the respiratory route, whereas avian influenza viruses are spread between birds primarily through direct contact via faecal-oral, faecal-faecal, and faecal-respiratory routes [Citation1]. Viruses replicate mainly in the epithelial cells lining the respiratory or intestinal tract [Citation1,Citation17]. Virus replication peaked at ~48 h after inoculation and declined slowly thereafter, with little shedding after 6–8 days [Citation17]. Mild influenza infections generally involve the upper respiratory tract and trachea, whereas the most severe and fatal human infections are associated with viral infections in the lower respiratory tract. Influenza virus causes the death of epithelial cells through various mechanisms. In addition, infected epithelial cells release cytokines and chemokines to attract infiltrating inflammatory cells such as neutrophils and macrophages and activate adjacent endothelial cells. These activated immune and non-immune cells produce even more inflammatory cytokines, such as IL − 6, IL − 1β, TNFα, and CCL − 2, to stimulate further infiltration, damage the epithelial-endothelial barrier, and cause more epithelial cell death. Zoonotic avian H5N1 viruses can cause a highly lethal disease in humans, which is associated with viral dissemination to broad tissue tropism, high viral titers in multiple organs [Citation18,Citation19], and a hyperinflammatory response [Citation18]. Thus, the pathogenesis of severe influenza disease is caused not only by direct viral cytopathic effects but also by exacerbated host inflammatory responses [Citation18,Citation20]. Therefore, effective therapeutics should aim to reduce both viral replication and pathogenic airway inflammation.
3. Mechanism of influenza virus evolution
3.1. History of influenza virus evolution
IAVs have 18 HA-and 11 NA-known subtypes. Except for H17N10 and H18N11 from bats, all other 16 HA and 9 NA subtypes were found in aquatic birds (ducks, geese, and shorebirds) [Citation21]. Avian influenza viruses can infect poultry (chickens, turkeys, and domestic ducks), causing major economic damage. A few of these avian-origin subtypes have established within-host infections in other species, such as H1N1, H2N2, and H3N2 in humans and pigs; H7N7 and H3N8 in equines; and H3N8 and H3N2 in canines.
Human influenza viruses have changed over the years [Citation4] (). Since the 1918 Spanish pandemic flu, the IAV subtype H1N1 persisted in humans until it was replaced in 1957 by an H2N2 subtype (Asian pandemic). H2N2 circulated in humans until 1968 when it was replaced by a H3N2 subtype (Hong Kong pandemic). The H1N1 virus reappeared in 1977 and was replaced by a new H1N1 strain in 2009 (swine pandemic). Currently, two IAV subtypes, H1N1 and H3N2, together with two lineages of IBVs (B/Yamagata and B/Victoria) [Citation22], co-circulate in humans. Therefore, annual flu vaccines are quadrivalent and contain two IAV subtypes and two IBV lineages.
Figure 2. Influenza virus evolution. Almost all IAV subtypes (H1 to H16, N1 to N9) have natural hosts in water birds, of which some have established infections in other species, such as H1N1, H2N2, and H3N2 in humans and pigs. Human influenza viruses have changed over the years mainly due to the emergence of four pandemic flu viruses. Currently, circulating influenza viruses include two IAV subtypes H1N1 and H3N2, and two IBV lineages (B/Yamagata and B/Victoria). Some animal IAVs, particularly bird and swine flu, can occasionally spill over to cause zoonotic infections in humans. In recent years, avian H5 and H7 viruses caused human infections with a high case fatality rate.
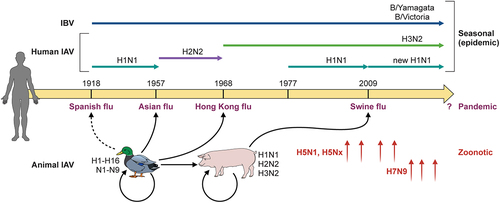
Humans are occasionally infected by influenza viruses from pigs and birds, which sometimes results in severe disease and death, but has not been established in humans. Avian influenza viruses of subtypes H5, H6, H7, and H9 are known to cause zoonotic infections. In particular, H5 and H7 have caused major outbreaks in birds, with significant losses in the poultry industry and thousands of human infections with a high case fatality rate (CFR) (reviewed in [Citation3]). The highly pathogenic avian influenza virus (HPAIV) H5N1 was first detected in Hong Kong in 1997 and has been circulating among domestic poultry and wild migratory birds in Asia, Europe, and Africa since 2003. The avian H5N1 virus has caused 868 laboratory-confirmed human infections and 457 deaths globally from 2003 to 2022 (CFR:53%) [Citation23]. Novel H7N9 viruses that emerged in China in 2013 have caused a total of 1,568 laboratory-confirmed human infections, including 616 fatal cases (CFR:39%) as of January 2023, according to the WHO avian influenza weekly update [Citation24]. These avian influenza viruses have yet to establish efficient human-to-human transmission, but their potential acquisition of airborne transmissibility through continuing viral evolution poses a potential pandemic threat [Citation25]. Since the fall of 2022, an HPAIV H5N1 of clade 2.3.4.4b was found to infect many mammalian species, including foxes, cats, ferrets, seals, grizzly bears, and humans, and more alarmingly, spread among minks in mink farms [Citation26]. Whether the mink virus can infect humans and cause efficient human-to-human infections is unknown.
3.2. Mechanisms of influenza virus evolution – antigenic drift and shift
The evolution of influenza viruses is driven by two major mechanisms: antigenic drift and antigenic shift. Antigenic drift is the accumulation of mutations in the viral genome during viral replication owing to the lack of proofreading activity of viral RNA-dependent RNA polymerase [Citation5]. Mutations located within the antibody epitopes of the surface envelope proteins, HA and NA, can reduce the recognition of pre-existing antibodies elicited against previous viral strains. Antigenic drift explains the occurrence of seasonal influenza infections and the need for annual influenza booster vaccines.
Antigenic shift is the result of the reassortment of viral genomic RNA segments of two different influenza viruses co-infecting the same cell to generate new strains and/or subtypes, which have the potential to cause severe disease and/or spread quickly in a population with no pre-existing immunity. The mechanism of influenza virus reassortment has been reviewed [Citation27]. Antigenic shift or reassortment is a major driver of the generation of pandemic or zoonotic strains. The 1957 pandemic influenza virus resulted from the reassortment of an H2N2 avian virus and a human H1N1 virus, while the 1968 pandemic virus was the result of reassortment of an H3 avian virus and a human H2N2 virus then. The 2009 H1N1 pandemic virus (2009 H1N1pdm) was generated after multiple reassortment events in swine. Avian H5N1 and H7N9 viruses, which cause zoonotic infections in humans with high mortality, are generated through reassortment with other subtypes of avian viruses, especially avian H9N2 viruses.
4. Viral determinants for pathogenicity and virulence
Influenza replication capacity, disease severity, and transmissibility are both virus- and host-specific. Low-pathogenic avian influenza viruses cause mild symptoms in wild birds and poultry, with little or no signs of illness, whereas highly pathogenic avian influenza viruses cause high mortality in infected poultry. Compared to seasonal influenza viruses, pandemic strains cause high morbidity, mortality, and rapid transmission in the human population. Even among the pandemic strains, the 1918 virus is unique in its extreme virulence (estimated 1% mortality) and its distinct “W-shaped” curve of age-specific death rates, with peak deaths observed in young adults in addition to the elderly and infants [Citation2]. Zoonotic infections by HPAIV H5N1 and H7N9 viruses led to 30% to 50% mortality in infected patients, but limited human-to-human transmission [Citation3].
Extensive studies have been conducted to identify and characterize viral determinants associated with increased pathogenicity, virulence, and transmission. In particular, the genetic changes required for adaptive replication and airborne transmissibility in mammalian hosts [Citation28–32] have been studied extensively (reviewed in [Citation33]). The major virulence determinants () include changes that affect viral entry, increase viral polymerase activity, and modulation of host responses. They function synergistically to cause efficient infection and sustained transmission in new hosts [Citation46,Citation68,Citation95]. For example, a set of five or six substitutions in three viral genes (HA, PB2, and PB1) are required for an avian H5N1 virus to acquire airborne transmissibility [Citation28,Citation32]. Identifying viral outbreaks of pandemic potential through active surveillance of the prevalence and evolution of influenza A viruses in avian, swine, and human hosts to identify molecular markers of virulence is an important component of the pandemic preparedness plan.
Table 1. Molecular markers associated with virulence and pathogenicity of influenza virus. HA substations are based on the H3 numbering system[Citation97].
4.1. HA variants expand tissue tropism and increase host cell susceptibility
HA is a major determinant of host range because of its essential role in host receptor binding and membrane fusion during viral entry. The HA trimer on the viral membrane consists of two domains: a globular head containing a receptor-binding site, and a membrane-proximal stem domain. The receptor-binding site consists of three secondary elements, the 130-loop, 190-helix, and 220-loop, and includes four highly conserved residues (Y98, W153, H183, and Y195) [Citation96]. All the HA positions were based on H3 numbering [Citation97]. HA mutants allow viruses to expand tissue tropism, switch receptor specificity, increase host receptor binding, and optimize membrane fusion at different temperatures or pH environments.
4.1.1. HA cleavage by host activating proteases
Low-pathogenic avian influenza virus (LPAIV) of H5 and H7 subtypes can mutate to HPAIV, mainly due to the acquisition of a multi-basic cleavage site [Citation60]. Host protease-mediated proteolytic cleavage of the HA precursor molecules HA0 to HA1 and HA2 activates a conformational change in HA, which is essential for membrane fusion and viral infectivity [Citation98]. Therefore, the distribution of activating proteases in the host is a major determinant of viral tissue tropism. The HA proteins of LPAIVs and mammalian influenza viruses contain a single basic residue at the cleavage site and can only be cleaved by extracellular host proteases, which restricts viral spread to tissues where specific proteases are present. HPAIV HA proteins acquire a multi-basic cleavage site, which allows intracellular cleavage by ubiquitously present proteases, resulting in the systemic dissemination of HPAIVs and lethal disease in poultry.
4.1.2. HA-dependent host adaptation
Except for bat influenza viruses (HA17 and HA18), HA proteins (HA1–16) recognize terminal sialic acids (SA) of cell surface glycoconjugates as receptors. Avian and equine influenza viruses preferentially bind SA linked to galactose via an α − 2,3 linkage (α2,3‐SA), whereas human influenza viruses prefer α − 2,6-linked SA and swine influenza viruses bind to both efficiently [Citation99]. Differential receptor-binding affinity is an important determinant of the influenza virus host range and viral pathogenicity. α2,3‐SA is prevalent in the intestinal epithelial cells of birds, allowing the efficient replication and shedding of avian influenza viruses. In humans, α2,6‐SA is more abundant in the upper respiratory tract (URT) than in the lower respiratory tract (LRT), whereas α2,3‐SA is more abundant in the lungs. Human influenza viruses primarily replicate in URT, causing mild disease, but efficient transmission among humans. Avian influenza virus infection in humans (zoonotic) replicates better in LRT than in URT, resulting in a more severe disease but limited ability for transmission [Citation99]. Therefore, avian influenza viruses must acquire human receptor-binding specificity for efficient infection and airborne transmission in humans. Pigs have both α2,3‐SA and α2,6‐SA in the respiratory tract and can act as mixing vessels for avian and human influenza viruses [Citation100].
