
ABSTRACT
Streptococcus sanguinis is a ubiquitous commensal species of the oral cavity commonly involved as an opportunistic pathogen in cardiovascular infections. In this study, we investigated the functions of endopeptidase O (PepO) and a C3-degrading protease (CppA) in the systemic virulence of S. sanguinis. Isogenic mutants of pepO and cppA obtained in strain SK36 showed increased susceptibility to C3b deposition and to opsonophagocytosis by human polymorphonuclear neutrophils (PMN). These mutants differ, however, in their profiles of binding to serum amyloid P component (SAP) and C1q, whereas both showed reduced interaction with C4b-binding protein (C4BP) and/or factor H (FH) regulators as compared to SK36. The two mutants showed defects in ex vivo persistence in human blood, serum-mediated invasion of HCAEC endothelial cells, and virulence in a Galleria mellonella infection model. The transcriptional activities of pepO and cppA, assessed by RT-qPCR in nine wild-type strains, further indicated strain-specific profiles of pepO/cppA expression. Moreover, non-conserved amino acid substitutions were detected among the strains, mostly in CppA. Phylogenetic comparisons with homologues of streptococcal species of the oral and oropharyngeal sites suggested that S. sanguinis PepO and CppA have independent ancestralities. Thus, this study showed that PepO and CppA are complement evasion proteins expressed by S. sanguinis in a strain-specific manner, which are required for multiple functions associated with cardiovascular virulence.
Introduction
Streptococcus sanguinis is an abundant commensal member of the human oral microbiome [Citation1–3]. This species colonizes the oral cavity early in life and contributes to microbiome homoeostasis through its competitive binding to dental surfaces and its ability to inhibit the growth of species promoting dental caries (Streptococcus mutans) and periodontitis (Porphyromonas gingivalis) [Citation1]. In contrast, S. sanguinis has been associated with infective endocarditis in susceptible hosts [Citation4–7]and may contribute to atherosclerosis [Citation7–9]. Once it gets access to the bloodstream from oral sites, S. sanguinis needs to adapt to blood conditions and avoid complement-mediated immunity, a major branch of innate immunity in blood clearance and host tissue defence [Citation10]. The fitness of S. sanguinis in heat-inactivated serum relies on the expression of genes involved in purine metabolism and metal transport [Citation11], consistent with the role of purB in S. sanguinis virulence in a rabbit model of infective endocarditis [Citation12]. However, the mechanisms by which S. sanguinis evades the heat-labile complement system are largely unknown [Citation10].
The complement system is activated on target microorganisms through three known pathways: classical (CP), lectin (LP), and alternative (AP) pathways. Although these pathways differ in the initial mechanisms of microbial recognition, all of them converge to the generation of C3 convertases, which cleave C3, an abundant protein in the blood and host tissues, into the effector fragments C3b and C3a (anaphylatoxin). C3b is a highly reactive molecule that covalently binds to nearby target microorganisms, functioning as a major opsonin for phagocytes and as a ligand for bacterial binding to erythrocytes, platelets, and other host cells expressing C3b receptors [Citation10]. Surface-bound C3b also triggers downstream events of the complement cascades, promoting the assembly of perforin-like membrane attack complexes (MACs) and the generation of a potent anaphylatoxin, C5a. Complement molecules further trigger multiple events such as inflammation, coagulation, and tissue repair, thus playing a crucial role in the pathogenesis of microbial and cardiovascular diseases [Citation10].
Microbial pathogens typically express functions to evade and/or subvert complement functions [Citation10,Citation13]. Among oral streptococci, S. sanguinis strains show lower susceptibility to C3b deposition than other species [Citation14], whereas diversity in C3b binding among S. sanguinis strains significantly influences susceptibility to opsonophagocytosis by neutrophils (PMN) in human peripheral blood [Citation14,Citation15]. The genome of S. sanguinis SK36 harbours two genes encoding putative proteases of the complement system, pepO (encoding endopeptidase O; PepO) and cppA (annotated as C3-complement degrading protease; CppA) [Citation16]. PepO homologues expressed by Streptococcus pneumoniae, Streptococcus pyogenes and/or Streptococcus mutans in secreted or cell-associated forms mediate complement evasion and/or invasion into host cells [Citation17–20]. Studies in S. pneumoniae and S. pyogenes further suggest that CppA might play roles in virulence, although the contribution of this protease to complement evasion remains to be addressed [Citation21–23]. In the present study, we investigated the roles of PepO and CppA in S. sanguinis evasion of complement activation and associated phenotypes of systemic virulence. We further assessed the transcriptional diversity and polymorphisms of these genes among S. sanguinis strains that differed in virulence phenotypes. Our findings establish the contribution of PepO and CppA to S. sanguinis evasion to complement immunity and the requirement of these proteases for the systemic virulence of S. sanguinis.
Material and methods
Bacterial strains, culture conditions and growth curves
lists the S. sanguinis strains used in this study [Citation24–26]. These strains were routinely grown from frozen stocks on brain heart infusion (BHI) agar (BD Difco, USA) (37°C in a 10% CO2 atmosphere). For phenotypic analyses, overnight (18 h) BHI cultures with adjusted absorbances were diluted into fresh BHI and incubated under the same conditions until they reached the mid-log (A550 nm 0.3) or late-log phases (A550 nm 0.7) of growth. When required, the BHI medium was supplemented with erythromycin (10 µg/ml) or spectinomycin (200 µg/ml). The Escherichia coli strain (DH5α) was grown in Luria-Bertani broth (BD Difco, USA) supplemented with ampicillin (100 µg/mL) for plasmid propagation.
Table 1. Streptococcal strains included in this study.
