
ABSTRACT
Viruses have developed different strategies to hijack mitophagy to facilitate their replication. However, whether and how African swine fever virus (ASFV) regulates mitophagy are largely unknown. Here, we found that the ASFV-encoded p17 induced mitophagy. Coimmunoprecipitation/mass spectrometry assays identified translocase of outer mitochondrial membrane 70 (TOMM70) as the protein that interacted with p17. The binding of TOMM70 to p17 promoted the binding of the mitophagy receptor SQSTM1 to TOMM70, led to engulfment of mitochondria by autophagosomes, and consequently decreased the number of mitochondria. Consistently, the levels of TOMM70 and TOMM20 decreased substantially after p17 expression or ASFV infection. Furthermore, p17-mediated mitophagy resulted in the degradation of mitochondrial antiviral signalling proteins and inhibited the production of IFN-α, IL-6 and TNFα. Overall, our findings suggest that ASFV p17 regulates innate immunity by inducing mitophagy via the interaction of SQSTM1 with TOMM70.
Introduction
African swine fever virus (ASFV) causes African swine fever (ASF) in swine; ASFV is contagious, and its infection is associated with high morbidity and mortality rates [Citation1]. To date, there is no effective vaccine or drug available, resulting in the rapid spread of ASFV in more than 60 countries and causing a substantial economic burden worldwide [Citation2]. ASFV is an enveloped icosahedral virus with a double-stranded DNA genome that ranges from 170 to 193 kb in length and encodes more than 150 proteins. Most virus proteins lack any known or predictable functional information, preventing the development of clinically effective drugs and vaccines against this virus [Citation3].
The cGAS/STING signalling pathway is critical for sensing and detecting intracellular microbial and host-derived DNA to activate innate immunity [Citation4]. As expected, the DNA virus ASFV stimulates this signalling pathway [Citation5,Citation6]. However, a number of ASFV virulence factors, such as DP96R, MGF-360-11 L, MGF-505-7 R, MGF-505-11 R and pS273R, mediate strategies that block activation of the cGAS/STING signalling pathway [Citation5,Citation7,Citation8]. Interestingly, the RNA sensor RIG-I-mediated signalling pathway is also activated by transcripts of AT-rich regions contained in the ASFV genome through DNA-dependent RNA polymerase III. However, this pathway is ultimately inhibited by ASFV protein I267L [Citation9]. Activation of TLR3, a sensor of dsRNA, is also inhibited by two ASFV-encoded proteins, A276R and I329L [Citation10,Citation11]. Mitochondria play key roles in the innate immune response via RLR signalling and mitochondrial DAMP (damage-associated molecular pattern; such as mtDNA and N-formylated peptide)-regulated signalling [Citation12]. Recently, a report showed that p17, encoded by the ASFV D117L gene, inhibits cGAS-STING signalling by binding to STING [Citation13]. However, whether and how ASFV mediates the mitochondria-mediated innate immune response is unclear.
Autophagy is one of the main pathways for the selective degradation of damaged or excessive mitochondria. This process is termed mitophagy. Mitochondrial outer membrane (MOM) proteins, including NIX, BNIP3, FUNDC1, BCL2L13, and FKBP8, serve as receptors for targeting mitochondria to autophagosomes for eventual autophagic degradation [Citation14]. Moreover, Parkin induces mitophagy by regulating the ubiquitination-mediated degradation of several mitochondrial proteins, such as mitochondrial fusion proteins Mfn1 and Mfn2, the mitochondrial input receptor subunits TOMM40, TOMM20 and TOMM70, hexokinase II, members of the Bcl-2 family, mitochondrial Rho GTPases and VDAC1 [Citation15–20]. Recently, mitochondrial import factors, including the Mim1–Mim2 complex and TOMM70, were reported to be crucial for mitophagy [Citation21]. Mitophagy plays critical roles in neurodegenerative diseases, ischaemia- or drug-induced tissue injury, and terminal differentiation of red blood cells [Citation22]. In particular, virus-triggered mitophagy benefits viral replication by inhibiting apoptosis, type I interferon responses and inflammation [Citation23]. However, whether a member of the TOMM core complex is an adaptor for mitophagy is still unknown.
p17 is encoded by the D117L gene and expressed late in the viral infection cycle. This protein majorly localizes at intracellular and extracellular mature virus particles, and is an integral membrane with a Singer type I topology [Citation24,Citation25]. p17 is highly abundant and essential for the assembly and maturation of the icosahedral capsid as well as general virus viability [Citation26]. Together with p72, H240R, M1249L, and p49, p17 forms the outer capsid shell [Citation27,Citation28]. The p17 protein appears to form trimers and is located at the interface of the centre gap region of three neighbouring pseudo-hexameric capsomers [Citation27]. P17 closely associates with the base domain of p72, and three copies of p17 encircle each p72 trimer capsomer in the inner capsid shell, firmly anchoring p72 capsomers on the inner membrane [Citation29].