Amino acids that determine receptor-binding specificity have been defined in many HA subtypes, which generally require two residue changes within the receptor-binding pocket, such as positions 190 and 225 for H1N1, 226 and 228 for H2N2 and H3N2 viruses [Citation96]. For H1N1 viruses, D190 and D225 are associated with α − 2,6 binding, whereas E190 and G225 are associated with α − 2,3 binding [Citation47,Citation48]. The 1918 H1N1 pandemic HA protein, containing D190 and D225 residues, binds α − 2,6 receptors exclusively. D225G substitution led to mixed α − 2,6 and α − 2,3 binding, whereas D190E substitution alone resulted in α − 2,3 binding. Consistent with the α − 2,3 receptor-binding specificity, the recombinant 1918 pandemic virus with D190E/D225G mutations lost respiratory transmission between ferrets [Citation49]. The recombinant 2009 H1N1pdm virus with D225G had dual receptor-binding specificity for α − 2,6 and α − 2,3-SAs and displayed increased binding to human LRT, but still retained aerosol transmission in mammalian hosts [Citation51], which helps explain the severe disease associated with D225G during the 2009 pandemic. For H2N2 and H3N2 viruses, L226 and S228 are associated with α − 2,6 binding, whereas Q226 and G228 are associated with α − 2,3 binding [Citation52]. Q226L and G228S substitutions switched receptor binding from α − 2,3 to α − 2,6, which contributed to the emergence of the 1957 H2N2 and 1968 H3N2 pandemic infections.
Q226L and G228S have also been shown to increase the α − 2,6 binding specificity of other avian HA subtypes (H5, H7, H9, H10, and H15) . However, switching these HA proteins to human receptor preference generally requires additional mutations [Citation33,Citation57] ().
Apart from substitutions to switch receptor-binding specificity, other mutations that increase HA stability and/or overall receptor binding are required for host adaptation. Site-directed mutagenesis and experimental adaptation of an avian H5N1 virus have shown that airborne transmissibility in ferrets requires only three to four substitutions in the H5 protein, apart from substitutions in polymerase genes [Citation28,Citation32]. Among the H5 substitutions, Q226L or G228S increased mammalian receptor-binding specificity, H110Y altered the acid and temperature stability of HA, and T160A increased overall receptor binding [Citation32,Citation36,Citation37]. Another experimental adaptation study [Citation31] showed that a laboratory-generated reassortant virus requires four substitutions (N158D, N224K, Q226L, and T318I) within the H5 protein to be transmitted among ferrets through aerosol or respiratory droplets. N158D, N224K, and Q226L allow the switch to human receptor binding [Citation31,Citation36], while T318I increases the heat stability and decreases the pH threshold of HA during membrane fusion [Citation37,Citation59]. As the human-receptor-binding mutations in H5 reduce the HA heat stability and increase the pH threshold for membrane fusion, compensatory mutations such as T318I that reverse the effects can facilitate avian virus respiratory transmission in humans [Citation101].
The emergence of H7N9 avian influenza viruses in China causing zoonotic infections with high CFR since 2013 has attracted considerable attention because of their continuing evolution and potential pandemic risk. Most zoonotic H7N9 isolates bind to both the human and avian receptors [Citation102,Citation103]. Q226L and G186V substitutions accounted for the increased α − 2,6 SA binding [Citation39,Citation56] but were not sufficient for the switch in receptor specificity. A combination of three substitutions, V186G/K-K193T-G228S or V186N-N224K-G228S, is sufficient to switch the receptor specificity of H7N9 from avian to human type [Citation44], of which, G228S is a known determinant for human receptor binding, V186G/K/N loses the α − 2,3 binding and increases α − 2,6 binding, and N224K increases overall receptor binding [Citation31,Citation35,Citation44].
4.2. Substitutions in the internal genes increase viral replication in target cells
Influenza virus internal genes, including three polymerase proteins (PB2, PB1, and PA), NP, and M1, contain important virulence markers that increase viral replication and progeny production. Polymerase genes play a critical role in the adaptive replication of avian influenza viruses [Citation72]. These adaptive mutations in polymerase proteins generally increase the polymerase activity in mammalian cells. The major virulence determinant and mammalian adaptive marker is PB2 E627K [Citation68,Citation69]. Avian influenza viruses generally have E627, while all human viruses and many zoonotic influenza viruses have K627, except for the 2009 H1N1pdm virus. Another mammalian-adaptive substitution is PB2 D701N, which has been shown to expand the host range of the avian H5N1 virus to mice and humans and to increase viral transmission in guinea pigs [Citation69,Citation71]. Surprisingly, 2009 H1N1pdm had neither E627K nor D701N; the introduction of K627 and N701 did not enhance virulence or transmission in ferrets or mice [Citation104].
Studies have shown that polymerase adaptive mutations, especially PB2 E627K, are caused by species-specific differences in ANP32 proteins, which are essential host factors for viral RNA synthesis [Citation105–107]. Compared to avian ANP32A, human ANP32A lacks a 33-amino acid insertion in the C-terminal disordered domain, which allows the binding of PB2 K627, but not PB2 E627 [Citation108,Citation109]. Both human ANP32A and ANP32B homologs can support viral RNA synthesis by human influenza viral polymerases or avian viral polymerases with the PB2 E627K adaptive mutation [Citation110,Citation111]. Other polymerase mutations, such as PB2 K562R, PA A343T, and K356R, can cooperate with PB2 E627K to synergistically increase viral polymerase activity [Citation40,Citation67,Citation80]. Another mechanism of adaptation has been shown for host importin-α isoforms, which mediate the nuclear import of vRNPs and viral proteins for viral RNA synthesis [Citation112]. The avian PB2 and NP depend on importin-α3, whereas the mammalian PB2 and NP primarily use importin-α7 [Citation112]. The PB2 D701N adaptive mutation enhances the interaction between PB2 and importin-α7 to promote vRNP nuclear import in mammalian cells [Citation112,Citation113]. Multiple virulence determinants or adaptive mutations have been identified in PB2, PB1, PA, and NP proteins, which function to increase avian viral polymerase activity in mammalian cells (), though their mechanisms of adaptation have not been well characterized.
As the major driver of viral budding and assembly, M1 also contributes to virulence by increasing viral replication. A previous study found that D30 and A215 contribute to the high pathogenicity of avian H5N1 viruses in mice and that the D30N/A215T double mutations significantly attenuated multiple H5N1 viruses in mice [Citation114]. The M1 A215T substitution eliminates the SUMOylation of M1, leading to a significant reduction in M1 stability, vRNP nuclear export, viral replication, and viral progeny production [Citation115]. The D30N substitution changes the shape of H5N1 viral particles from filamentous to spherical, although the mechanism by which the virion shape affects viral pathogenicity is unknown [Citation115]. M1 T37A of the H9N2 virus abolishes the phosphorylation site to stabilize the M1 protein and increases viral replication in mice and human cells [Citation116].
4.3. Viral antagonism of host antiviral immunity
Like all other viruses, influenza viruses must overcome host antiviral immunity for efficient replication. The viral NS1 protein is a key virulence factor known to impair host antiviral responses through various mechanisms (reviewed in [Citation117]). NS1 has been shown to interact with many viral and host factors through its functional domains, including an N-terminal RNA-binding domain (RBD) (aa 1–73), a linker region (aa 74–88), an effector domain (ED) (aa 89–202), and a C-terminal tail of variable length [Citation118]. Many of the NS1 functions are strain-specific owing to the sequence variations of different NS1 proteins. A single substitution of P42S in the NS1 RBD prevented the dsRNA-mediated activation of the NF-κB and IRF − 3 pathways and drastically increased the pathogenicity of an H5N1 virus in mice [Citation119]. The NS1 I106M substitution in an H7N9 virus increased its binding to CPSF30 and blocked host gene expression, leading to a more virulent infection in vivo [Citation120]. Two NS1 mutations, D189N and V194I, present in circulating human H3N2 viruses impair their ability to inhibit host gene expression and attenuate viral virulence in mice, while NS1 V194I affects the thermosensitivity of viral replication and further attenuated viral virulence [Citation120,Citation121]. Recombinant influenza viruses expressing human NS1 (from human H1N1 or H3N2) induced higher levels of type I and III interferons (IFN-I/III) than those expressing avian NS1 (from avian H5N1, H7N9, and H7N2) in dendritic cells (DCs), suggesting that avian NS1 has an increased ability to antagonize IFN-I/III production [Citation122].
PA-X protein is expressed from PA mRNAs via ribosomal frameshifting [Citation123]. It has the same N-terminal 191 residues as PA, containing the endonuclease domain and a unique C-terminal domain, which is 61 aa long for most viral isolates, including the 1918 H1N1pdm and avian viruses, but is shorter (41 aa) in the 2009 H1N1pdm virus [Citation123]. PA-X selectively targets cellular mRNAs, but not viral mRNAs for degradation, leading to host protein shutoff and inhibition of cellular antiviral responses [Citation118,Citation123]. PA-X also modulates host responses such as inflammation, immune responses, and apoptosis. The functional mechanism of PA-X in innate immune suppression is reviewed in detail [Citation118]. Studies on PA-X-deficient influenza viruses have suggested that the biological roles of PA-X in viral replication and pathogenicity are host- and strain-specific [Citation118]. PA-X was shown to decrease pathogenicity in mice by modulating host responses following infection with the 1918 H1N1pdm virus, the 2009 H1N1pdm strain (A/Beijing/16/2009 (BJ/09), HPAI H5N1 viruses, and circulating H1N1 strains [Citation123–125]. Other studies, however, showed a pro-virulence role of PA-X in the 2009 H1N1pdm strain A/California/04/09, avian H9N2, and A/PR8 in mice [Citation126–129].
PB1-F2 is a small protein expressed in the PB1 gene through the + 1 reading frame shift [Citation130] and is a major virulence factor that modulates host innate immune responses (reviewed in [Citation131]). PB1-F2 expression is detected in many, but not all, influenza A strains and is not detected in influenza B viruses [Citation130]. PB1-F2 proteins vary in length and sequence. As such, the role of PB1-F2 in viral virulence is strain- and host-specific [Citation131]. Different PB1-F2 proteins can differentially modify IFN-I responses, inflammatory responses, and immune cell death, contributing to viral virulence and pathogenicity [Citation131]. A single mutation N66S in the PB1-F2 protein is associated with high pathogenicity of the 1918 H1N1pdm and HPAIV H5N1 viruses [Citation94]. PB1-F2 of the 1918 H1N1pdm virus, but not of the A/PR8 virus, binds to DDX3 and causes its degradation [Citation132], which may explain the severe pathogenicity of the 1918 pandemic virus. PB1-F2 of the highly pathogenic H7N9 virus, but not of the A/WSN virus, is a potent inhibitor of MAVS, an essential mediator of antiviral signalling, by forming aggregates in the mitochondria to prevent MAVS aggregation and activation, resulting in suppression of IFN-I production and MAVS-mediated NLRP3 activation [Citation133]. H7N9 PB1-F2, however, does not affect extracellular NLRP3 inflammasome maturation, in sharp contrast to PB1-F2 of the A/WSN virus, which effectively suppresses IL − 1β processing and secretion by all stimuli [Citation134]. Thus, the differential function of PB1-F2 may account for the highly elevated cytokine storm observed in the H7N9 infection but in not the WSN infection [Citation134].
5. Host immune responses: immune protection or immunopathogenesis
The host immune response against influenza viral infections plays an important role in determining the disease pathogenesis [Citation135]. A potent and fine-tuned immune response can effectively control and eliminate viral infection without significant damage, whereas an excessive and/or prolonged inflammatory response has been shown to cause tissue damage and disease exacerbation. Elucidating the mechanisms of immune protection and immunopathogenesis is crucial for the development of effective therapeutics against influenza.