Construction of isogenic mutants and complemented strains
The pepO and cppA non-polar isogenic mutants were obtained from S. sanguinis SK36 using a PCR-ligation strategy, as previously described [Citation16], using primers shown in . For pepO deletion, a recombinant allele was constructed in which a 1,679 bp internal sequence of pepO was replaced with an erythromycin resistance cassette (ErmR) obtained from plasmid pVA838. The cppA recombinant allele was obtained by replacing a 505 bp internal sequence of cppA with ErmR. The recombinant alleles were then transformed into SK36 cells, as previously described [Citation27]. The transformants recovered in BHI agar with erythromycin were then tested by PCR and DNA sequencing analyses. The confirmed mutants of pepO and cppA were designated SKpepO and SKcppA, respectively. To obtain the complemented mutants, SKpepO and SKcppA were transformed with the shuttle plasmid pDL278 [Citation28] harbouring a full-length copy pepO or cppA, respectively, which were amplified from the SK36 genomic DNA using primers C1 and C2 for pepO (C1pepO and C2pepO) or cppA (C1cppA and C2cppA) (). The respective complemented mutants, SKpepO+ and SKcppA+, were grown in BHI agar supplemented with spectinomycin (200 µg/ml).
Table 2. Oligonucleotides used in this study.
Human blood and serum samples
Blood and serum samples were collected as described in previous studies [Citation29], from two healthy volunteers with good general health, according to protocols previously approved by the Ethics Committee of the Piracicaba Dental School, University of Campinas (CEP/FOP-UNICAMP; no. CAAE: 83140418.0.0000.5418). Commercial human serum (Millipore Sigma) was also used for Human Coronary Artery Endothelial Cells (HCAEC) invasion assays.
Bacterial binding to C3b, C1q, serum amyloid P component (SAP), C4b-binding protein (C4BP), and factor H (FH)
Bacterial binding to complement proteins was determined as previously described [Citation15]. Briefly, approximately 107 CFU of strains at A550 nm 0.3 were washed twice with PBS (pH 7.4), resuspended in 20 µL of 20% human serum (in PBS), and incubated at 37°C °C for 30 min. Bacterial cells were washed twice with PBS-Tween 0.05% (PBST) and incubated with specific antibodies for each complement protein diluted in PBST. These included fluorescein isothiocyanate (FITC)-conjugated polyclonal goat anti-human C3 IgG antibody (1:300; on ice for 40 min) (ICN, USA), FITC-conjugated polyclonal goat anti-human C1q (1:300; 37°C for 60 min) (LSBio, USA) [Citation30], FITC-conjugated polyclonal anti-human SAP (1:200; 37°C for 60 min) (LSBio, EUA) [Citation31], FITC-conjugated polyclonal rabbit anti-human C4BP (1:225 in PBST; 25°C for 60 min). (LSBio, USA) [Citation30]. To probe surface-bound factor H (FH), goat anti-human FH IgG (1:100, 37°C for 30 min) (Calbiochem), followed by incubation with fluorescein isothiocyanate (FITC)-conjugated anti-goat IgG (1:1.000; 4°C, 40 min) was used. Cells were then washed twice with PBST and fixed in 3% paraformaldehyde in PBS for flow cytometry analysis using a FACSCalibur flow cytometer (BD Biosciences). A total of 25,000 bacterial cells were gated using the forward and side scatter parameters. The results were analysed using the geometric mean fluorescence intensity (MFI) of the positive cells for each complement protein. The relative intensities of bacterial binding to each protein were expressed using the fluorescence index (FI), which was calculated by multiplying the MFI values by the respective percentages of positive cells [Citation15]. Preliminary experiments were performed using heat-inactivated serum (56°C for 20 min.) as negative controls for each tested strain and antibody, indicating irrelevant effects on comparative analyses of the strains. Cells treated with PBS instead of serum were used as controls in all experiments.
PMN isolation and phagocytosis assays
PMN were isolated from fresh heparinized human blood samples by centrifugation over a double gradient composed of 1119 and 1083 density Histopaque (Sigma-Aldrich) as previously described [Citation29]. Isolated PMN were suspended in RPMI 1640 medium (GIBCO, Life Technologies, NY, USA) supplemented with inactivated 10% foetal bovine serum. Cell viability (>98%) and purity (>95%) were monitored using trypan blue exclusion and May-Grunwald Giemsa staining. The bacteria used in phagocytosis assays were previously labelled with FITC, as described elsewhere [Citation29]. The frequency of phagocytosis was assessed in 96-well plates containing 2 × 105 PMNs per well (in 50 µL of RPMI medium with 20% human serum) and approximately 107 CFU/well of FITC-labelled bacteria [multiplicity of infection (MOI) of 200 bacteria per PMN], which were incubated for 5 min. (37°C °C, 10% CO2, and gentle shaking). The PMN were then washed twice with PBS to remove extracellular bacteria, fixed by the addition of 100 µL/well of 3% paraformaldehyde, and analysed by flow cytometry using FACSCalibur (BD Biosciences). The frequency of phagocytosis was expressed as the number of PMN cells with associated bacteria within a total of 10,000 PMN analysed [Citation29]. Parallel assays were performed in RPMI medium not supplemented with human serum.
Ex vivo persistence in human blood
Ex vivo persistence of S. sanguinis in human blood was analysed as previously described [Citation29]. Briefly, S. sanguinis strains were harvested by centrifugation (11,000 × g, 2 min) of BHI cultures (A550 nm 0.3), washed twice with PBS, and resuspended in 1 mL of fresh peripheral human blood. An aliquot of the blood suspension was immediately collected to determine the initial blood counts (log CFU/ml at time 0), and suspensions were incubated (37°C in a 10% CO2 atmosphere) in an orbital shaker for 42 h. Samples of these suspensions were collected at selected intervals for serial dilution and determination of bacterial counts on BHI agar plates. Three independent experiments were performed in triplicates.
Bacterial invasion into primary human coronary artery endothelial cells (HCAEC)
Antibiotic protection assays were performed to assess S. sanguinis invasion of HCAEC, as described previously [Citation15]. Briefly, primary HCAEC (Lonza) were cultured in basal medium (EBM-2, Lonza) supplemented with EGM-2 MV (Lonza) and seeded (1 × 105 cells/well) in 24-well culture plates (Corning). The medium was removed, the cells were washed three times with pre-warmed HEPES buffer, and then incubated (37°C, 5% CO2) for 2 h with 1 ml of bacterial suspension (1 × 107 CFU/ml) previously treated with 20% human serum (37°C °C, 20 min.) in antibiotic-free EGM-2 medium with 2% of foetal bovine serum, to a MOI of 100:1. Afterwards, the culture supernatants were removed and the HCAEC cells were washed three times with pre-warmed HEPES buffer and then incubated for 1 h with EBM-2 medium containing penicillin G (50 µg/ml) and gentamycin (300 µg/ml) to kill extracellular bacteria. The HCAEC were then washed three times with pre-warmed HEPES buffer and lysed in cold ultrapure type I H2O for 20 min at room temperature. Serial dilutions of cell lysates were plated on BHI agar for bacterial counting after 48 h of growth at 37°C (10% CO2 in air). Bacterial counts of the initial inoculum were used as a reference to calculate the percentage of invasion. As controls, similar assays were performed with bacteria that were previously treated (37°C , 20 min) with heat-inactivated serum and PBS.