In this study, we found that p17 promoted mitophagy by facilitating the interaction between TOMM70 and SQSTM1 and inhibited the innate immune response by degrading MAVS, suggest a role for p17-mediated mitophagy in immune escape by ASFV.
Results
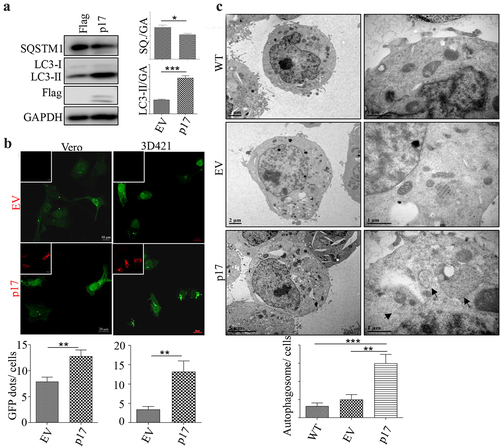
AFSV p17 promotes autophagy
To determine whether ASFV p17 regulates autophagy, HEK-293T cells were transfected with a vector expressing p17 for 36 h, and the expression of the autophagy markers LC3 and SQSTM1 was measured by Western blotting (WB) assay. As shown in , the conversion of LC3-I to LC3-II increased, and the level of SQSTM1 decreased greatly, indicating that p17 induced autophagy. We then tested whether p17 induced the formation of autophagosomes by confocal analysis of GFP-LC3 puncta formation. As shown in , p17 expression increased the number of GFP-LC3 puncta in Vero cells and in 3D4/21 cells. We further measured autophagic activity by quantifying autophagosome number in wild-type cells or cells transfected with an empty vector or a vector expressing p17 by transmission electron microscopy (TEM). Autophagosomes are identified by the presence of two distinct membrane bilayers that are separated by an electron-translucent aperture. These structures typically measure between 0.5–1.5 μm in size and contain materials intended for degradation [Citation30]. As shown in , few autophagosomes were observed in WT cells or cells harbouring empty vector, while the number of bilayer membrane structures in cells expressing p17 increased significantly (). The above data suggest that p17 induced autophagy.
Figure 1. AFSV p17 promotes autophagy.
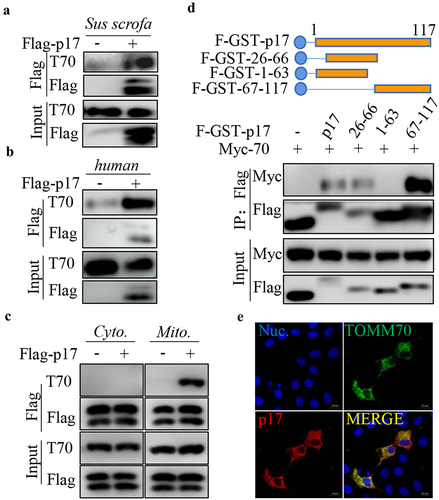
P17 interacts with the host protein TOMM70
Our IP-MS data showed that p17 potentially interacted with TOMM70 [Citation31]. Here, we used a coimmunoprecipitation (CO-IP) assay to evaluate the interaction between p17 and TOMM70. 3D4/21 cells or HEK-293T cells were transfected with vectors expressing Flag-p17. Anti-Flag IP was then performed. As shown in , p17 precipitated together with endogenous TOMM70 from human or Sus scrofa samples. To examine whether p17 specifically interacts with mitochondrial TOMM70, mitochondrial and cytoplasmic fractions extracted from HEK-293T cells transfected with an empty vector or a vector expressing Flag-p17 were subjected to anti-Flag IP. As shown in , p17 interacted with TOMM70 from mitochondria rather than from the cytoplasm. Moreover, to map the segments of p17 that interact with TOMM70, HEK-293T cells were cotransfected with vectors expressing Flag-GST-tagged truncated p17 and Myc-TOMM70, and GST pull down was performed. WB analysis showed that segments 26 to 66 and 67 to 117, but not segments 1 to 63, interacted with Myc-TOMM70, suggesting that theC-terminus of p17 is required for binding to TOMM70 (). We also tested the colocalization between p17 and TOMM70 by confocal analysis. As shown in a showed that p17 potentially inter 2E, p17 overlapped with TOMM70, confirming the interaction between p17 and TOMM70.