5.1. Early activation of innate immunity correlates with protection
Upon infection, influenza viruses are recognized by the innate immune system through different classes of pattern recognition receptors (PRRs), membrane-bound Toll-like receptors (TLR3, TLR7, and TLR8), cytosolic receptor RIG-I, and NOD-like receptor family NLRP3 [Citation136]. Activation of the TLR and RIG-I signalling pathways leads to the upregulation of IFN-I/III and proinflammatory cytokines. IFNs act through paracrine and autocrine signalling to induce the expression of interferon-stimulating genes (ISGs), many of which exert antiviral activities to restrict viral replication. Proinflammatory cytokines activate phagocytic cells, recruit additional immune cells to the infection site, and contribute to the initiation of adaptive immunity. The NLRP3 inflammasome triggers the proteolytic cleavage and release of cytokines IL − 1β and IL − 18 and elicits pyroptosis in infected cells [Citation136]. The protective roles of these innate immune pathways against influenza virus infection have been demonstrated in various knockout animal models. Some ISGs have demonstrated strong antiviral activity. Mx is a well-known restriction factor for the influenza virus. IFITMs block virus-host membrane fusion. The 2’−5’-oligoadenylate synthase (OAS) and RNase L degrade viral RNA. PKR binds to dsRNA and blocks protein translation by phosphorylating eukaryotic translation initiation factor 2α (EIF2α). Other antiviral factors have been identified that block influenza virus replication through various mechanisms (reviewed in [Citation136]).
Early activation of robust innate immunity correlates with host protection against influenza viral infections in animal models. A highly pathogenic H7N9 virus caused complete lethality in mice with a high lung virus titer, whereas the H9N2 virus caused a mild and self-resolved infection. Compared to H7N9, H9N2 infection resulted in early and transient induction of innate immunity, as evidenced by the upregulation of IFN-Is, ISGs, immune cell markers, and proinflammatory genes [Citation137,Citation138]. Macrophages were recruited to the lungs at a much earlier time point (as early as 6 h) for H9N2 infection than for H7N9 infection (day 3) [Citation137]. Co-infection with H9N2 resulted in effective protection against both H7N9 and PR8 (H1N1) infections, supporting the protective role of early innate immunity [Citation137].
5.2. Pathogenic effect of accelerated activation of proinflammatory responses
Studies on severe influenza viral diseases in both humans and animals have demonstrated the pathological effect of hyperactivation of immune responses against elevated viral replication. The 1918 H1N1pdm virus is highly virulent and causes an abnormally high death rate in young adults, the elderly, and infants [Citation2]. Mice and non-human primates infected with the reconstructed 1918 H1N1pdm virus have shown marked and prolonged activation of proinflammatory and cell death pathways, suggesting that accelerated activation of the host immune response contributes to severe pulmonary pathology [Citation95,Citation139]. Virological and immunological studies of human fatal cases of avian H5N1 and 2009 H1N1pdm infections suggest that a high viral load and the resulting intense inflammatory responses are central to the disease pathogenesis [Citation18,Citation140–142]. The avian H7N9 virus caused a more severe phenotype in animals than H9N2 and seasonal H3N2, and the disease severity correlated with increased infectivity and elevated induction of proinflammatory cytokines [Citation143–146]. Excessive levels of proinflammatory cytokines, such as IL − 6, IL − 8, IP10, G-CSF, MCP − 1, and MIP − 1α, are associated with pulmonary inflammation and tissue damage in acute lung injury caused by influenza virus and SARS-CoV −2 infections, as well as in asthma and chronic obstructive pulmonary disease [Citation95,Citation145,Citation147,Citation148]. The immunopathogenic roles of cytokine storms and innate immune cells have been reviewed [Citation149].
5.3. Antiviral and immunopathological roles of macrophages
Macrophages are important innate immune cells that protect against viral infections by secreting cytokines, initiating adaptive immunity, and clearing debris and dead cells [Citation150]. Alveolar macrophages are tissue-resident macrophages that play an essential role in the protection from influenza virus-induced morbidity and mortality [Citation86]. However, the excessive activation of cytokines in macrophages contributes to lung injury and disease severity [Citation141,Citation151]. Thus, the controlled activation of macrophages is critical for their protective role following influenza viral infections. Recent studies have shown that cellular Wnt/β-catenin/HIF −1α signalling and the transcriptional factor PPAR-γ function to promote the inflammatory activity of alveolar macrophages [Citation90,Citation152], and that downregulation of PPAR-γ following IFN-I antiviral signalling is critical for the suppression of exaggerated inflammatory response [Citation152].
Macrophages are susceptible to influenza viral infection; however, most infections are abortive, and only a small number of viruses can produce infectious particles by overcoming cellular blocks [Citation87,Citation88,Citation153]. Viruses that productively infect primary murine alveolar macrophages include a subset of highly pathogenic H5N1 viruses and A/WSN viruses. Even though productive replication in macrophages does not solely determine viral virulence in vivo, it decreases the phagocytic function of macrophages and thus may contribute to disease development [Citation88].
Influenza virus strains may also differentially modulate macrophage polarization, leading to either antiviral or pathogenic state [Citation89]. Macrophages are highly heterogeneous and include two major subclasses: proinflammatory M1 and anti-inflammatory M2 macrophages. M1 macrophages are activated early in infection to secrete inflammatory cytokines, such as IL − 1, IL − 6, and TNF-α, and trigger antiviral responses. M2 macrophages are activated later during infection to terminate inflammation, repair tissue damage, and produce TGF-β and IL − 10 [Citation154–156]. Recent studies have suggested that M2 macrophages are beneficial for influenza infection, while M1 polarization is associated with acute lung injury in severe influenza disease [Citation155,Citation157,Citation158]. Thus, switching macrophage polarization from M1 to M2 May be a novel therapy for influenza diseases [Citation159,Citation160].
5.4. Antiviral and immunopathogenic roles of T cells
Both CD4 and CD8 T cells are important components of adaptive immunity to clear viral infections and protect the host from severe disease [Citation161,Citation162]. Pre-existing influenza-specific CD4 T cells are associated with lower viral shedding and less severe disease in healthy volunteers following influenza viral infection [Citation91]. The adoptive transfer of memory CD4 T cells protects unprimed mice [Citation92]. Memory CD8 T cells have also been shown to provide cross-reactive protection [Citation93,Citation163]. A comparison of host immunity in hospitalized patients after zoonotic H7N9 infection with diverse disease outcomes revealed an important protective role of memory CD8 T cells against severe influenza disease [Citation164,Citation165]. Patients with early recovery showed an early robust H7N9-specific CD8 T cell response, while those with prolonged hospitalization showed late recruitment of CD4 and CD8 T cells. In contrast, deceased patients showed minimal influenza-specific immunity and little T-cell activation [Citation164]. These studies suggest that an early strong memory T cell response can help reduce disease severity. In contrast, overactive T cells can be harmful because they cause tissue damage and/or trigger inflammatory responses [Citation162,Citation166].
Tissue-resident memory T cells (TRM) play an important role in the first-line defence against re-infection [Citation167]. Lung CD8 TRM cells mediate protection against respiratory infections by producing immediate effector responses at the site of pathogen entry; however, heightened responses may also cause inflammation and tissue damage [Citation168]. Lung CD8 + TRM cells have been shown to adopt exhausted-like phenotypes to avoid post-infection inflammation and fibrotic sequelae after influenza viral infection [Citation169]. In aged hosts, however, lung CD8 TRM cells exhibit malfunction in antiviral response and support chronic lung inflammation and fibrotic sequelae [Citation170], which helps explain severe pneumonia in the elderly after influenza viral infection.
6. Host factors and cell signalling contribute to viral replication and pathogenesis
6.1. Pro-viral host factors
Like all other viral infections, influenza viruses rely on host factors and cellular machinery to reproduce (). Hundreds of pro-viral host factors have been identified through genome-wide gene knockdown or knockout by CRISPR or RNAi, and through proteomic analysis of viral protein-associated cellular proteins [Citation171]. Validating the functional roles of each candidate in influenza virus infection and characterizing their molecular mechanisms require tremendous effort and resources. Limited by space, only some proviral host factors are briefly summarized here.
Figure 3. Pro-viral host factors and host signalling pathways important for influenza viral replication. Host factors and cellular signalling pathways are hijacked by the influenza virus to promote viral replication at different steps of the viral life cycle. Host factors are listed inside light green boxes next to the specific step of viral replication. Receptor tyrosine kinases (EGFR, c-Met, TrkA) on plasma membranes are activated by influenza viral infection and function to promote viral replication. EGFR, c-Met, and PLC-γ1 enhance viral uptake. TrkA signalling is important for several steps of the viral life cycle: viral RNA synthesis, vRNP nuclear export, and viral budding and release. NF-κB signalling enhances viral RNA synthesis and induces the expression of proinflammatory genes. PI3K/Akt and Raf/MEK/ERK pathways also strongly increase viral replication.
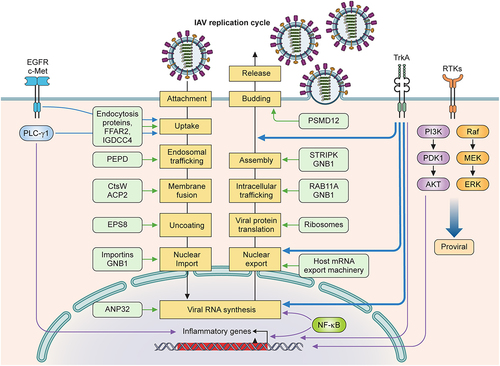
Viral entry includes attachment, uptake, and fusion, and each step involves multiple host factors. Viral attachment requires sialic acids of cell surface glycoproteins or glycolipids. Viral uptake by endocytosis occurs through coordinated actions of cellular endocytic proteins. Host factors that promote influenza virus internalization include free fatty acid receptor 2 (FFAR2) and transmembrane protein immunoglobulin superfamily DCC subclass member 4 (IGDCC4) [Citation172,Citation173]. Once internalized, viral particles move from early to late endosomes, where fusion and uncoating occur to release the vRNPs. Prolidase (PEPD) is important for the early endosomal trafficking of viral particles [Citation174]. The cysteine protease cathepsin W (CtsW) and lysosomal acid phosphatase 2 (ACP2) are required for viral fusion [Citation175,Citation176]. Cellular factor epidermal growth factor receptor pathway substrate 8 (EPS8) has been shown to promote influenza viral uncoating [Citation177].
Influenza virus components must travel between the cytoplasm and the nucleus, as viral RNA synthesis occurs in the nucleus, while viral protein synthesis and viral particle assembly occur in the cytoplasm. The influenza virus utilizes the cellular nucleocytoplasmic trafficking machinery for the nuclear import and export of vRNPs, viral mRNAs, and viral proteins [Citation178]. Nuclear importation depends on cellular importin proteins [Citation179] and is enhanced by G protein subunit β1 (GNB1), which increases the interaction between PB2 and importin proteins [Citation180], and by BinCARD1, an isoform of Bcl10-interacting protein with CARD (BinCARD), which increased the binding of NP with importin α7 [Citation181]. The nuclear export of viral mRNAs utilizes the host mRNA export machinery to translocate through the nuclear pore complex for translation in the cytoplasm [Citation182]. Many cellular factors are involved in different steps of viral RNA synthesis and have been reviewed [Citation183]. Newly synthesized vRNPs must accumulate at the plasma membrane for assembly. Cytoplasmic trafficking of progeny vRNPs depends on their association with the cellular Rab11 GTPase isoform RAB11A [Citation184] in recycling endosomes along the microtubule network, alternative vesicles, or liquid-phase organelles (reviewed in [Citation185]).
The viral matrix proteins M1 and M2 play a central role in viral assembly and budding. M1-interacting host factors have been shown to modulate their functions. GNB1 binds to M1 via intracellular trafficking to the plasma membrane and promotes viral release [Citation186]. PSMD12, a 26S proteasome regulatory subunit, mediates K63-linked ubiquitination of M1 at K102 to promote viral budding [Citation187]. M1 phosphorylation at the T108 site increases multimerization at the cell membrane and controls its binding affinity to the cellular striatin-interacting phosphatase and kinase (STRIPAK) complex, which is important for M1 polymerization and viral replication [Citation188].