Galleria mellonella infection model
A G. mellonella infection model was established as previously described [Citation20]. Briefly, S. sanguinis strains were grown for 24 h at 37°C (5% CO2) in BHI medium (with or without erythromycin or spectinomycin). The cultures were then adjusted to 108 cells/mL (A600 nm 0.42), and the bacterial suspensions in PBS (10 µL) were inoculated into the left proleg of G. mellonella larvae at the final larval stage [Citation32,Citation33] using micro syringes (Hamilton Inc., USA). Larvae inoculated with PBS or not receiving the injection (to monitor the quality of larvae rearing) were used as controls. After infection, larvae were kept at 37°C in the dark and analysed daily over the course of 7 days to evaluate the survival curve and the health index score based on the Health Index Scoring System (HISS) considering movement activity, cocoon formation, melanization, and survival [Citation34].
Natural transformation
S. sanguinis strains were grown in THB medium until the A660nm 0.7 to 0.8. Volumes of 300 µL of these cultures were then mixed with 1 µg of purified shuttle plasmid pDL278 (containing a spectinomycin resistance gene) and incubated at 37°C with shaking (160 rpm) for 90 min. Afterwards, samples were plated on agar BHI supplemented or not with spectinomycin (200 µg/ml) and incubated (37°C; 10% CO2 atmosphere) for determination of bacterial counts. The bacterial counts recovered in BHI agar supplemented with spectinomycin were determined and expressed as the number of transformants per microgram of transforming DNA.
Analysis of pepO and cppA polymorphisms in S. sanguinis genomes
Sequences of pepO and cppA of S. sanguinis strains were obtained by retrieving genomic nucleotide sequences from the FTP directory for GenBank assembly (assembly IDs: GCA_000014205.1, GCA_000191125.1, GCA_000195025.1, GCA_000212835.1, GCA_000191085.1, GCA_000192185.1, GCA_000192205.1, GCA_000192275.1, and GCA_000212815.1). Genomes were annotated with PROKKA [Citation33] and the annotated protein sequences were searched (using BLASTP 2.13.0+) against PepO (GenBank: ABN43722.1) and CppA (GenBank: ABN43788.1) sequences of SK36 genome (GenBank; NC_009009.1). The protein and gene sequences homologous to pepO and cppA were extracted from the genome annotation. Conserved sequences of pepO and cppA targeted in RT-qPCR assays () were identified by multiple sequence alignments using ClustalW (http://www.ebi.ac.uk/tools/msa/clustalw2/). ClustalW analysis was also performed with PepO and CppA amino acid sequences to identify protein polymorphisms. Phylogenetic comparisons of PepO and CppA sequences of the S. sanguinis strains and with the sequences of homologues of other streptococcal species of the oral cavity and oropharynx (available at GenBank) were performed using PhyML (https://ngphylogeny.fr/) [Citation35].
RNA isolation and transcription analysis
RNA was purified from a normalized number of bacterial cells collected from BHI cultures (A550 nm 0.3) using an RNeasy kit (Qiagen, USA) and treated with Turbo DNase (Ambion, USA), as previously described [Citation16]. Next, 1 μg of total RNA was used to obtain cDNA using random primers [Citation16] and the SuperScript III system (Life Technologies, USA), according to the manufacturer’s instructions. Quantitative PCR (qPCR) was performed using primers targeting conserved sequences of pepO and cppA. The qPCR reactions were carried out in a total volume of 10 μl, using a StepOne™ Real-Time PCR System (Life Technologies, USA) with cDNA (10 ng), 10 μM of each primer, and 1× Power SYBR® Green PCR Master Mix (Life Technologies, USA). Ten-fold serial dilutions of genomic DNA (0.003–300 ng) were used to generate standard curves for the quantification of RNA expression levels. The expression levels of the tested genes were normalized to that of the 16S rRNA gene [Citation16].
Data analysis
Data obtained by flow cytometry (FI and frequencies of phagocytosis by PMN), natural transformation, and transcription analyses were compared between strains using Kruskal-Wallis with post hoc Dunn’s test or Mann-Whitney U-test. Ex vivo persistence in blood was compared between strains at each time point using the Kruskal – Wallis test with post hoc Dunn’s test, with correction for repeated measures. Pearson’s correlation analysis was used to identify associations between transcript levels of pepO and cppA or between gene transcript levels and relative measures of strain binding to complement proteins. The Kaplan-Meier survival curves of G. mellonella were compared using the log-rank test (Mantel-Cox). An Unpaired t test was used to analyse the health index of G. mellonella larvae. Differences were considered significant when a p-value of lesser than 0.05 was obtained.
Results
PepO and CppA impair C3b deposition on S. sanguinis and modulate its interactions with pattern recognition proteins and complement regulators
Studies in S. pneumoniae and S. mutans indicate that PepO impairs C3b deposition by binding to C4BP, a fluid-phase downregulator of the CP and LP, and by preventing C1q recognition, a major recognition protein of the CP [Citation17,Citation20]. On the other hand, CppA roles in complement evasion are still elusive [Citation21,Citation36]. Thus, we applied flow cytometry analyses to assess the effects of deletion of pepO and cppA on C3b deposition in S. sanguinis SK36, as well as on SK36 interactions with complement proteins that modulate complement activation. As shown in , deletion of pepO or cppA clearly increased C3b deposition in SK36 cells treated with human serum, whereas C3b-binding phenotypes were fully restored in the complemented mutants. The pepO mutant also showed reduced binding to C4BP, a phenotype that was restored in the respective complemented mutant (). A slight reduction in C4BP binding was detected in the cppA mutant (p > 0.05) (). To assess whether cppA (and pepO) also affect AP activation, we analysed bacterial binding to factor H, a major downregulator of this pathway. As shown in , both pepO and cppA mutants showed defects in FH binding, whereas these defects were eliminated in the respective complemented mutants.