Figure 2. P17 interacts with TOMM70.
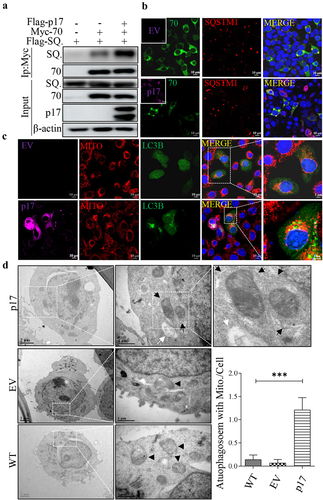
Viral protein p17 promotes autophagic engulfment of mitochondria by facilitating the interaction of SQSTM1 with TOMM70
SQSTM1 is a classic autophagic receptor located throughout the cell and leads to the autophagic degradation of mitochondria [Citation32]. We therefore tested whether p17 promotes the interaction between SQSTM1 and TOMM70. HEK-293T cells were cotransfected with vectors expressing Myc-TOMM70 and Flag-SQSTM1, with or without Flag-p17 expression. As shown in , p17 obviously enhanced the interaction between SQSTM1 and TOMM70. Next, we tested whether p17 promoted the colocalization of TOMM70 and SQSTM1 by confocal microscopy. As shown in , p17 enhanced the overlay between TOMM70 and SQSTM1. The above data suggested that p17 promoted the binding of SQSTM1 to TOMM70. Given that SQSTM1 is an autophagy cargo receptor, we wondered whether p17 regulates the inclusion of TOMM70 in the autophagosome. Confocal analysis suggested that colocalization between p17 and GFP-LC3B increased greatly in the presence of Flag-p17 (). According to transmission electron microscopy, p17 promoted the engulfment of mitochondria by autophagic vesicles, and the mitochondria are more damaged and swollen than the crest of the control group (), strongly suggesting that p17 promotes autophagic degradation of mitochondria.
Figure 3. P17 promotes autophagic engulfment of mitochondria by facilitating the binding of SQSTM1 to TOMM70.
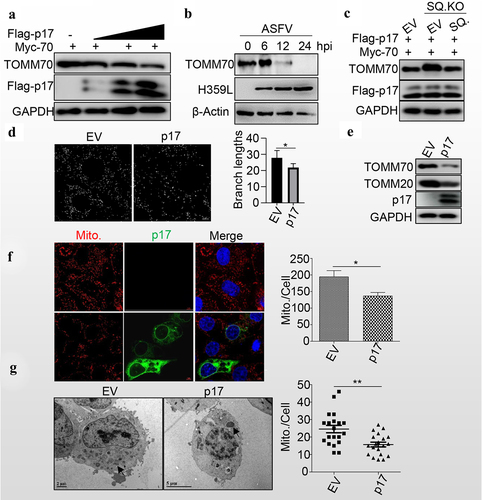
Viral protein p17 promotes mitochondrial degradation
Given that p17 promotes autophagy, we wondered whether p17 regulates the degradation of TOMM70. Interestingly, the levels of TOMM70 gradually decreased with the expression of p17 in a dose-dependent manner (). Similarly, the level of TOMM70 gradually decreased as the ASFV infection progressed (). Consistently, SQSTM1 knockout increased the level of TOMM70, while SQSTM1 expression in SQSTM1 KO cell lines rescued the degradation of TOMM70 (). Moreover, mitochondrial dynamics were analysed by using ImageJ-MiNA software, and the results showed that p17 expression significantly reduced the branch length, suggesting that p17 resulted in the fission of mitochondria (). Consequently, we observed that p17 expression decreased the levels of TOMM20 and TOMM70 (). Moreover, MitoTracker Red CMXRos staining showed that p17 expression significantly decreased the number of mitochondria (). Furthermore, the number of mitochondria was counted in empty vector- or p17-expressing cells by TEM analysis, and the data showed that p17 expression strongly decreased the number of mitochondria (), confirming that p17 promotes SQSTM1-mediated autophagic degradation of mitochondria.