6.2. Pro-viral host signalling pathways
In addition to antiviral responses, various cellular pathways are activated by influenza virus infections and exploited to support viral replication. Receptor-tyrosine kinases (RTKs) and their downstream signalling pathways (Raf/MEK/ERK, phosphatidylinositol − 3-kinase (PI3K)/Akt, JAK/STAT, PLC-γ1, NF-κB) have been found to facilitate viral replication through different mechanisms [Citation189]. Influenza virus infection activates many RTKs, such as epidermal growth factor receptor (EGFR), c-Met, and tropomyosin receptor kinase A (TrkA), possibly through clustering of lipid rafts induced by multivalent virus binding [Citation190,Citation191]. Activated EGFR and c-Met promote efficient viral uptake [Citation190]. EGFR inhibition by small compounds or the protein antagonist SOCS5 reduces influenza virus replication [Citation192,Citation193]. In addition to directly promoting viral entry, EGFR activation has been shown to suppress IFN-I/III production and antiviral response [Citation194,Citation195], further increasing viral production. However, the role of EGFR signalling in influenza disease could be complex. EGFR has been predicted to be a key regulator of viral pathogenicity by network-based analyses of transcriptomic and proteomic data from mice infected with six influenza virus strains with differential disease severity [Citation196]. Treatment of virus-infected mice with the EGFR inhibitor gefitinib caused more body weight loss after non-lethal infections, but not after lethal infections [Citation196], suggesting an antiviral role of EGFR during non-lethal infections.
TrkA is another RTK activated by influenza virus infection but has post-entry functional roles in the viral replication cycle [Citation191]. TrkA and its high-affinity ligand, nerve growth factor (NGF), are essential for neuron cell development and functions [Citation197]. TrkA is also expressed in a wide variety of non-neuronal tissues and cell types, including human lung epithelial and endothelial cells [Citation198], and plays an important role in allergic airway inflammation [Citation199,Citation200]. TrkA inhibitors block influenza virus replication at multiple steps after viral entry, viral RNA synthesis, vRNP nuclear export, and viral budding and release [Citation201,Citation202]. Using a TrkAF592A knock-in mouse model to specifically control TrkA kinase activity [Citation203], Verma et al. showed that TrkA not only promotes influenza virus replication in airway epithelial cells but also contributes to airway inflammation and lung pathology by activating the expression of proinflammatory cytokines/chemokines in infected cells [Citation191]. Thus, targeting TrkA could be an effective treatment against influenza viral diseases by blocking both viral replication and airway inflammation.
NF-κB is a major mediator of proinflammatory cytokines, which can exert antiviral activities [Citation204,Citation205] or mediate pathogenic inflammatory responses [Citation206,Citation207]. Interestingly, many studies have demonstrated a pro-viral role for NF-κB in influenza virus replication [Citation208–212], specifically in promoting viral RNA replication [Citation208,Citation209,Citation213,Citation214], suggesting that influenza viruses can convert this antiviral and proinflammatory pathway for viral replication. Similar findings were observed for PI3K, whose activation by influenza viral infection [Citation215–217] was shown to promote IFN-I production through RIG-I signalling [Citation217]. PI3K inhibitors, however, have been found to strongly suppress influenza virus replication at multiple steps of the viral life cycle [Citation218–220], demonstrating a pro-viral role of PI3K signalling. In addition, inhibitors of the Raf/MEK/ERK pathway have been shown to strongly inhibit IAVs and IBVs in vitro and in vivo [Citation221–224]. Phospholipase C gamma 1 (PLC-γ1) signalling is virus- and cell-type-specific. Activated PLC-γ1 has an important pro-viral role in the cellular uptake of H1N1, but not H3N2 virus, in epithelial cells [Citation225], while it exerts an antiviral and inflammatory role in H1N1-infected macrophages by activating NF-κB to express inflammatory cytokines in a positive feedback loop [Citation226].
Viral infection is expected to activate the host signalling network, leading to various cellular responses, such as stress, inflammation, and cell death, which can restrict viral infection (antiviral) and cause disease symptoms (proinflammatory). Influenza viruses not only antagonize antiviral responses but also hijack the activated signalling network to efficiently accomplish different steps of the viral life cycle (pro-viral role). Thus, targeting the pro-vial and/or proinflammatory pathways in the influenza virus-infected cells can be developed as effective anti-influenza therapeutics [Citation191,Citation210–212,Citation227], which, unlike inhibitors of single viral or host targets, generally has a high barrier to resistance [Citation193,Citation201,Citation210]. The roles of host signalling pathways in viral replication and disease severity, however, can be virus- and cell-type-specific and need to be carefully evaluated for therapeutic development.
Summary
The continuing evolution of influenza viruses, driven by antigenic drift (mutation) and shift (reassortment), is a major barrier to the development of effective vaccines and antiviral therapeutics. Severe influenza is caused by direct viral cytopathic effects and immunopathogenesis. This review summarizes viral determinants and host components that affect influenza virus pathogenicity and virulence. Further studies are needed to characterize the molecular mechanisms of viral virulence determinants and pro-viral host factors, elucidate the complex roles of host immunity and cellular pathways in viral replication and disease development, and identify the control mechanisms of the protective and pathogenic roles of host signalling and immune responses. This knowledge will facilitate the development of new classes of antiviral drugs targeting different viral and host components as well as novel therapeutics that exploit antiviral roles and dampen the pathogenic effects of host signalling and immune responses.
Data sharing statement
Data sharing does not apply to this article, as no new data were created or analysed in this study.
Disclosure statement
No potential conflict of interest was reported by the author.
Additional information
Funding
References
- Krammer F, Smith GJD, Fouchier RAM, et al. Influenza. Nat Rev Dis Primers. 2018;4(1):3. doi: 10.1038/s41572-018-0002-y
- Taubenberger JK, Morens DM. 1918 influenza: the mother of all pandemics. Emerg Infect Dis. 2006;12(1):15–21. doi: 10.3201/eid1209.05-0979.
- Shi J, Zeng X, Cui P, et al. Alarming situation of emerging H5 and H7 avian influenza and effective control strategies. Emerg Microbes Infect. 2023;12(1):2155072. doi: 10.1080/22221751.2022.2155072
- Smyk JM, Szydłowska N, Szulc W, et al. Evolution of influenza viruses—drug resistance, treatment options, and prospects. IJMS. 2022;23(20):12244. doi: 10.3390/ijms232012244.
- Wright PF, Neumann G, Kawaoka Y. Orthomyxoviruses. In: Knipe DM, Howley PM, editor. Fields Virology. Lippincott Williams & Wilkins; 2013. pp. 1146–1243.
- Liu R, Sheng Z, Huang C, et al. Influenza D virus. Curr Opin Virol. 2020;44:154–161. doi: 10.1016/j.coviro.2020.08.004
- Chauhan RP, Gordon ML. An overview of influenza a virus genes, protein functions, and replication cycle highlighting important updates. Virus Genes. 2022;58(4):255–269. doi: 10.1007/s11262-022-01904-w.
- Huang TS, Palese P, Krystal M. Determination of influenza virus proteins required for genome replication. J Virol. 1990;64(11):5669–5673. doi: 10.1128/jvi.64.11.5669-5673.1990.
- Pleschka S, Jaskunas R, Engelhardt OG, et al. A plasmid-based reverse genetics system for influenza a virus. J Virol. 1996;70(6):4188–4192. doi: 10.1128/jvi.70.6.4188-4192.1996
- Te Velthuis AJW, Fodor E. Influenza virus RNA polymerase: insights into the mechanisms of viral RNA synthesis. Nat Rev Microbiol. 2016;14(8):479–493. doi: 10.1038/nrmicro.2016.87.
- Nayak DP, Hui EK-W, Barman S. Assembly and budding of influenza virus. Virus Res. 2004;106(2):147–165. doi: 10.1016/j.virusres.2004.08.012.
- Noda T, Murakami S, Nakatsu S, et al. Importance of the 1+7 configuration of ribonucleoprotein complexes for influenza a virus genome packaging. Nat Commun. 2018;9(1):54. doi: 10.1038/s41467-017-02517-w
- Dong G, Peng C, Luo J, et al. Adamantane-resistant influenza a viruses in the world (1902–2013): frequency and distribution of M2 gene mutations. PLoS ONE. 2015;10(3):e0119115. doi: 10.1371/journal.pone.0119115.
- Chesnokov A, Patel MC, Mishin VP, et al. Replicative fitness of seasonal influenza a viruses with decreased susceptibility to baloxavir. J Infect Dis. 2020;221:367–371. doi: 10.1093/infdis/jiz472
- Jones JC, Kumar G, Barman S, et al. Identification of the I38T PA substitution as a resistance marker for next-generation influenza virus endonuclease inhibitors. MBio. 2018;9(2):e00430–18. doi: 10.1128/mBio.00430-18.
- Govorkova EA, Takashita E, Daniels RS, et al. Global update on the susceptibilities of human influenza viruses to neuraminidase inhibitors and the cap-dependent endonuclease inhibitor baloxavir, 2018-2020. Antiviral Res. 2022;200:105281. doi: 10.1016/j.antiviral.2022.105281
- Taubenberger JK, Morens DM. The pathology of influenza virus infections. Annu Rev Pathol. 2008;3(1):499–522. doi: 10.1146/annurev.pathmechdis.3.121806.154316.
- de Jong MD, Simmons CP, Thanh TT, et al. Fatal outcome of human influenza a (H5N1) is associated with high viral load and hypercytokinemia. Nat Med. 2006;12:1203–1207. doi: 10.1038/nm1477
- Gu J, Xie Z, Gao Z, et al. H5N1 infection of the respiratory tract and beyond: a molecular pathology study. Lancet. 2007;370(9593):1137–1145. doi: 10.1016/S0140-6736(07)61515-3
- Flerlage T, Boyd DF, Meliopoulos V, et al. Influenza virus and SARS-CoV-2: pathogenesis and host responses in the respiratory tract. Nat Rev Microbiol. 2021;19(7):425–441. doi: 10.1038/s41579-021-00542-7
- Olsen B, Munster VJ, Wallensten A, et al. Global patterns of influenza a virus in wild birds. Science. 2006;312(5772):384–388. doi: 10.1126/science.1122438.
- Caini S, Kusznierz G, Garate VV, et al. The epidemiological signature of influenza B virus and its B/Victoria and B/Yamagata lineages in the 21st century. PLoS ONE. 2019;14(9):e0222381. doi: 10.1371/journal.pone.0222381
- WHO. Cumulative number of confirmed human cases for avian influenza A(H5N1) reported to WHO. 5January 2023 [Internet]. [cited 2023 February 2]; Available from: https://www.who.int/publications/m/item/cumulative-number-of-confirmed-human-cases-for-avian-influenza-ah5n1-reported-to-who-2003-2022-5-jan-2023
- WHO. Avian influenza weekly update [Internet]. 2023. Available from: https://www.who.int/docs/default-source/wpro—documents/emergency/surveillance/avian-influenza/ai_20230113.pdf?sfvrsn=5bc7c406_19
- Sutton TC. The pandemic threat of emerging H5 and H7 avian influenza viruses. Viruses. 2018;10(9):461. doi: 10.3390/v10090461.
- Kupferschmidt K. Bird flu spread between mink is a ‘warning bell’. Science. 2023;379(6630):316–317. doi: 10.1126/science.adg8342.
- Lowen AC, Spindler KR. It’s in the mix: reassortment of segmented viral genomes. PLOS Pathog. 2018;14(9):e1007200. doi: 10.1371/journal.ppat.1007200.
- Herfst S, Schrauwen EJA, Linster M, et al. Airborne transmission of influenza A/H5N1 virus between ferrets. Science. 2012;336(6088):1534–1541. doi: 10.1126/science.1213362
- Chen L-M, Blixt O, Stevens J, et al. In vitro evolution of H5N1 avian influenza virus toward human-type receptor specificity. Virology. 2012;422(1):105–113. doi: 10.1016/j.virol.2011.10.006.