Figure 1. Effects of pepO and cppA on complement activation. Bacterial binding to C3b, C4BP, fator H, C1q and SAP were compared between pepO or cppA mutants (grey columns on the left side) with the parent strain SK36 (black columns) and the respective complemented strains (hatched columns). Intensities of protein binding were determined by flow cytometry in strains treated with 20% human serum or PBS (negative controls) and expressed as fluorescence index (FI). Columns represent means of three independent experiments. Bars indicate standard deviations. Asterisks indicate significant differences in relation to SK36 (Kruskal-Wallis with post hoc Dunn’s test; *p < 0.05).
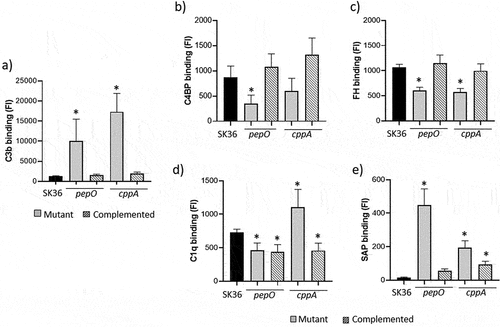
The effects of pepO and cppA on S. sanguinis binding to C1q were assessed next. The pepO mutant showed a slight, although statistically significant, reduction in binding to C1q; however, this phenotype was not restored in the complemented strain (). Conversely, the cppA mutant showed a clear increase in C1q binding, which was fully restored in the complemented mutant (). We also explored the roles of PepO and CppA in evasion of CP by assessing the effects of gene deletion in S. sanguinis binding to the short pentraxin SAP, which promotes CP activation in S. pneumoniae [Citation31]. Interestingly, the pepO mutant showed a significant increase in SAP binding, whereas a smaller increase was observed in the cppA mutant (). Both phenotypes were restored in the complemented mutants. Therefore, PepO and CppA prevent C3b deposition on S. sanguinis, reducing S. sanguinis recognition by SAP or C1q and contributing to bacterial binding to C4BP and FH.
Deletion of pepO and cppA increases S. sanguinis susceptibility to opsonophagocytosis by human PMN and reduces its persistence in human blood
To determine whether changes in C3b deposition observed in the mutant strains could reflect S. sanguinis susceptibility to opsonophagocytosis by human PMN, we compared the frequencies of phagocytosis by PMN between strains. As shown in , no relevant changes in the frequency of phagocytosis were observed among the strains in the absence of human serum. In contrast, significant increases in serum-mediated phagocytosis were observed in the SKpepO and SKcppA mutants when compared to SK36. Importantly, the opsonophagocytic phenotypes were completely restored in the respective complemented mutants, strengthening the requirement of PepO and CppA for evasion of serum-mediated phagocytosis ().
Figure 2. Effects of pepO and cppA on S. sanguinis susceptibility to phagocytosis by PMN and persistence in human blood. Mutants of pepO (SKpepO) and cppA (SKcppA) were compared to SK36 and the respective complemented strains (+). a) Phagocytosis by PMN isolated from blood were assessed by flow cytometry after incubation with FITC-labelled strains in the absence (PBS) or presence of 20% human serum (HS). Asterisks indicate significant differences in relation to SK36 at the same condition (Kruskal-Wallis with post hoc Dunn’s test; *p < 0.05). b) Comparisons of bacterial counts (log UFC/ml) in blood suspensions. Initial counts (time 0) were determined just after bacterial suspension in human blood. Asterisks indicate significant differences in bacterial counts in relation to SK36 at each time point. Kruskal–Wallis with post hoc Dunn’s test (p < 0.05).
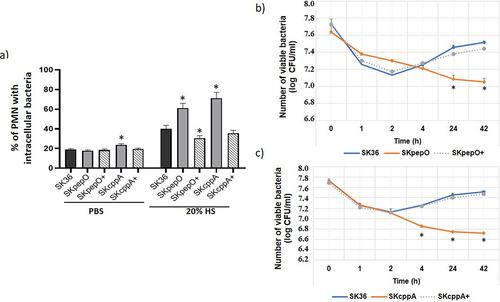
Next, we assessed bacterial persistence in human blood using ex vivo assays. As shown in , the SKpepO strain showed a reduction of 0.5 to 0.9 log in the number of viable cells when compared to SK36, after 24 to 42 h of incubation in human blood. A reduction in viability was more evident in the SKcppA mutant, which showed lower counts in blood when compared to SK36 after 4 h of incubation (). Strengthening the defects in blood fitness observed in the SKpepO and SKcppA mutants, the complemented mutants SKpepO+ and SKcppA+ showed blood persistence phenotypes similar to those observed in SK36 (). Therefore, PepO and CppA promote evasion of opsonophagocytosis by PMN and contribute to S. sanguinis persistence in human blood.
PepO and CppA contribute to invasion of S. sanguinis into HCAEC in a serum-dependent way
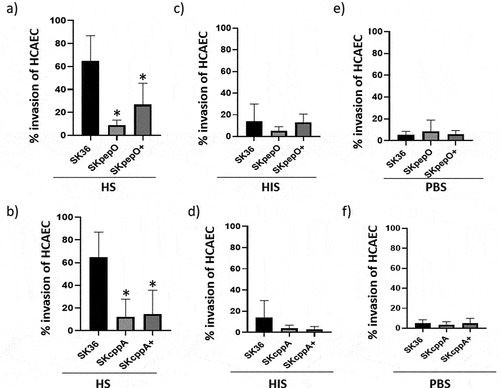
Previously, we found that S. sanguinis depends on serum to invade endothelial cells [Citation15]. Thus, we investigated the effects of pepO and cppA deletion on the S. sanguinis capacity to invade primary HCAEC in the presence or absence of human serum (HS). To this end, we performed antibiotic protection assays with mutant, parent, and complemented strains previously treated with 20% HS, heat-inactivated human serum (HIS), or PBS. As shown in the SKpepO and SKcppA mutants pretreated with HS showed defects in HCAEC invasion compared to SK36. No differences between these mutants and SK36 were detected when the strains were treated with HIS () or PBS (). The serum-dependent invasion phenotypes were not restored in the SKcppA+ complemented mutant, suggesting defects in episomal cppA expression in these assays. Therefore, PepO and CppA contribute to S. sanguinis invasiveness.