Figure 4. P17 promotes mitochondrial degradation.
Viral protein p17 inhibits the innate immune response by degrading MAVS
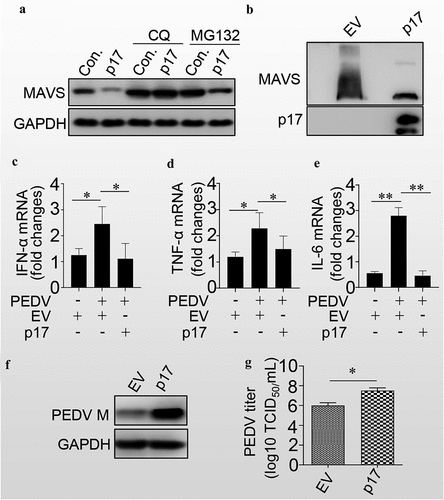
Since p17 induced the degradation of mitochondria and mitochondrial antiviral-signalling protein (MAVS) localized at the outer membrane of mitochondria, we next tested whether p17 promoted the degradation of MAVS. As shown in , p17 expression enhanced MAVS degradation. However, treatment with CQ, but not MG132, blocked the p17-mediated degradation of MAVS, suggesting that p17 induced the degradation of MAVS through autophagy. We also found that p17 expression significantly reduced the aggregation of MAVS (). We next tested the effect of p17 on the level of IFN-α. As shown in , PEDV indeed significantly increased the levels of IFN-α, TNF-α and IL-6, while p17 expression counteracted the effect of PEDV-mediated induction of these innate immunity factors. Consequently, as expected, p17 indeed increased the level of PEDV M (), as well as the virus titres (). The above data strongly suggest that p17 is beneficial for RNA virus replication by promoting the degradation of MAVS, MDA5 and RIGI.
Figure 5. P17 inhibits the innate immune response by autophagic degradation of MAVS.
Discussion
ASFV causes severe leucopenia and leads to massive destruction of lymphoid organs and tissues. In general, the above pathogenesis is believed to result from cell death [Citation33–36]. However, similar to other viruses, ASFV exploits multiple strategies to inhibit apoptotic pathways to allow complete virus replication. Several ASFV-encoding viral proteins, such as pA179L, pA224L, pEP153R and pDP71L, play critical roles in inhibiting apoptosis [Citation37–41]. p17 has no effect on inducing apoptosis. In our study, we showed that p17 promoted the degradation of TOMM70, which has been reported to promote SeV-induced apoptosis [Citation42], suggesting that the removal of TOMM70 and damaged mitochondria are likely to prevent apoptosis during ASFV infection.
TOMM70 not only acts as an input receptor for the outer membrane protein of the mitochondria, which relies on maintaining the basic morphological structure of the mitochondria, but also plays a variety of roles in innate immunity and maintenance of cellular homoeostasis [Citation43]. The interaction of TOMM70 and Hsp90 activates IRF3 and IRF-mediated antiviral responses [Citation44]. TOMM70 functions as a receptor of the mitochondrial antiviral-signalling protein (MAVS) and thereby participates in the corresponding innate immune response against viral infections [Citation45]. Additionally, the TOMM core complex, which includes TOMM70, 40 and 22, is critical for Parkin-induced mitophagy [Citation15,Citation16]. In the present study, we found that ASFV p17 promotes mitophagy by facilitating the binding of SQSTM1 to TOMM70, which is a novel role for p17 as an adaptor for mitophagy. Hence, we might expect that TOMM subunits act as adaptors for mitophagy.
The mitochondrial outer membrane provides MAVS, an essential component of the innate immune pathway, mediates innate immunity against RNA viruses. Several viruses have consequently developed their own strategies to regulate mitophagy to dampen host antiviral immunity. HPIV3, measles virus (MeV), IAV, EBV, and HTNV block the host type I IFN response by inducing mitophagy and facilitating MAVS degradation [Citation46–50]. In our study, p17-induced mitophagy facilitated the inhibition of the type I IFN response induced by PEDV infection, implying the possibility that viruses benefit each other during infection.