- Zhang Y, Zhang Q, Kong H, et al. H5N1 hybrid viruses bearing 2009/H1N1 virus genes transmit in guinea pigs by respiratory droplet. Science. 2013;340(6139):1459–1463. doi: 10.1126/science.1229455
- Imai M, Watanabe T, Hatta M, et al. Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature. 2012;486(7403):420–428. doi: 10.1038/nature10831
- Linster M, van Boheemen S, de Graaf M, et al. Identification, characterization, and natural selection of mutations driving airborne transmission of A/H5N1 virus. Cell. 2014;157(2):329–339. doi: 10.1016/j.cell.2014.02.040
- Thompson AJ, Paulson JC. Adaptation of influenza viruses to human airway receptors. J Biol Chem. 2021;296:100017. doi: 10.1074/jbc.REV120.013309
- Yamada S, Suzuki Y, Suzuki T, et al. Haemagglutinin mutations responsible for the binding of H5N1 influenza a viruses to human-type receptors. Nature. 2006;444(7117):378–382. doi: 10.1038/nature05264
- Schrauwen EJA, Richard M, Burke DF, et al. Amino acid substitutions that affect receptor binding and stability of the hemagglutinin of influenza A/H7N9 virus. J Virol. 2016;90(7):3794–3799. doi: 10.1128/JVI.03052-15
- de Vries RP, Zhu X, McBride R, et al. Hemagglutinin receptor specificity and structural analyses of respiratory droplet-transmissible H5N1 viruses. J Virol. 2014;88:768–773. doi: 10.1128/JVI.02690-13
- Mair CM, Meyer T, Schneider K, et al. A histidine residue of the influenza virus hemagglutinin controls the pH dependence of the conformational change mediating membrane fusion. J Virol. 2014;88(22):13189–13200. doi: 10.1128/JVI.01704-14
- Wang W, Lu B, Zhou H, et al. Glycosylation at 158N of the hemagglutinin protein and receptor binding specificity synergistically affect the antigenicity and immunogenicity of a live attenuated H5N1 A/Vietnam/1203/2004 vaccine virus in ferrets. J Virol. 2010;84(13):6570–6577. doi: 10.1128/JVI.00221-10
- Shi Y, Zhang W, Wang F, et al. Structures and receptor binding of hemagglutinins from human-infecting H7N9 influenza viruses. Science. 2013;342(6155):243–247. doi: 10.1126/science.1242917
- Peng X, Liu F, Wu H, et al. Amino acid substitutions HA A150V, PA A343T, and PB2 E627K increase the virulence of H5N6 influenza virus in mice. Front Microbiol. 2018;9:453. doi: 10.3389/fmicb.2018.00453
- Auewarakul P, Suptawiwat O, Kongchanagul A, et al. An avian influenza H5N1 virus that binds to a human-type receptor. J Virol. 2007;81(18):9950–9955. doi: 10.1128/JVI.00468-07
- Crusat M, Liu J, Palma AS, et al. Changes in the hemagglutinin of H5N1 viruses during human infection–influence on receptor binding. Virology. 2013;447(1–2):326–337. doi: 10.1016/j.virol.2013.08.010
- Zhu X, Viswanathan K, Raman R, et al. Structural basis for a switch in receptor binding specificity of two H5N1 hemagglutinin mutants. Cell Rep. 2015;13(8):1683–1691. doi: 10.1016/j.celrep.2015.10.027
- de Vriesx RP, Peng W, Grant OC, et al. Three mutations switch H7N9 influenza to human-type receptor specificity. PLOS Pathog. 2017;13(6):e1006390. doi: 10.1371/journal.ppat.1006390
- Yang H, Carney PJ, Chang JC, et al. Structural and molecular characterization of the hemagglutinin from the fifth-epidemic-wave A(H7N9) influenza viruses. J Virol. 2018;92(16):e00375–18. doi: 10.1128/JVI.00375-18
- Liu K, Guo Y, Zheng H, et al. Enhanced pathogenicity and transmissibility of H9N2 avian influenza virus in mammals by hemagglutinin mutations combined with PB2-627K. Virol Sin. 2022;38(1):S47–55. doi: 10.1016/j.virs.2022.09.006
- Glaser L, Stevens J, Zamarin D, et al. A single amino acid substitution in 1918 influenza virus hemagglutinin changes receptor binding specificity. J Virol. 2005;79(17):11533–11536. doi: 10.1128/JVI.79.17.11533-11536.2005
- Stevens J, Blixt O, Glaser L, et al. Glycan microarray analysis of the hemagglutinins from modern and pandemic influenza viruses reveals different receptor specificities. J Mol Biol. 2006;355(5):1143–1155. doi: 10.1016/j.jmb.2005.11.002
- Tumpey TM, Maines TR, Van Hoeven N, et al. A two-amino acid change in the hemagglutinin of the 1918 influenza virus abolishes transmission. Science. 2007;315(5812):655–659. doi: 10.1126/science.1136212
- Watanabe Y, Ibrahim MS, Ellakany HF, et al. Acquisition of human-type receptor binding specificity by new H5N1 influenza virus sublineages during their emergence in birds in Egypt. PLOS Pathog. 2011;7(5):e1002068. doi: 10.1371/journal.ppat.1002068
- Chutinimitkul S, Herfst S, Steel J, et al. Virulence-associated substitution D222G in the hemagglutinin of 2009 pandemic influenza A(H1N1) virus affects receptor binding. J Virol. 2010;84(22):11802–11813. doi: 10.1128/JVI.01136-10
- Vines A, Wells K, Matrosovich M, et al. The role of influenza a virus hemagglutinin residues 226 and 228 in receptor specificity and host range restriction. J Virol. 1998;72(9):7626–7631. doi: 10.1128/JVI.72.9.7626-7631.1998
- Stevens J, Blixt O, Tumpey TM, et al. Structure and receptor specificity of the hemagglutinin from an H5N1 influenza virus. Science. 2006;312(5772):404–410. doi: 10.1126/science.1124513
- Chutinimitkul S, van Riel D, Munster VJ, et al. In vitro assessment of attachment pattern and replication efficiency of H5N1 influenza a viruses with altered receptor specificity. J Virol. 2010;84:6825–6833. doi: 10.1128/JVI.02737-09
- Zhang W, Shi Y, Lu X, et al. An airborne transmissible avian influenza H5 hemagglutinin seen at the atomic level. Science. 2013;340(6139):1463–1467. doi: 10.1126/science.1236787
- Xiong X, Martin SR, Haire LF, et al. Receptor binding by an H7N9 influenza virus from humans. Nature. 2013;499(7459):496–499. doi: 10.1038/nature12372
- Tzarum N, de Vries RP, Peng W, et al. The 150-loop restricts the host specificity of human H10N8 influenza virus. Cell Rep. 2017;19(2):235–245. doi: 10.1016/j.celrep.2017.03.054
- Sun X, Belser JA, Maines TR. Adaptation of H9N2 influenza viruses to mammalian hosts: a review of molecular markers. Viruses. 2020;12(5):541. doi:10.3390/v12050541.
- Xiong X, Coombs PJ, Martin SR, et al. Receptor binding by a ferret-transmissible H5 avian influenza virus. Nature. 2013;497(7449):392–396. doi: 10.1038/nature12144
- de Bruin ACM, Funk M, Spronken MI, et al. Hemagglutinin subtype specificity and mechanisms of highly pathogenic avian influenza virus genesis. Viruses. 2022;14:1566. doi: 10.3390/v14071566
- Steinhauer DA. Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology. 1999;258(1):1–20. doi:10.1006/viro.1999.9716.
- Zaraket H, Bridges OA, Russell CJ. The pH of activation of the hemagglutinin protein regulates H5N1 influenza virus replication and pathogenesis in mice. J Virol. 2013;87(9):4826–4834. doi:10.1128/JVI.03110-12.
- Steinhauer DA, Wharton SA, Skehel JJ, et al. Amantadine selection of a mutant influenza virus containing an acid-stable hemagglutinin glycoprotein: evidence for virus-specific regulation of the pH of glycoprotein transport vesicles. Proc Natl Acad Sci U S A. 1991;88(24):11525–11529. doi: 10.1073/pnas.88.24.11525
- Kim JH, Hatta M, Watanabe S, et al. Role of host-specific amino acids in the pathogenicity of avian H5N1 influenza viruses in mice. J Gen Virol. 2010;91(5):1284–1289. doi: 10.1099/vir.0.018143-0
- Graef KM, Vreede FT, Lau Y-F, et al. The PB2 subunit of the influenza virus RNA polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J Virol. 2010;84(17):8433–8445. doi: 10.1128/JVI.00879-10
- Bussey KA, Bousse TL, Desmet EA, et al. PB2 residue 271 plays a key role in enhanced polymerase activity of influenza a viruses in mammalian host cells. J Virol. 2010;84(9):4395–4406. doi: 10.1128/JVI.02642-09
- Song W, Wang P, Mok BW-Y, et al. The K526R substitution in viral protein PB2 enhances the effects of E627K on influenza virus replication. Nat Commun. 2014;5(1):5509. doi: 10.1038/ncomms6509
- Van Hoeven N, Pappas C, Belser JA, et al. Human HA and polymerase subunit PB2 proteins confer transmission of an avian influenza virus through the air. Proc Natl Acad Sci U S A. 2009;106(9):3366–3371. doi: 10.1073/pnas.0813172106
- Steel J, Lowen AC, Mubareka S, et al. Transmission of influenza virus in a mammalian host is increased by PB2 amino acids 627K or 627E/701N. PLOS Pathog. 2009;5(1):e1000252. doi: 10.1371/journal.ppat.1000252
- Arai Y, Kawashita N, Ibrahim MS, et al. PB2 mutations arising during H9N2 influenza evolution in the Middle East confer enhanced replication and growth in mammals. PLOS Pathog. 2019;15(7):e1007919. doi: 10.1371/journal.ppat.1007919
- Li Z, Chen H, Jiao P, et al. Molecular basis of replication of duck H5N1 influenza viruses in a mammalian mouse model. J Virol. 2005;79(18):12058–12064. doi: 10.1128/JVI.79.18.12058-12064.2005
- Gabriel G, Dauber B, Wolff T, et al. The viral polymerase mediates adaptation of an avian influenza virus to a mammalian host. Proc Natl Acad Sci U S A. 2005;102(51):18590–18595. doi: 10.1073/pnas.0507415102
- Elgendy EM, Arai Y, Kawashita N, et al. Identification of polymerase gene mutations that affect viral replication in H5N1 influenza viruses isolated from pigeons. J Gen Virol. 2017;98(1):6–17. doi: 10.1099/jgv.0.000674
- Li J, Liang L, Jiang L, et al. Viral RNA-binding ability conferred by SUMOylation at PB1 K612 of influenza a virus is essential for viral pathogenesis and transmission. PLOS Pathog. 2021;17(2):e1009336. doi: 10.1371/journal.ppat.1009336
- Feng X, Wang Z, Shi J, et al. Glycine at position 622 in PB1 contributes to the virulence of H5N1 avian influenza virus in mice. J Virol. 2016;90(4):1872–1879. doi: 10.1128/JVI.02387-15
- Yamayoshi S, Yamada S, Fukuyama S, et al. Virulence-affecting amino acid changes in the PA protein of H7N9 influenza a viruses. J Virol. 2014;88(6):3127–3134. doi: 10.1128/JVI.03155-13
- Hu M, Chu H, Zhang K, et al. Amino acid substitutions V63I or A37S/I61T/V63I/V100A in the PA N-terminal domain increase the virulence of H7N7 influenza a virus. Sci Rep. 2016;6(1):37800. doi: 10.1038/srep37800
- Bussey KA, Desmet EA, Mattiacio JL, et al. PA residues in the 2009 H1N1 pandemic influenza virus enhance avian influenza virus polymerase activity in mammalian cells. J Virol. 2011;85(14):7020–7028. doi: 10.1128/JVI.00522-11
- Song J, Xu J, Shi J, et al. Synergistic effect of S224P and N383D substitutions in the PA of H5N1 avian influenza virus contributes to mammalian adaptation. Sci Rep. 2015;5(1):10510. doi: 10.1038/srep10510
- Xu G, Zhang X, Gao W, et al. Prevailing PA mutation K356R in avian influenza H9N2 virus increases mammalian replication and pathogenicity. J Virol. 2016;90(18):8105–8114. doi: 10.1128/JVI.00883-16
- Mehle A, Dugan VG, Taubenberger JK, et al. Reassortment and mutation of the avian influenza virus polymerase PA subunit overcome species barriers. J Virol. 2012;86(3):1750–1757. doi: 10.1128/JVI.06203-11
- Zhu W, Zou X, Zhou J, et al. Residues 41V and/or 210D in the NP protein enhance polymerase activities and potential replication of novel influenza (H7N9) viruses at low temperature. Virol J. 2015;12(1):71. doi: 10.1186/s12985-015-0304-6
- Gabriel G, Herwig A, Klenk H-D, et al. Interaction of polymerase subunit PB2 and NP with importin α1 is a determinant of host range of influenza a virus. PLOS Pathog. 2008;4(2):e11. doi:10.1371/journal.ppat.0040011.