Figure 3. Contribution of pepO and cppA to the S. sanguinis capacity to invade human coronary artery endothelial cells. Bacterial invasion into primary HCAEC was determined in antibiotic-protection assays with pepO and cppA mutants, SK36 and complemented strains (+). Strains were treated with human serum (HS) (a,b), heat-inactivated serum (HIS) (c,d) or PBS (e,f) before co-cultivation with HCAEC. Invasion rates were expressed as the percentage of intracellular bacteria in relation to the initial inoculum. Columns represent means of three independent experiments. Bars indicate standard deviation values. Asterisks indicate significant differences in relation to SK36 (Kruskal-Wallis with post hoc Dunn’s test; *p < 0.05).
Deletion of pepO and cppA abolishes the virulence of S. sanguinis in a Galleria mellonela infection model
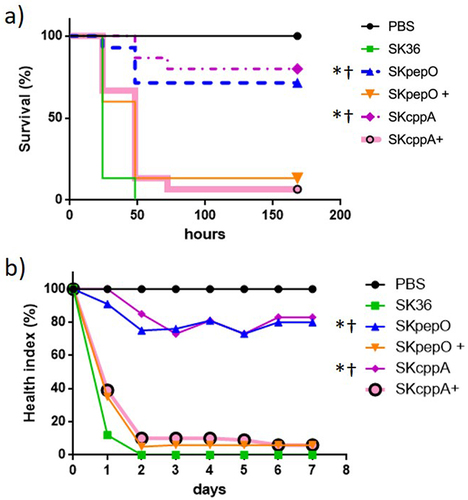
Deletion of pepO or PepO blockage with PepO antiserum impairs the S. mutans capacity to kill G. mellonella [Citation20]. Thus, we investigated the effects of pepO and cppA deletions on the virulence of SK36 using the G. mellonella infection model. As shown in , the mutants SKpepO and SKcppA showed an impaired capacity to kill G. mellonella larvae as compared to SK36. In addition, the virulence of the complemented strains SKpepO+ and SKcppA+ in this model was restored. Analysis of health indices of the infected larvae () was further compatible with survival curves, confirming that pepO and cppA are required for S. sanguinis virulence.
Figure 4. Contribution of pepO and cppA to S. sanguinis virulence. Galleria mellonella larvae were infected with the S. sanguinis strains SK36, mutants of pepO (SKpepO) and cppA (SKcppA), and the respective complemented strains (+). Larvae inoculated with PBS were used as negative controls. a) Kaplan–Meier survival curves. Percents of larvae survival were compared between the mutant strains with SK36 (indicated by asterisks) or with the respective complemented mutant (indicated by crosses) at p < 0.001 (log rank test). b) Curves of health index scores of larvae infected with S. sanguinis strains or with PBS (negative control). Significant increases in health index scores were observed in larvae infected with SKpepO or SKcppA in relation to SK36 (indicated by asterisks) or to the respective complemented mutant (indicated by crosses) (Unpaired t test; p < 0.001).
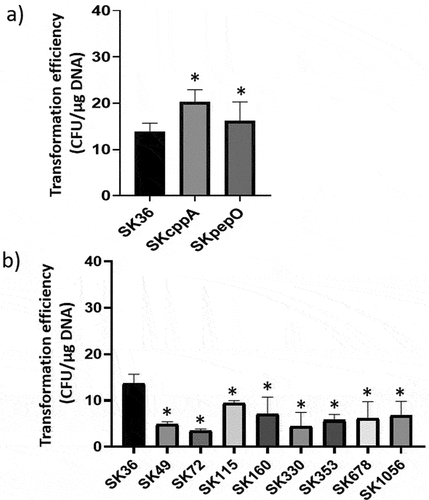
Deletion of pepO and/or cppA modestly contribute to transformation efficiency of S. sanguinis
In S. pyogenes, PepO intracellularly degrades quorum-sensing short hydrophobic peptides (SHP) involved in the allosteric activation of regulators of virulence and competence [Citation37,Citation38]. Thus, we investigated the effects of pepO and cppA deletions on the efficiency of the natural transformation of SK36. As shown in , SKcppA and SKpepO showed a slight, although significant, increase in transformation efficiency as compared to SK36. We further compared the transformation efficiency of SK36 with other S. sanguinis wild-type strains, confirming that SK36 is naturally competent and achieves the highest transformation efficiency among all tested strains (). Thus, pepO and cppA modestly influenced the competence phenotypes of S. sanguinis SK36.
Figure 5. Transformation efficiency of S. sanguinis strains. Strains at A660 nm 0.7 to 0.8 were incubated with plasmid pDL278 during 90 min. and the transformants recovered in agar BHI with spectinomycin. The number of transformants obtained per µg of plasmid DNA was expressed as transformation efficiency. a) Comparisons of the transformation efficiency of mutant strains (SKpepO and SKcppA) with SK36. b) Comparisons of the transformation efficiency of eight clinical strains with SK36. Columns represent means of four independent experiments; bars represent standard deviations. Asterisks indicate significant differences in relation to the reference strain SK36 (Mann Whitney test; p < 0.05).