Autophagy is used by cells to break down viral particles. ASFV downregulates the autophagy-related genes Atg2A, Atg9A, Atg101, Atg4B and Atg7; upregulates nuclear protein 1 (NUPR1), which inhibits autophagy; affects bnip3 expression in infected cells [Citation51] activates the mTORC1-AKT pathway, which negatively regulates autophagy [Citation52] and inhibits autophagy during the early stage of ASFV infection [Citation53], suggesting that autophagy probably plays an antiviral role during ASFV infection. However, evidence that autophagy inhibits ASFV infection is still lacking, and whether p17-induced autophagy mediates the antiviral response is also unknown. Therefore, further study on the interaction between autophagy and ASFV is necessary to deepen our understanding of the immune response to ASFV infection.
Materials and methods
Cell cultures
HEK-293T cells, Vero cells and 3D4/21 cells were cultured in Dulbecco’s modified Eagle medium (DMEM; Gibco, Carlsbad, CA USA) supplemented with 10% foetal bovine serum (FBS; 1616756, Biological Industries, Israel).
Western blotting
Cells were harvested at indicated time points and lysed immediately in lysis buffer containing 2% sodium dodecyl sulphate [SDS], 50 mM Tris-HCl, 1% Triton X-100, 150 mM NaCl with pH 7.5. The lysate proteins were separated though SDS-polyacrylamide gel electrophoresis (PAGE), and the proteins were then transferred onto nitrocellulose blotting membranes (NC) (10600001; GE Healthcare Life Science). The membranes were blocked with 5% non-fat dry milk and 0.1% Tween 20 for 30 min at 37 °C, and then incubated with primary antibodies for 2 h at 37 °C, followed by incubating with horseradish peroxidase-conjugated anti-mouse/rabbit IgG (Kirkegaard & Perry Laboratories, Inc., 074–1506), finally imaged using AI680 Images (GE Healthcare, USA) after using enhanced chemiluminescence (ECL).
Cell transfection and treatment
For cell transfection, HEK-293T or 3D4/21 cells were seeded on the designated plates or glass coverslips according to the experimental scheme. When cells were 70–80% confluent, they were transfected with ExFect™ Transfection Reagent. After transfection with vectors for 24 h, the cells were harvested for immunoblotting to measure the protein levels.
Immunofluorescence staining and confocal microscopy
The indicated HEK-293T or 3D4/21 cells were seeded on 35 mm glass-bottomed cell culture dishes for 24 h, and then transfected with the indicated plasmids, in the presence or absence of the indicated drugs, or cultured under starvation conditions for indicated time. After fixing using 4% paraformaldehyde for 10 min at room temperature, the cells were then blocked and permeabilized with 5% non-fat milk with 0.2% Triton X-100 for 1 h. The cell samples were then incubated with the indicated antibodies for 2 h at 37 °C after washing three times with PBST, and with another three times of washing with PBST, the samples were then incubated with DyLight 405-, FITC-, Alexa Fluor 647-, or Alexa Fluor 546-labelled IgG antibodies for 1 h. After washing three times with PBST and stained with DAPI indicating nuclei, and the cells were observed under an N-structured illumination microscopy (SIM) Super-Resolution microscope (Nikon, Tokyo, JPN) or a Nikon A1R/A1 laser scanning confocal microscope (Nikon, Tokyo, JPN). Only cells with more than GFP dots or ring-like structures were scored as positive.
Coimmunoprecipitation assay
Cell lines were separately transfected with various vectors together or singly for indicated time and then lysed using NP40 lysis buffer with phenylmethanesulfonyl fluoride (PMSF, 1 mM) for 4 h at 4 °C. Then, the supernatants from the cell lysis after centrifuging at 12,000 × g for 10 min, followed by incubation with anti-Flag antibody and Protein A/G beads for 4 h at 4 °C. The supernatants were removed after centrifugation, and the pellets were suspended in washing buffer. This step were repeated for 5 times. The pellets were finally lysed in lysis buffer for WB analysis.
RNA extraction and quantitative real-time PCR
Total RNA was extracted following the instructions of the TRIzol reagent, and the cDNA were generated after reverse transcription of RNA a reverse transcription kit. Quantitative real-time PCR was performed using AceQ qPCR SYBR Green Master Mix with specific primers and a Roche LC96 real-time PCR system.