- Zhu W, Feng Z, Chen Y, et al. Mammalian-adaptive mutation NP-Q357K in Eurasian H1N1 Swine influenza viruses determines the virulence phenotype in mice. Emerg Microbes Infect. 2019;8(1):989–999. doi: 10.1080/22221751.2019.1635873
- Kamal RP, Alymova IV, York IA. Evolution and virulence of influenza a virus protein PB1-F2. IJMS. 2017;19(1):96. doi:10.3390/ijms19010096.
- Schneider C, Nobs SP, Heer AK, et al. Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLOS Pathog. 2014;10(4):e1004053. doi: 10.1371/journal.ppat.1004053
- van Riel D, Leijten LME, van der Eerden M, et al. Highly pathogenic avian influenza virus H5N1 infects alveolar macrophages without virus production or excessive TNF-alpha induction. PLOS Pathog. 2011;7(6):e1002099. doi:10.1371/journal.ppat.1002099.
- Marvin SA, Russier M, Huerta CT, et al. Influenza virus overcomes cellular blocks to productively replicate, impacting macrophage function. J Virol. 2017;91(2):e01417–16. doi: 10.1128/JVI.01417-16
- Yu S, Ge H, Li S, et al. Modulation of macrophage polarization by viruses: turning off/on host antiviral responses. Front Microbiol. 2022;13:839585. doi: 10.3389/fmicb.2022.839585
- Zhu B, Wu Y, Huang S, et al. Uncoupling of macrophage inflammation from self-renewal modulates host recovery from respiratory viral infection. Immunity. 2021;54(6):1200–1218.e9. doi: 10.1016/j.immuni.2021.04.001
- Wilkinson TM, Li CKF, Chui CSC, et al. Preexisting influenza-specific CD4+ T cells correlate with disease protection against influenza challenge in humans. Nat Med. 2012;18(2):274–280. doi: 10.1038/nm.2612
- McKinstry KK, Strutt TM, Kuang Y, et al. Memory CD4+ T cells protect against influenza through multiple synergizing mechanisms. J Clin Invest. 2012;122(8):2847–2856. doi: 10.1172/JCI63689
- Hayward AC, Wang L, Goonetilleke N, et al. Natural T cell–mediated protection against seasonal and pandemic influenza. Results of the flu watch cohort study. Am J Respir Crit Care Med. 2015;191(12):1422–1431. doi: 10.1164/rccm.201411-1988OC
- Conenello GM, Zamarin D, Perrone LA, et al. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza a viruses contributes to increased virulence. PLOS Pathog. 2007;3(10):1414–1421. doi: 10.1371/journal.ppat.0030141
- Kash JC, Tumpey TM, Proll SC, et al. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature. 2006;443(7111):578–581. doi: 10.1038/nature05181
- Shi Y, Wu Y, Zhang W, et al. Enabling the ’host jump’: structural determinants of receptor-binding specificity in influenza a viruses. Nat Rev Microbiol. 2014;12(12):822–831. doi:10.1038/nrmicro3362.
- Burke DF, Smith DJ, Digard P. A recommended numbering scheme for influenza a HA subtypes. PLoS ONE. 2014;9(11):e112302. doi:10.1371/journal.pone.0112302.
- Chen J, Lee KH, Steinhauer DA, et al. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell. 1998;95(3):409–417. doi: 10.1016/S0092-8674(00)81771-7
- de Graaf M, Fouchier RAM. Role of receptor binding specificity in influenza a virus transmission and pathogenesis. Embo J. 2014;33(8):823–841. doi:10.1002/embj.201387442.
- Ma W, Kahn RE, Richt JA. The pig as a mixing vessel for influenza viruses: human and veterinary implications. J Mol Genet Med. 2008;3(1):158–166. doi:10.1038/nchina.2008.185.
- Hanson A, Imai M, Hatta M, et al. Identification of stabilizing mutations in an H5 hemagglutinin influenza virus protein. J Virol. 2016;90(6):2981–2992. doi: 10.1128/JVI.02790-15
- Richard M, Schrauwen EJA, de Graaf M, et al. Limited airborne transmission of H7N9 influenza a virus between ferrets. Nature. 2013;501(7468):560–563. doi: 10.1038/nature12476
- Xu R, de Vries RP, Zhu X, et al. Preferential recognition of avian-like receptors in human influenza a H7N9 viruses. Science. 2013;342:1230–1235. doi: 10.1126/science.1243761
- Herfst S, Chutinimitkul S, Ye J, et al. Introduction of virulence markers in PB2 of pandemic swine-origin influenza virus does not result in enhanced virulence or transmission. J Virol. 2010;84(8):3752–3758. doi: 10.1128/JVI.02634-09
- Long JS, Giotis ES, Moncorgé O, et al. Species difference in ANP32A underlies influenza a virus polymerase host restriction. Nature. 2016;529(7584):101–104. doi: 10.1038/nature16474
- Park YH, Chungu K, Lee SB, et al. Host-specific restriction of avian influenza virus caused by differential dynamics of ANP32 family members. J Infect Dis. 2020;221(1):71–80. doi: 10.1093/infdis/jiz506
- Liang L, Jiang L, Li J, et al. Low polymerase activity attributed to PA drives the acquisition of the PB2 E627K mutation of h7n9 avian influenza virus in mammals. MBio. 2019;10(3):e01162–19. doi: 10.1128/mBio.01162-19
- Camacho-Zarco AR, Kalayil S, Maurin D, et al. Molecular basis of host-adaptation interactions between influenza virus polymerase PB2 subunit and ANP32A. Nat Commun. 2020;11(1):3656. doi: 10.1038/s41467-020-17407-x
- Carrique L, Fan H, Walker AP, et al. Host ANP32A mediates the assembly of the influenza virus replicase. Nature. 2020;587(7835):638–643. doi: 10.1038/s41586-020-2927-z
- Zhang H, Zhang Z, Wang Y, et al. Fundamental contribution and host range determination of ANP32A and ANP32B in influenza a virus polymerase activity. J Virol. 2019;93(13):e00174–19. doi: 10.1128/JVI.00174-19
- Long JS, Idoko-Akoh A, Mistry B, et al. Species specific differences in use of ANP32 proteins by influenza a virus. Elife. 2019;8:e45066. doi: 10.7554/eLife.45066
- Gabriel G, Klingel K, Otte A, et al. Differential use of importin-α isoforms governs cell tropism and host adaptation of influenza virus. Nat Commun. 2011;2(1):156. doi: 10.1038/ncomms1158
- Sediri H, Schwalm F, Gabriel G, et al. Adaptive mutation PB2 D701N promotes nuclear import of influenza vRnps in mammalian cells. Eur J Cell Biol. 2015;94(7–9):368–374. doi: 10.1016/j.ejcb.2015.05.012
- Fan S, Deng G, Song J, et al. Two amino acid residues in the matrix protein M1 contribute to the virulence difference of H5N1 avian influenza viruses in mice. Virology. 2009;384(1):28–32. doi:10.1016/j.virol.2008.11.044.
- Guo J, Chen J, Li Y, et al. Sumoylation of matrix protein M1 and filamentous morphology collectively contribute to the replication and virulence of highly pathogenic H5N1 avian influenza viruses in mammals. J Virol. 2022;96(4):e0163021. doi: 10.1128/jvi.01630-21
- Wang C, Qu R, Zong Y, et al. Enhanced stability of M1 protein mediated by a phospho-resistant mutation promotes the replication of prevailing avian influenza virus in mammals. PLOS Pathog. 2022;18(7):e1010645. doi: 10.1371/journal.ppat.1010645
- Ayllon J, García-Sastre A. The NS1 protein: a multitasking virulence factor. Curr Top Microbiol Immunol. 2015;386:73–107.
- Nogales A, Martinez-Sobrido L, Topham DJ, et al. Modulation of innate immune responses by the influenza a NS1 and PA-X proteins. Viruses. 2018;10(12):708. doi: 10.3390/v10120708
- Jiao P, Tian G, Li Y, et al. A single-amino-acid substitution in the NS1 protein changes the pathogenicity of H5N1 avian influenza viruses in mice. J Virol. 2008;82(3):1146–1154. doi: 10.1128/JVI.01698-07
- Ayllon J, Domingues P, Rajsbaum R, et al. A single amino acid substitution in the novel H7N9 influenza a virus NS1 protein increases CPSF30 binding and virulence. J Virol. 2014;88(20):12146–12151. doi: 10.1128/JVI.01567-14
- Nogales A, Martinez-Sobrido L, Topham DJ, et al. NS1 protein amino acid changes D189N and V194I affect interferon responses, thermosensitivity, and virulence of circulating H3N2 human influenza a viruses. J Virol. 2017;91(5):e01930–16. doi: 10.1128/JVI.01930-16
- Monteagudo PL, Muñoz-Moreno R, Fribourg M, et al. Differential modulation of innate immune responses in human primary cells by influenza a viruses carrying human or avian nonstructural protein 1. J Virol. 2019;94(1):e00999–19. doi: 10.1128/JVI.00999-19
- Jagger BW, Wise HM, Kash JC, et al. An overlapping protein-coding region in influenza a virus segment 3 modulates the host response. Science. 2012;337(6091):199–204. doi: 10.1126/science.1222213
- Gao H, Sun Y, Hu J, et al. The contribution of PA-X to the virulence of pandemic 2009 H1N1 and highly pathogenic H5N1 avian influenza viruses. Sci Rep. 2015;5(1):8262. doi: 10.1038/srep08262
- Nogales A, Martinez-Sobrido L, Chiem K, et al. Functional evolution of the 2009 pandemic H1N1 influenza virus NS1 and PA in humans. J Virol. 2018;92(19):e01206–18. doi: 10.1128/JVI.01206-18
- Hayashi T, MacDonald LA, Takimoto T, et al. Influenza A virus protein PA-X contributes to viral growth and suppression of the host antiviral and immune responses. J Virol. 2015;89(12):6442–6452. doi: 10.1128/JVI.00319-15
- Gao H, Xu G, Sun Y, et al. PA-X is a virulence factor in avian H9N2 influenza virus. J Gen Virol. 2015;96(9):2587–2594. doi: 10.1099/jgv.0.000232
- Lee J, Yu H, Li Y, et al. Impacts of different expressions of PA-X protein on 2009 pandemic H1N1 virus replication, pathogenicity and host immune responses. Virology. 2017;504:25–35. doi: 10.1016/j.virol.2017.01.015
- Qin T, Chen Y, Huangfu D, et al. PA-X protein of H1N1 subtype influenza virus disables the nasal mucosal dendritic cells for strengthening virulence. Virulence. 2022;13(1):1928–1942. doi:10.1080/21505594.2022.2139474.