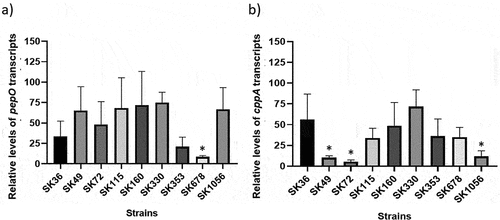
Streptococcus sanguinis strains differ in transcriptional activities of pepO and cppA, and harbour multiple PepO/CppA polymorphisms
Transcription analysis of pepO and cppA in nine selected strains (including SK36) showed that all the strains expressed pepO. However, strains SK353 and SK678 showed reduced pepO transcript levels (). Diversity in cppA transcriptional activities was also detected among strains (); strains SK49, SK72, and SK1056 showed the lowest levels of cppA transcripts (). Transcript levels of pepO did not correlate with transcript levels of cppA (Pearson correlation; r = 0.03; p > 0.05). Moreover, the transcript levels of pepO or cppA determined in the nine strains did not correlate with previously published data on strain binding to complement proteins (Alves et al., 2022) (Pearson correlation; p > 0.05). However, although not achieving statistical significance at p < 0.05, the relative measures of strain binding to FH tended to positively correlate with cppA transcript levels (Pearson correlation: r = 0.626, p = 0.071).
Figure 6. Transcription analysis of pepO and cppA in S. sanguinis strains. Levels of gene transcripts were determined by RT-qPCR in samples with equal number of bacterial cells at A550 nm 0.3. a) Levels of pepO transcripts normalized by the respective levels of 16S rRNA gene. b) Levels of cppA transcripts normalized by the respective levels of 16S rRNA gene. Columns represent means of three independent experiments; bars indicate standard deviation. Asterisks indicate significant differences in relation to the reference strain SK36 (Kruskal-Wallis with post hoc Dunn’s test; p < 0.05).
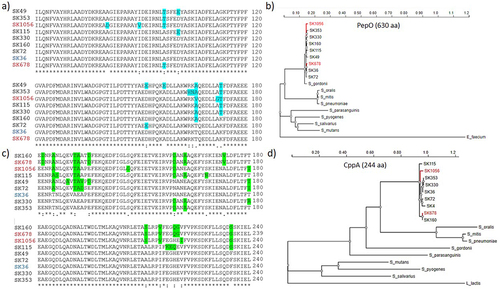
We further assessed gene and protein polymorphisms in eight clinical strains using SK36 sequences as a reference. A total of 288 single nucleotide polymorphisms (SNPs) were found in pepO (1,893 bp) encoding PepO (630 amino acids). These included 246 silent mutations and 42 missense mutations resulting in amino acid changes, which were mostly located in the N-terminal half of PepO (). The C-terminal part of PepO, containing a HEXXH motif of zinc metalloproteases and peptidases (amino acid position: 478 … 482), was conserved among strains (data not shown). The CppA protein (244 amino acids) showed higher diversity than PepO. A total of 100 SNPs were found in cppA (735 bp), including 35 missense mutations accounting for amino acid changes mostly associated with the C-terminal part of CppA. Notably, several non-conservative amino acid substitutions, potentially affecting the protein structure, were found in the C-terminal portion of CppA (). Phylogenetic comparisons of these S. sanguinis proteases with homologues of streptococcal species of the oral cavity and oropharynx showed clustering of S. sanguinis PepO sequences with the homologue of S. gordonii, a closely related species of the Sanguinis group. However, S. sanguinis PepO was more distantly related to other species of the same group, Streptococcus parasanguinis, when compared to homologues of Mitis group streptococci, including S. pneumoniae, S. mitis and S. oralis (). In contrast, S. sanguinis CppA showed higher phylogenetic similarity to homologues of the Mitis group streptococci than to the Sanguinis group species (S. gordonii and S. parasanguinis) (). The two proteases were also more distantly related to homologues of S. pyogenes, S. mutans and S. salivarius as compared to those of Sanguinis and Mitis streptococci.
Figure 7. Polymorphism and phylogenetic analysis of PepO and CppA. CppA and PepO protein sequences obtained from the genomes of SK36 (reference) and of eight S. sanguinis strains (partial genomes) available that the GenBank were compared by multiple sequence alignment using ClustalW (http://www.Ebi.ac.uk/Tools/msa/clustalw2/). Phylogenetic analyses of PepO and CppA homologues were performed using the PhyML platform (https://ngphylogeny.Fr/). a) Multiple alignment of a polymorphic region (position 61…180) of PepO including conservative and non-conservative amino acid substitutions (marked in blue). b) Phylogram of streptococcal PepO. A PepO homologue of Enterococcus faecium was used as outgroup. c) the polymorphic region (position 121…240) of CppA including non-conservative amino acid substitutions (marked in green). c) Phylogram of streptococcal CppA. A CppA homologue of Lactococcus lactis was used as outgroup. S. sanguinis strains isolated from blood samples are marked in red. Protein accession numbers of PepO homologues: S. gordonii (MBN2959327.1); S. parasanguinis (MBS5358381.1); S. pneumoniae (WP_061759435.1); S. mitis (WP_125455933.1); S. oralis (WP_125440898.1); S. salivarius (WP_195968955.1); S. mutans (WP_002262350); S. pyogenes (WP_009880612.1); Enterococcus faecium (EGP5219851.1). Protein accession numbers of CppA homologues: S. gordonii (KXT70936); S. parasanguinis (WP_272157479.1); S. mitis (WP_049502209.1); S. pneumoniae (WP_054394392.1); S. oralis (KXT93124.1); S. mutans UA159 (WP_002262622); S. salivarius (WP_195413538.1); S. pyogenes (WP_032462135.1); Lactococcus lactis (WP_259742941.1).
Discussion
The complement system plays critical roles not only in controlling microorganisms accessing the bloodstream and internal tissues but also in maintaining the homoeostasis of the human microbiome, including the microbial communities of the oral cavity [Citation10]. Thus, as a pioneer member of dental surfaces bathed by serum components present in whole saliva and gingival crevicular fluid, S. sanguinis likely evolved the capacity to evade complement immunity, which might account for its systemic virulence at certain conditions. Thus, addressing the mechanisms of S. sanguinis evasion of complement immunity could provide important information for understanding the commensal and pathogenic lifestyles of this species. A particular trait of S. sanguinis as compared to other streptococci, is the large number of cell wall-associated proteins [Citation26]. Deletion of srtA encoding sortase A transpeptidase, which anchors precursor proteins with an LPxTG-motif to the peptidoglycan, was reported to enhance S. sanguinis phagocytosis by PMN in the presence of serum [Citation39], and to impair bacterial capacity to aggregate platelets [Citation40]. These two functions are significantly amplified by the complement system and are involved in atherogenesis, as well as in the formation of vegetation in infective endocarditis [Citation41,Citation42]. Additional proteins of S. sanguinis reported to promote platelet aggregation and/or cardiovascular virulence include PcsB, murein hydrolases mur2, RTX-like protein SSA_1099, and serine-rich repeat adhesin [Citation43] however, the roles of these proteins in complement evasion have not been addressed. In Streptococcus mitis, PcsB (also known as glucan-binding protein B; GbpB) has been shown to contribute to complement evasion by promoting bacterial binding to sucrose-derived glucan [Citation44,]]. Here, we report two additional virulence factors, PepO and CppA, that individually impair C3b deposition and virulence-associated phenotypes in S. sanguinis. We also provide evidence that, as typical complement evasion proteins [Citation10], these proteases are multi-functional, contributing to the recruitment of complement down-regulators of the CP/LP (C4BP) and/or AP (FH) to the S. sanguinis surface and impairing bacterial recognition by SAP and/or C1q.