TEM sample preparation
The cells were fixed with 2.5% glutaraldehyde at 4 °C overnight. The cell pellets were then acquired for agarose embedding. After the agarose cooled, the cell samples were cut into small pieces and then rinsed using phosphate buffer (0.1 M, pH 7.0) 3 times. The samples were fixed using 1% osmium acid solution for 1.5 h. Then, the osmium acid solution was discarded, and the samples were washed with phosphoric acid buffer 3 times for 15 min each. The samples were then dehydrated with different concentrations of ethanol solutions, namely, 30%, 50%, 70%, 80%, for 15 min each, and then, 90% and 95% acetone solutions were added and incubated with the samples to dehydrate them for 15 min each. Finally, the samples were treated twice with pure acetone solution for 20 min each. The samples were treated with Spur embedding agent and acetone at a ratio of 1:1 at room temperature for 1 h, the samples were treated with Spur embedding agent and acetone at a ratio of 3:1 at room temperature for 3 h, and finally, the samples were treated with pure embedding agent overnight at room temperature. Finally, the samples were labelled for observation.
Then, these samples were observed using a Hitachi H-9500 transmission electron microscope (Hitachi High-Technologies Corporation) at 80 kV.
Statistical analysis
The western blotting and Co-IP results are representative of three independent experiments. All the statistical analyses were performed using Prism 8 (Graph Pad). Student’s t test was used to evaluate the statistical significance, and p < 0.05 was considered statistically significant.
Acknowledgements
We would like to acknowledge Dr. Changjiang Wen and Dr. Li Huang for kindly providing inactive ASFV infection samples.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data Availability statement
The authors confirm that the data supporting the findings of this study are available within the article.
Additional information
Funding
References
- Dixon LK, Stahl K, Jori F, et al. African swine fever epidemiology and control. Ann Rev Anim Biosci. 2020;8:221–11. doi: 10.1146/annurev-animal-021419-083741
- Gaudreault NN, Madden DW, Wilson WC, et al. African swine fever virus: an emerging DNA arbovirus. Front Vet Sci. 2020;7:215. doi: 10.3389/fvets.2020.00215
- Dixon LK, Chapman DA, Netherton CL, et al. African swine fever virus replication and genomics. Virus Res. 2013;173:3–14. doi: 10.1016/j.virusres.2012.10.020
- Decout A, Katz JD, Venkatraman S, et al. The Cgas-STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol. 2021;21:548–569. doi: 10.1038/s41577-021-00524-z
- Wang X, Wu J, Wu Y, et al. Inhibition of Cgas-STING-TBK1 signaling pathway by DP96R of ASFV China 2018/1. Biochem Biophys Res Commun. 2018;506(3):437–443. doi: 10.1016/j.bbrc.2018.10.103
- Garcia-Belmonte R, Perez-Nunez D, Pittau M, et al. African swine fever virus armenia/07 virulent strain controls interferon beta production through the Cgas-STING pathway. J Virol. 2019;93(12). doi: 10.1128/JVI.02298-18
- Yang K, Huang Q, Wang R, et al. African swine fever virus MGF505-11R inhibits type I interferon production by negatively regulating the Cgas-STING-mediated signaling pathway. Vet Microbiol. 2021;263:109265. doi: 10.1016/j.vetmic.2021.109265
- Luo J, Zhang J, Ni J, et al. The African swine fever virus protease pS273R inhibits DNA sensing Cgas-STING pathway by targeting IKKepsilon. Virulence. 2022;13:740–756. doi: 10.1080/21505594.2022.2065962
- Ran Y, Li D, Xiong MG, et al. African swine fever virus I267L acts as an important virulence factor by inhibiting RNA polymerase III-RIG-I-mediated innate immunity. PLOS Pathog. 2022;18(1):e1010270. doi: 10.1371/journal.ppat.1010270