- Chen W, Calvo PA, Malide D, et al. A novel influenza a virus mitochondrial protein that induces cell death. Nat Med. 2001;7(12):1306–1312. doi: 10.1038/nm1201-1306
- Cheung P-H-H, Lee T-WT, Chan C-P, et al. Influenza a virus PB1-F2 protein: an ambivalent innate immune modulator and virulence factor. J Leukocyte Biol. 2020;107(5):763–771. doi:10.1002/JLB.4MR0320-206R.
- Park E-S, Byun YH, Park S, et al. Co-degradation of interferon signaling factor DDX3 by PB1-F2 as a basis for high virulence of 1918 pandemic influenza. Embo J. 2019;38(10):e99475. doi: 10.15252/embj.201899475
- Cheung P-H-H, Lee T-WT, Kew C, et al. Virus subtype-specific suppression of MAVS aggregation and activation by PB1-F2 protein of influenza a (H7N9) virus. PLOS Pathog. 2020;16(6):e1008611. doi:10.1371/journal.ppat.1008611.
- Cheung P-H-H, Ye Z-W, Lee T-WT, et al. PB1-F2 protein of highly pathogenic influenza a (H7N9) virus selectively suppresses RNA-induced NLRP3 inflammasome activation through inhibition of MAVS-NLRP3 interaction. J Leukocyte Biol. 2020;108(5):1655–1663. doi:10.1002/JLB.4AB0420-694R.
- Koutsakos M, Kedzierska K, Subbarao K. Immune responses to avian influenza viruses. J Immunol. 2019;202(2):382–391. doi:10.4049/jimmunol.1801070.
- Iwasaki A, Pillai PS. Innate immunity to influenza virus infection. Nat Rev Immunol. 2014;14(5):315–328. doi:10.1038/nri3665.
- Zhu C, Zhang M, Fu W, et al. Comparison of H7N9 and H9N2 influenza infections in mouse model unravels the importance of early innate immune response in host protection. Front Cell Infect Microbiol. 2022;12:941078. doi: 10.3389/fcimb.2022.941078
- He Y, Fu W, Cao K, et al. IFN-κ suppresses the replication of influenza a viruses through the IFNAR-MAPK-Fos-CHD6 axis. Sci Signal. 2020;13(626):eaaz3381. doi: 10.1126/scisignal.aaz3381
- Kobasa D, Jones SM, Shinya K, et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. 2007;445(7125):319–323. doi: 10.1038/nature05495
- Yuen KY, Chan PK, Peiris M, et al. Clinical features and rapid viral diagnosis of human disease associated with avian influenza a H5N1 virus. Lancet. 1998;351(9101):467–471. doi: 10.1016/S0140-6736(98)01182-9
- Cheung CY, Poon LLM, Lau AS, et al. Induction of proinflammatory cytokines in human macrophages by influenza a (H5N1) viruses: a mechanism for the unusual severity of human disease? Lancet. 2002;360(9348):1831–1837. doi: 10.1016/S0140-6736(02)11772-7
- Gao R, Bhatnagar J, Blau DM, et al. Cytokine and chemokine profiles in lung tissues from fatal cases of 2009 pandemic influenza a (H1N1): role of the host immune response in pathogenesis. Am J Pathol. 2013;183(4):1258–1268. doi: 10.1016/j.ajpath.2013.06.023
- Belser JA, Gustin KM, Pearce MB, et al. Pathogenesis and transmission of avian influenza a (H7N9) virus in ferrets and mice. Nature. 2013;501(7468):556–559. doi: 10.1038/nature12391
- Mok CKP, Lee HHY, Chan MCW, et al. Pathogenicity of the novel A/H7N9 influenza virus in mice. MBio. 2013;4(4):e00362–13. doi:10.1128/mBio.00362-13.
- Meliopoulos VA, Karlsson EA, Kercher L, et al. Human H7N9 and H5N1 influenza viruses differ in induction of cytokines and tissue tropism. J Virol. 2014;88(22):12982–12991. doi: 10.1128/JVI.01571-14
- Bi Y, Xie Q, Zhang S, et al. Assessment of the internal genes of influenza a (H7N9) virus contributing to high pathogenicity in mice. J Virol. 2015;89(1):2–13. doi: 10.1128/JVI.02390-14
- Li X, Shao M, Zeng X, et al. Signaling pathways in the regulation of cytokine release syndrome in human diseases and intervention therapy. Sig Transduct Target Ther. 2021;6:1–16. doi: 10.1038/s41392-021-00764-4
- Fajgenbaum DC, June CH, Longo DL. Cytokine storm. N Engl J Med. 2020;383(23):2255–2273. doi:10.1056/NEJMra2026131.
- Gu Y, Zuo X, Zhang S, et al. The mechanism behind influenza virus cytokine storm. Viruses. 2021;13(7):1362. doi: 10.3390/v13071362
- Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–737. doi:10.1038/nri3073.
- Qi F, Liu M, Li F, et al. Interleukin-37 ameliorates influenza pneumonia by attenuating macrophage cytokine production in a MAPK-dependent manner. Front Microbiol. 2019;10:2482. doi: 10.3389/fmicb.2019.02482
- Huang S, Zhu B, Cheon IS, et al. PPAR-γ in macrophages limits pulmonary inflammation and promotes host recovery following respiratory viral infection. J Virol. 2019;93(9):e00030–19. doi: 10.1128/JVI.00030-19
- Cline TD, Karlsson EA, Seufzer BJ, et al. The hemagglutinin protein of highly pathogenic H5N1 influenza viruses overcomes an early block in the replication cycle to promote productive replication in macrophages. J Virol. 2013;87(3):1411–1419. doi: 10.1128/JVI.02682-12
- Zhao X, Dai J, Xiao X, et al. PI3K/Akt signaling pathway modulates influenza virus induced mouse alveolar macrophage polarization to M1/M2b. PLoS ONE. 2014;9(8):e104506. doi: 10.1371/journal.pone.0104506
- Chida J, Hara H, Uchiyama K, et al. Prion protein signaling induces M2 macrophage polarization and protects from lethal influenza infection in mice. PLOS Pathog. 2020;16(8):e1008823. doi: 10.1371/journal.ppat.1008823
- Arora S, Dev K, Agarwal B, et al. Macrophages: their role, activation and polarization in pulmonary diseases. Immunobiology. 2018;223:383–396. doi: 10.1016/j.imbio.2017.11.001
- Cole SL, Dunning J, Kok WL, et al. M1-like monocytes are a major immunological determinant of severity in previously healthy adults with life-threatening influenza. JCI Insight. 2017;2(7):e91868. doi: 10.1172/jci.insight.91868
- Yao D, Bao L, Li F, et al. H1N1 influenza virus dose dependent induction of dysregulated innate immune responses and STAT1/3 activation are associated with pulmonary immunopathological damage. Virulence. 2022;13(1):1558–1572. doi:10.1080/21505594.2022.2120951.
- Geng P, Zhu H, Zhou W, et al. Baicalin inhibits influenza a virus infection via promotion of M1 macrophage polarization. Front Pharmacol. 2020;11:01298. doi: 10.3389/fphar.2020.01298
- Halstead ES, Umstead TM, Davies ML, et al. GM-CSF overexpression after influenza a virus infection prevents mortality and moderates M1-like airway monocyte/macrophage polarization. Respir Res. 2018;19(1):3. doi: 10.1186/s12931-017-0708-5
- Hufford MM, Kim TS, Sun J, et al. The effector T cell response to influenza infection. Curr Top Microbiol Immunol. 2015;386:423–455.
- McMichael AJ. Legacy of the influenza pandemic 1918: the host T cell response. Biomed J. 2018;41(4):242–248. doi:10.1016/j.bj.2018.08.003.
- McMichael AJ, Gotch FM, Noble GR, et al. Cytotoxic T-cell immunity to influenza. N Engl J Med. 1983;309(1):13–17. doi:10.1056/NEJM198307073090103.
- Wang Z, Wan Y, Qiu C, et al. Recovery from severe H7N9 disease is associated with diverse response mechanisms dominated by CD8+ T cells. Nat Commun. 2015;6(1):6833. doi: 10.1038/ncomms7833
- Guan W, Yang Z, Wu NC, et al. Clinical correlations of transcriptional profile in patients infected with avian influenza H7N9 virus. J Infect Dis. 2018;218(8):1238–1248. doi: 10.1093/infdis/jiy317
- Duan S, Thomas PG. Balancing immune protection and immune pathology by CD8(+) T-Cell responses to influenza infection. Front Immunol. 2016;7:25. doi: 10.3389/fimmu.2016.00025
- Masopust D, Soerens AG. Tissue-resident T cells and other resident leukocytes. Annu Rev Immunol. 2019;37(1):521–546. doi:10.1146/annurev-immunol-042617-053214.
- Behr FM, Chuwonpad A, Stark R, et al. Armed and ready: transcriptional regulation of tissue-resident memory CD8 T cells. Front Immunol. 2018;9:1770. doi: 10.3389/fimmu.2018.01770
- Wang Z, Wang S, Goplen NP, et al. PD-1hi CD8+ resident memory T cells balance immunity and fibrotic sequelae. Sci Immunol. 2019;4(36):eaaw1217. doi: 10.1126/sciimmunol.aaw1217
- Goplen NP, Wu Y, Son YM, et al. Tissue-resident CD8+ T cells drive age-associated chronic lung sequelae after viral pneumonia. Sci Immunol. 2020;5(53):eabc4557. doi: 10.1126/sciimmunol.abc4557
- Trimarco JD, Heaton NS. From high-throughput to therapeutic: host-directed interventions against influenza viruses. Curr Opin Virol. 2022;53:101198. doi: 10.1016/j.coviro.2021.12.014
- Wang G, Jiang L, Wang J, et al. The G protein-coupled receptor FFAR2 promotes internalization during influenza a virus entry. J Virol. 2020;94(2):e01707–19. doi: 10.1128/JVI.01707-19
- Song Y, Huang H, Hu Y, et al. A genome-wide CRISPR/Cas9 gene knockout screen identifies immunoglobulin superfamily DCC subclass member 4 as a key host factor that promotes influenza virus endocytosis. PLOS Pathog. 2021;17(12):e1010141. doi: 10.1371/journal.ppat.1010141
- Pohl MO, Edinger TO, Stertz S, et al. Prolidase is required for early trafficking events during influenza a virus entry. J Virol. 2014;88(19):11271–11283. doi: 10.1128/JVI.00800-14
- Edinger TO, Pohl MO, Yángüez E, et al. Cathepsin W is required for escape of influenza a virus from late endosomes. MBio. 2015;6(3):e00297. doi:10.1128/mBio.00297-15.
- Lee J, Kim J, Son K, et al. Acid phosphatase 2 (ACP2) is required for membrane fusion during influenza virus entry. Sci Rep. 2017;7(1):43893. doi:10.1038/srep43893.
- Larson GP, Tran V, Yú S, et al. EPS8 facilitates uncoating of influenza a virus. Cell Reports. 2019;29(8):2175–2183.e4. doi:10.1016/j.celrep.2019.10.064.