The capacity of S. sanguinis PepO to bind C4BP, a major downregulator of CP and LP, has also been reported for homologues expressed by S. pneumoniae [Citation17] and S. mutans [Citation20]. PepO homologues further bind to C1q [Citation17,Citation19,Citation20], and different explanations have been proposed to link this property with the inhibition of CP. First, secreted PepO likely sequesters fluid-phase C1q, reducing C1q availability for CP activation on the bacterial surface [Citation17]. Second, PepO binding to the globular part of C1q blocks C1q interaction with target molecules at the bacterial surface, mostly the Fc regions of IgG and IgM antibodies or short pentraxins, such as C-reactive protein (CRP) [Citation19]. The classical ligand of CRP, phosphocholine of S. pneumoniae teichoic acids (C-polysaccharide), is absent in the S. sanguinis cell wall, explaining the lack of S. sanguinis recognition by CRP [Citation14,Citation45]. However, the diversity in the S. sanguinis strain binding to another short pentraxin, SAP, was significantly associated with C3 deposition [Citation15].
Human SAP has approximately 66% homology with CRP, and although the functions of SAP as a molecular pattern recognition protein are controversial, it binds to S. pneumoniae, activating CP for complement deposition and opsonophagocytosis [Citation31]. In this study, we could not detect a clear effect of S. sanguinis pepO on C1q binding. However, we found that pepO deletion in SK36 promoted a clear increase in bacterial binding to SAP, indicating that PepO prevents SAP binding to the S. sanguinis surface, which could indirectly influence C1q binding. We also provide new evidence that PepO (and CppA) might contribute to evasion of AP, because loss of pepO and cppA reduced S. sanguinis binding to FH, a major down-regulator of AP.
There is limited information on the function of CppA proteases in complement evasion. Although annotated as a C3-degrading protease, the proteolytic functions of CppA could not be demonstrated in S. pyogenes [Citation21] or reported in other streptococci [Citation36]. On the other hand, genetic screenings using mice and/or ferret models of S. pneumoniae infection have consistently identified cppA as required for streptococcal fitness in host environments, virulence, and transmissibility from mothers to offspring [Citation22,Citation23]. Our analysis of CppA domain architecture using SMART (Simple Modular Architecture Research Tool, http://smart.embl211heidelberg.de/) could not identify functional domains previously characterized (data not shown). The role of CppA in S. sanguinis fitness in human blood might be related to its function in complement evasion because our cppA isogenic mutant showed increased susceptibility to C3b deposition and opsonophagocytosis by human PMN. Consistently, cppA was not detected in the genetic screening of S. sanguinis genes that are required for persistence in heat-inactivated human serum [Citation11]. Our findings indicate that CppA interference in C3b deposition involves, at least in part, recruitment of the complement down-regulators C4BP and FH, as well as inhibition of C1q binding, impairing CP activation. Further studies investigating the proteolytic activity of CppA may provide additional information on complement evasion.
There is significant diversity in the capacity of S. sanguinis strains to invade primary cardiac endothelial cells, which for most strains depends on the presence of serum [Citation15]. Consistent with findings reported for S. mutans PepO [Citation20], we did not detect the influence of pepO on S. sanguinis invasion of HCAEC in the absence of serum. In contrast, we showed that deletion of pepO and cppA impaired S. sanguinis serum-dependent invasion of HCAEC, implying the role of these proteases in S. sanguinis invasiveness. Multiple serum components can influence S. sanguinis invasion mediated by PepO and/or CppA, and several of them may be produced locally by vascular endothelial cells, including C1q, C3, factor H, and fibronectin [Citation46–49]. For example, S. pneumoniae binding to factor H promotes invasion into human cells expressing factor H receptors [Citation50], and because endothelial cells also bind to factor H [Citation51], it is possible that PepO- and CppA-mediated recruitment of this downregulator could contribute to S. sanguinis invasiveness. PepO binding to fibronectin has also been reported in S. mutans and Streptococcus suis, and at least in S. suis, this function is required for bacterial invasion of human endothelial cells [Citation20,Citation52]. S. mutans PepO further binds to fibrinogen, plasminogen, and laminin, but the binding profiles of S. sanguinis PepO and CppA to plasma and extracellular matrix proteins remain to be determined. Of note, the strains of S. sanguinis included in this study more clearly differed in binding to soluble complement components (C1q, SAP, C3b, C4BP, and factor H) than to other plasma and/or extracellular matrix glycoproteins, including fibronectin, fibrinogen, plasminogen, and type I collagen [Citation15].
The diversity in S. sanguinis strain invasiveness in HCAEC cells is compatible with strain virulence in a rabbit model of infective endocarditis [Citation15,Citation53]. Here, by applying a Galleria mellonella infection model, we strengthened the important roles of PepO and CppA in the systemic virulence of S. sanguinis, because the pepO and cppA mutants showed clear defects in their capacity to kill G. mellonella larvae and compromised the health index scores of infected larvae. The impact of PepO and CppA on virulence observed in this model could reflect their functions in complement evasion because the G. mellonella immune system includes complement-like proteins and phagocytic cells (haemocytes) [Citation54]. However, additional functions of these proteases in S. sanguinis fitness and virulence may also exist. In S. pyogenes PepO plays an intracellular role by degrading SHP required for allosteric activation of Rgg transcriptional regulators and downstream quorum-sensing responses [Citation37]. Deletion of pepO and cppA in SK36 promoted slight increases in transformation efficiency, although no clear associations between pepO or cppA transcriptional activities and the efficiency of natural transformation could be detected among the studied S. sanguinis strains. However, because S. sanguinis has an Rgg homologue, the molecular roles of PepO and CppA in S. sanguinis quorum-sensing might be of further interest.