- de Oliveira VL, Almeida SC, Soares HR, et al. A novel TLR3 inhibitor encoded by African swine fever virus (ASFV). Arch Virol. 2011;156:597–609. doi: 10.1007/s00705-010-0894-7
- Correia S, Ventura S, Parkhouse RM. Identification and utility of innate immune system evasion mechanisms of ASFV. Virus Res. 2013;173(1):87–100. doi: 10.1016/j.virusres.2012.10.013
- West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nat Rev Immunol. 2011;11(6):389–402. doi: 10.1038/nri2975
- Zheng W, Xia N, Zhang J, et al. African Swine fever virus structural protein p17 inhibits Cgas-STING signaling pathway through interacting with STING. Front Immunol. 2022;13:941579. doi: 10.3389/fimmu.2022.941579
- Kirkin V, Rogov VV. A diversity of selective autophagy receptors determines the specificity of the autophagy pathway. Mol Cell. 2019;76:268–285. doi: 10.1016/j.molcel.2019.09.005
- Chan NC, Salazar AM, Pham AH, et al. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet. 2011;20:1726–1737. doi: 10.1093/hmg/ddr048
- Geisler S, Holmstrom KM, Treis A, et al. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy. 2010;6(7):871–878. doi: 10.4161/auto.6.7.13286
- Lazarou M, Narendra DP, Jin SM, et al. PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J Cell Bio. 2013;200(2):163–172. doi: 10.1083/jcb.201210111
- Okatsu K, Iemura S, Koyano F, et al. Mitochondrial hexokinase HKI is a novel substrate of the Parkin ubiquitin ligase. Biochem Biophys Res Commun. 2012;428(1):197–202. doi: 10.1016/j.bbrc.2012.10.041
- Rakovic A, Shurkewitsch K, Seibler P, et al. Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J Biol Chem. 2013;288(4):2223–2237. doi: 10.1074/jbc.M112.391680
- Yoshii SR, Kishi C, Ishihara N, et al. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem. 2011;286(22):19630–19640. doi: 10.1074/jbc.M110.209338
- Fukuda T, Ebi Y, Saigusa T, et al. Atg43 tethers isolation membranes to mitochondria to promote starvation-induced mitophagy in fission yeast. Elife. 2020;9. doi: 10.7554/eLife.61245.
- Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012;393(7):547–564. doi: 10.1515/hsz-2012-0119
- Li Y, Wu K, Zeng S, et al. The role of mitophagy in viral infection. Cells. 2022;11(4):11. doi: 10.3390/cells11040711
- Rouiller I, Brookes SM, Hyatt AD, et al. African swine fever virus is wrapped by the endoplasmic reticulum. J Virol. 1998;72(3):2373–2387. doi: 10.1128/JVI.72.3.2373-2387.1998
- Alejo A, Matamoros T, Guerra M, et al. A proteomic atlas of the African swine fever virus particle. J Virol. 2018;92(23). doi: 10.1128/JVI.01293-18
- Suarez C, Gutierrez-Berzal J, Andres G, et al. African swine fever virus protein p17 is essential for the progression of viral membrane precursors toward icosahedral intermediates. J Virol. 2010;84:7484–7499. doi: 10.1128/JVI.00600-10
- Liu S, Luo Y, Wang Y, et al. Cryo-EM structure of the African swine fever virus. Cell Host Microbe. 2019;26:836–43 e3. doi: 10.1016/j.chom.2019.11.004
- Xia N, Wang H, Liu X, et al. African swine fever virus structural protein p17 inhibits cell proliferation through ER stress—ROS mediated cell cycle arrest. Viruses. 2020;13:13. doi: 10.3390/v13010021
- Wang N, Zhao D, Wang J, et al. Architecture of African swine fever virus and implications for viral assembly. Science. 2019;366(6465):640–644. doi: 10.1126/science.aaz1439
- Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy. 2021;17:1–382. doi: 10.1080/15548627.2020.1797280
- Zhong G, Deng T, Xu K, et al. Identification of host proteins interacting with African swine fever virus inner envelope protein p17. Chin J Biotech. 2022;38:8.