- Yarbrough ML, Mata MA, Sakthivel R, et al. Viral subversion of nucleocytoplasmic trafficking. Traffic. 2014;15(2):127–140. doi: 10.1111/tra.12137
- Resa-Infante P, Thieme R, Ernst T, et al. Importin-α7 is required for enhanced influenza a virus replication in the alveolar epithelium and severe lung damage in mice. J Virol. 2014;88(14):8166–8179. doi: 10.1128/JVI.00270-14
- Zheng H, Ma L, Gui R, et al. G protein subunit β1 facilitates influenza a virus replication by promoting the nuclear import of PB2. J Virol. 2022;96(12):e0049422. doi: 10.1128/jvi.00494-22
- Wang X, Jiang L, Wang G, et al. Influenza a virus use of BinCARD1 to facilitate the binding of viral NP to importin α7 is counteracted by TBK1-p62 axis-mediated autophagy. Cell Mol Immunol. 2022;19(10):1168–1184. doi: 10.1038/s41423-022-00906-w
- Esparza M, Bhat P, Fontoura BM. Viral-host interactions during splicing and nuclear export of influenza virus mRnas. Curr Opin Virol. 2022;55:101254. doi: 10.1016/j.coviro.2022.101254
- Peacock TP, Sheppard CM, Staller E, et al. Host determinants of influenza RNA synthesis. Annu Rev Virol. 2019;6(1):215–233. doi: 10.1146/annurev-virology-092917-043339
- Eisfeld AJ, Kawakami E, Watanabe T, et al. RAB11A is essential for transport of the influenza virus genome to the plasma membrane. J Virol. 2011;85(13):6117–6126. doi: 10.1128/JVI.00378-11
- Staller E, Barclay WS. Host cell factors that interact with influenza virus ribonucleoproteins. Cold Spring Harb Perspect Med. 2021;11(11):a038307. doi:10.1101/cshperspect.a038307.
- Li F, Liu J, Yang J, et al. H9N2 virus-derived M1 protein promotes H5N6 virus release in mammalian cells: mechanism of avian influenza virus inter-species infection in humans. PLOS Pathog. 2021;17(12):e1010098. doi: 10.1371/journal.ppat.1010098
- Hui X, Cao L, Xu T, et al. PSMD12-mediated M1 ubiquitination of influenza a virus at K102 regulates viral replication. J Virol. 2022;96(15):e0078622. doi: 10.1128/jvi.00786-22
- Liu L, Weber A, Linne U, et al. Phosphorylation of influenza a virus matrix protein 1 at threonine 108 controls its multimerization state and functional association with the STRIPAK complex. MBio. 2023;14(1):e0323122. doi:10.1128/mbio.03231-22.
- Meineke R, Rimmelzwaan GF, Elbahesh H. Influenza virus infections and cellular kinases. Viruses. 2019;11(2):171. doi:10.3390/v11020171.
- Eierhoff T, Hrincius ER, Rescher U, et al. The epidermal growth factor receptor (EGFR) promotes uptake of influenza a viruses (IAV) into host cells. PLOS Pathog. 2010;6(9):e1001099. doi: 10.1371/journal.ppat.1001099
- Verma V, Dileepan M, Huang Q, et al. Influenza a virus activates cellular tropomyosin receptor kinase a (TrkA) signaling to promote viral replication and lung inflammation. PLOS Pathog. 2022;18(9):e1010874. doi: 10.1371/journal.ppat.1010874
- Kedzierski L, Tate MD, Hsu AC, et al. Suppressor of cytokine signaling (SOCS)5 ameliorates influenza infection via inhibition of EGFR signaling. Elife. 2017;6:e20444. doi: 10.7554/eLife.20444
- O’Hanlon R, Leyva-Grado VH, Sourisseau M, et al. An influenza virus entry inhibitor targets class II PI3 kinase and synergizes with oseltamivir. ACS Infect Dis. 2019;5(10):1779–1793. doi: 10.1021/acsinfecdis.9b00230
- Ueki IF, Min-Oo G, Kalinowski A, et al. Respiratory virus–induced EGFR activation suppresses IRF1-dependent interferon λ and antiviral defense in airway epithelium. J Exp Med. 2013;210(10):1929–1936. doi:10.1084/jem.20121401.
- Wang Q, Pan W, Wang S, et al. Protein tyrosine phosphatase SHP2 suppresses host innate immunity against influenza a virus by regulating EGFR-mediated signaling. J Virol. 2021;95(6):e02001–20. doi: 10.1128/JVI.02001-20
- Mitchell HD, Eisfeld AJ, Stratton KG, et al. The role of EGFR in influenza pathogenicity: multiple network-based approaches to identify a key regulator of non-lethal infections. Front Cell Dev Biol. 2019;7:200. doi: 10.3389/fcell.2019.00200
- Huang EJ, Reichardt LF. Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem. 2003;72(1):609–642. doi:10.1146/annurev.biochem.72.121801.161629.
- Ricci A, Felici L, Mariotta S, et al. Neurotrophin and neurotrophin receptor protein expression in the human lung. Am J Respir Cell Mol Biol. 2004;30(1):12–19. doi: 10.1165/rcmb.2002-0110OC
- Dileepan M, Ge XN, Bastan I, et al. Regulation of eosinophil recruitment and allergic airway inflammation by tropomyosin receptor kinase A. J Immunol. 2020;204(3):682–693. doi: 10.4049/jimmunol.1900786
- Yang Y-G, Tian W-M, Zhang H, et al. Nerve growth factor exacerbates allergic lung inflammation and airway remodeling in a rat model of chronic asthma. Exp Ther Med. 2013;6(5):1251–1258. doi: 10.3892/etm.2013.1284
- Kumar N, Sharma NR, Ly H, et al. Receptor tyrosine kinase inhibitors that block replication of influenza a and other viruses. Antimicrob Agents Chemother. 2011;55(12):5553–5559. doi: 10.1128/AAC.00725-11
- Kumar N, Liang Y, Parslow TG, et al. Receptor tyrosine kinase inhibitors block multiple steps of influenza a virus replication. J Virol. 2011;85(6):2818–2827. doi: 10.1128/JVI.01969-10
- Chen X, Ye H, Kuruvilla R, et al. A chemical-genetic approach to studying neurotrophin signaling. Neuron. 2005;46(1):13–21. doi:10.1016/j.neuron.2005.03.009.
- Chen L, Xing C, Ma G, et al. N-myc downstream-regulated gene 1 facilitates influenza a virus replication by suppressing canonical NF-κB signaling. Virus Res. 2018;252:22–28. doi: 10.1016/j.virusres.2018.05.001
- Dam S, Kracht M, Pleschka S, et al. The influenza a virus genotype determines the antiviral function of NF-κB. J Virol. 2016;90(17):7980–7990. doi: 10.1128/JVI.00946-16
- Lv C, Li Y, Wang T, et al. Taurolidine improved protection against highly pathogenetic avian influenza H5N1 virus lethal-infection in mouse model by regulating the NF-κB signaling pathway. Virol Sin. 2022;38(1):S119–127. doi: 10.1016/j.virs.2022.11.010
- Xu Y, Liu L. Curcumin alleviates macrophage activation and lung inflammation induced by influenza virus infection through inhibiting the NF-κB signaling pathway. Influenza Other Respir Viruses. 2017;11(5):457–463. doi:10.1111/irv.12459.
- Wurzer WJ, Ehrhardt C, Pleschka S, et al. NF-κB-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Fas/FasL is crucial for efficient influenza virus propagation. J Biol Chem. 2004;279(30):30931–30937. doi:10.1074/jbc.M403258200.
- Nimmerjahn F, Dudziak D, Dirmeier U, et al. Active NF-κB signalling is a prerequisite for influenza virus infection. J Gen Virol. 2004;85(8):2347–2356. doi: 10.1099/vir.0.79958-0
- Ehrhardt C, Rückle A, Hrincius ER, et al. The NF-κB inhibitor SC75741 efficiently blocks influenza virus propagation and confers a high barrier for development of viral resistance. Cell Microbiol. 2013;15(7):1198–1211. doi: 10.1111/cmi.12108
- Droebner K, Haasbach E, Dudek SE, et al. Pharmacodynamics, pharmacokinetics, and antiviral activity of BAY 81-8781, a novel NF-κB inhibiting anti-influenza drug. Front Microbiol. 2017;8:2130. doi: 10.3389/fmicb.2017.02130
- Haasbach E, Reiling SJ, Ehrhardt C, et al. The NF-kappaB inhibitor SC75741 protects mice against highly pathogenic avian influenza a virus. Antiviral Res. 2013;99(3):336–344. doi: 10.1016/j.antiviral.2013.06.008
- Kumar N, Xin Z-T, Liang Y, et al. NF-κB signaling differentially regulates influenza virus RNA synthesis. J Virol. 2008;82(20):9880–9889. doi:10.1128/JVI.00909-08.
- Kuwahara T, Yamayoshi S, Noda T, et al. G protein pathway suppressor 1 promotes influenza virus polymerase activity by activating the NF-κB signaling pathway. MBio. 2019;10(6):e02867–19. doi:10.1128/mBio.02867-19.
- Hale BG, Jackson D, Chen Y-H, et al. Influenza a virus NS1 protein binds p85β and activates phosphatidylinositol-3-kinase signaling. Proc Natl Acad Sci U S A. 2006;103(38):14194–14199. doi:10.1073/pnas.0606109103.
- Shin Y-K, Liu Q, Tikoo SK, et al. Influenza a virus NS1 protein activates the phosphatidylinositol 3-kinase (PI3K)/Akt pathway by direct interaction with the p85 subunit of PI3K. J Gen Virol. 2007;88(1):13–18. doi: 10.1099/vir.0.82419-0
- Hrincius ER, Dierkes R, Anhlan D, et al. Phosphatidylinositol-3-kinase (PI3K) is activated by influenza virus vRNA via the pathogen pattern receptor Rig-I to promote efficient type I interferon production. Cell Microbiol. 2011;13(12):1907–1919. doi: 10.1111/j.1462-5822.2011.01680.x
- Ehrhardt C, Marjuki H, Wolff T, et al. Bivalent role of the phosphatidylinositol-3-kinase (PI3K) during influenza virus infection and host cell defence. Cell Microbiol. 2006;8(8):1336–1348. doi: 10.1111/j.1462-5822.2006.00713.x
- Shin Y-K, Liu Q, Tikoo SK, et al. Effect of the phosphatidylinositol 3-kinase/Akt pathway on influenza a virus propagation. J Gen Virol. 2007;88(3):942–950. doi: 10.1099/vir.0.82483-0
- Deinhardt-Emmer S, Jäckel L, Häring C, et al. Inhibition of phosphatidylinositol 3-kinase by pictilisib blocks influenza virus propagation in cells and in lungs of infected mice. Biomolecules. 2021;11(6):808. doi: 10.3390/biom11060808
- Pleschka S, Wolff T, Ehrhardt C, et al. Influenza virus propagation is impaired by inhibition of the Raf/MEK/ERK signalling cascade. Nat Cell Biol. 2001;3(3):301–305. doi: 10.1038/35060098
- Laure M, Hamza H, Koch-Heier J, et al. Antiviral efficacy against influenza virus and pharmacokinetic analysis of a novel MEK-inhibitor, ATR-002, in cell culture and in the mouse model. Antiviral Res. 2020;178:104806. doi: 10.1016/j.antiviral.2020.104806
- Ludwig S, Wolff T, Ehrhardt C, et al. MEK inhibition impairs influenza B virus propagation without emergence of resistant variants. FEBS Lett. 2004;561(1–3):37–43. doi: 10.1016/S0014-5793(04)00108-5
- Meineke R, Stelz S, Busch M, et al. FDA-Approved inhibitors of RTK/Raf signaling potently impair multiple steps of in vitro and ex vivo influenza a virus infections. Viruses. 2022;14(9):2058. doi: 10.3390/v14092058
- Zhu L, Ly H, Liang Y. PLC-γ1 signaling plays a subtype-specific role in postbinding cell entry of influenza a virus. J Virol. 2014;88(1):417–424. doi:10.1128/JVI.02591-13.
- Zhu L, Yuan C, Ding X, et al. PLC-γ1 is involved in the inflammatory response induced by influenza a virus H1N1 infection. Virology. 2016;496:131–137. doi: 10.1016/j.virol.2016.06.003
- Pinto R, Herold S, Cakarova L, et al. Inhibition of influenza virus-induced NF-kappaB and Raf/MEK/ERK activation can reduce both virus titers and cytokine expression simultaneously in vitro and in vivo. Antiviral Res. 2011;92(1):45–56. doi: 10.1016/j.antiviral.2011.05.009