- Yamada T, Dawson TM, Yanagawa T, et al. SQSTM1/P62 promotes mitochondrial ubiquitination independently of PINK1 and PRKN/parkin in mitophagy. Autophagy. 2019;15(11):2012–2018. doi: 10.1080/15548627.2019.1643185
- Blome S, Gabriel C, Beer M. Pathogenesis of African swine fever in domestic pigs and European wild boar. Virus Res. 2013;173(1):122–130. doi: 10.1016/j.virusres.2012.10.026
- Galindo I, Hernaez B, Munoz-Moreno R, et al. The ATF6 branch of unfolded protein response and apoptosis are activated to promote African swine fever virus infection. Cell Death Dis. 2012;3(7):e341. doi: 10.1038/cddis.2012.81
- Gomez Del Moral M, Ortuno E, Fernandez-Zapatero P, et al. African swine fever virus infection induces tumor necrosis factor alpha production: implications in pathogenesis. J Virol. 1999;73:2173–2180. doi: 10.1128/JVI.73.3.2173-2180.1999
- Zsak L, Neilan JG. Regulation of apoptosis in African swine fever virus-infected macrophages. ScientificWorldjournal. 2002;2:1186–1195. doi: 10.1100/tsw.2002.214
- Galindo I, Hernaez B, Diaz-Gil G, et al. A179L, a viral Bcl-2 homologue, targets the core Bcl-2 apoptotic machinery and its upstream BH3 activators with selective binding restrictions for Bid and Noxa. Virology. 2008;375(2):561–572. doi: 10.1016/j.virol.2008.01.050
- Nogal ML, Gonzalez de Buitrago G, Rodriguez C, et al. African swine fever virus IAP homologue inhibits caspase activation and promotes cell survival in mammalian cells. J Virol. 2001;75:2535–2543. doi: 10.1128/JVI.75.6.2535-2543.2001
- Hurtado C, Granja AG, Bustos MJ, et al. The C-type lectin homologue gene (EP153R) of African swine fever virus inhibits apoptosis both in virus infection and in heterologous expression. Virology. 2004;326(1):160–170. doi: 10.1016/j.virol.2004.05.019
- Zhang F, Moon A, Childs K, et al. The African swine fever virus DP71L protein recruits the protein phosphatase 1 catalytic subunit to dephosphorylate eIf2alpha and inhibits CHOP induction but is dispensable for these activities during virus infection. J Virol. 2010;84:10681–10689. doi: 10.1128/JVI.01027-10
- Hernaez B, Diaz-Gil G, Garcia-Gallo M, et al. The African swine fever virus dynein-binding protein p54 induces infected cell apoptosis. FEBS Lett. 2004;569:224–228. doi: 10.1016/j.febslet.2004.06.001
- Wei B, Cui Y, Huang Y, et al. Tom70 mediates Sendai virus-induced apoptosis on mitochondria. J Virol. 2015;89(7):3804–3818. doi: 10.1128/JVI.02959-14
- Backes S, Bykov YS, Flohr T, et al. The chaperone-binding activity of the mitochondrial surface receptor Tom70 protects the cytosol against mitoprotein-induced stress. Cell Rep. 2021;35(1):108936. doi: 10.1016/j.celrep.2021.108936
- Yang K, Shi H, Qi R, et al. Hsp90 regulates activation of interferon regulatory factor 3 and TBK-1 stabilization in Sendai virus-infected cells. Mol Biol Cell. 2006;17(3):1461–1471. doi: 10.1091/mbc.e05-09-0853
- Kreimendahl S, Rassow J. The mitochondrial outer membrane protein Tom70-mediator in protein traffic, membrane contact sites and innate immunity. Int J Mol Sci. 2020;21(19):21. doi: 10.3390/ijms21197262
- Xia M, Gonzalez P, Li C, et al. Mitophagy enhances oncolytic measles virus replication by mitigating DDX58/RIG-I-like receptor signaling. J Virol. 2014;88(9):5152–5164. doi: 10.1128/JVI.03851-13
- Ding B, Zhang L, Li Z, et al. The matrix protein of human parainfluenza virus type 3 induces mitophagy that suppresses interferon responses. Cell Host Microbe. 2017;21:538–47 e4. doi: 10.1016/j.chom.2017.03.004
- Wang K, Ma H, Liu H, et al. The glycoprotein and nucleocapsid protein of hantaviruses manipulate autophagy flux to restrain host innate immune responses. Cell Rep. 2019;27:2075–91 e5. doi: 10.1016/j.celrep.2019.04.061
- Vilmen G, Glon D, Siracusano G, et al. BHRF1, a BCL2 viral homolog, disturbs mitochondrial dynamics and stimulates mitophagy to dampen type I IFN induction. Autophagy. 2021;17(6):1296–1315. doi: 10.1080/15548627.2020.1758416
- Wang R, Zhu Y, Ren C, et al. Influenza a virus protein PB1-F2 impairs innate immunity by inducing mitophagy. Autophagy. 2021;17(2):496–511. doi: 10.1080/15548627.2020.1725375
- Zhu JJ, Ramanathan P, Bishop EA, et al. Mechanisms of African swine fever virus pathogenesis and immune evasion inferred from gene expression changes in infected swine macrophages. PLoS One. 2019;14(11):e0223955. doi: 10.1371/journal.pone.0223955
- Wang RC, Wei Y, An Z, et al. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012;338(6109):956–959. doi: 10.1126/science.1225967
- Shimmon GL, Hui JYK, Wileman TE, et al. Autophagy impairment by African swine fever virus. J Gen Virol. 2021;102(8). doi: 10.1099/jgv.0.001637