The contribution of PepO and CppA to S. sanguinis virulence reported in this study emphasizes the need for further studies to investigate the mechanisms controlling cppA and pepO expression in S. sanguinis strains. Because transcript levels of cppA did not correlate with pepO transcript levels in the studied strains, we speculate that these genes might be involved in different regulatory circuits. In S. pyogenes and S. mutans, pepO is regulated by the CovR response regulator [Citation20,Citation55]. On the other hand, the S. sanguinis cppA was identified as a potential target of the two-component system SptRS [Citation16], also known as RelRS, in S. mutans [Citation56]. The SptRS system is required for S. sanguinis fitness in human whole saliva and regulates genes involved in cell wall homoeostasis, H2O2 production, immune evasion, and competence [Citation16]. Apart from the diversity in cppA transcriptional activities, CppA polymorphisms involving non-conservative amino acid changes are also of further interest in light of the evidence that positive selection of mutations in complement evasion genes plays an important role in shaping host-pathogen interactions [Citation57,Citation58].
In summary, we report two proteases expressed by S. sanguinis strains, PepO and CppA, which are required for bacterial evasion of complement-mediated immunity, persistence in human blood, invasion into human endothelial cells and virulence. We showed that pepO and cppA play multiple functions in modulating complement evasion, including inhibition of SAP or C1q binding and recruitment of fluid-phase complement down-regulators of the CP, LP, and AP. Moreover, by reporting diversity in pepO and cppA transcription levels and protein polymorphisms, we further highlighted the need for studies addressing the influence of these proteases on S. sanguinis strain-specific behaviour as a commensal or pathogen.
Ethical statement
This study was conducted in strict accordance with the National Commission on Ethics in Experimentation (CONEP), Brazil.
Acknowledgements
This study was supported by the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; grant no. 2018/02054-4 and 2021/13074-9). JCJ and ROMG were supported by the National Council of Scientific and Technological Development (CNPq; grant no. 306330/2018-0 and no. 303896/2022-1, respectively). DB was supported by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brazil (CAPES; NPD 8887.352647/2019–0). TLSA was supported by the FAPESP (grant no. 2018/13739-8 and fellowship no. 2019/20435-8). LAA and VAF were supported by FAPESP (fellowship no. 2017/19899-4 and 2018/12248-0, respectively). HN and EMF are supported by CAPES. This study was also partially financed by CAPES Finance Code 001.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data that support the findings of this study are openly available in https://doi.org/10.25824/redu/U4IJL0.
Additional information
Funding
References
- Kreth J, Giacaman RA, Raghavan R, et al. The road less traveled - defining molecular commensalism with Streptococcus sanguinis. Mol Oral Microbiol. 2017;32(3):181–16. doi: 10.1111/omi.12170
- Huttenhower C, Gevers D, Knight R, et al. Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214.
- Bin Z, Lorna CM, Todd K, et al. Streptococcus sanguinis Biofilm Formation & Interaction with Oral Pathogens. Future Microbiol. 2018;13. doi: 10.2217/fmb-2018-0043
- von Reyn CF, VON Reyn CF, Levy BS, et al. Infective endocarditis: An analysis based on strict case definitions. Ann Intern Med. 1981;94:505. doi: 10.7326/0003-4819-94-4-505
- Douglas CWI, Heath J, Hampton KK, et al. Identity of viridans streptococci isolated from cases of infective endocarditis. J Med Microbiol. 1993;39(3):179–182. doi: 10.1099/00222615-39-3-179
- Di Filippo S, Delahaye F, Semiond B, et al. Current patterns of infective endocarditis in congenital heart disease. Heart. 2006;92:1490–1495. doi: 10.1136/hrt.2005.085332
- Nakano K, Inaba H, Nomura R, et al. Detection of cariogenic Streptococcus mutans in extirpated heart valve and atheromatous plaque specimens. J Clin Microbiol. 2006;44(9):3313–3317. doi: 10.1128/JCM.00377-06
- Hashizume-Takizawa T, Yamaguchi Y, Kobayashi R, et al. Oral challenge with Streptococcus sanguinis induces aortic inflammation and accelerates atherosclerosis in spontaneously hyperlipidemic mice. Biochem Biophys Res Commun. 2019;520(3):507–513. doi: 10.1016/j.bbrc.2019.10.057
- Chiu B. Multiple infections in carotid atherosclerotic plaques. Am Heart J. 1999;138(5):S534–S536. doi: 10.1016/S0002-8703(99)70294-2
- Mattos-Graner RO, Klein MI, Alves LA. The complement system as a key modulator of the oral microbiome in health and disease. Crit Rev Microbiol. 2023;1–30. doi: 10.1080/1040841X.2022.2163614
- Zhu B, Green SP, Ge X, et al. Genome-wide identification of Streptococcus sanguinis fitness genes in human serum and discovery of potential selective drug targets. Mol Microbiol. 2021;115(4):658–671. doi: 10.1111/mmi.14629
- Paik S, Senty L, Das S, et al. Identification of virulence determinants for endocarditis in Streptococcus sanguinis by signature-tagged mutagenesis. Infect Immun. 2005;73(9):6064–6074. doi: 10.1128/IAI.73.9.6064-6074.2005
- Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol. 2008 6;6(2):132–142. doi: 10.1038/nrmicro1824
- Alves LA, De Carli TR, Chu ENH, et al. Oral streptococci show diversity in resistance to complement immunity. J Med Microbiol. 2019;68(4):600–608. doi: 10.1099/jmm.0.000955
- Alves LA, Salvatierra GC, Freitas VA, et al. Diversity in Phenotypes Associated with Host Persistence and Systemic Virulence in Streptococcus sanguinis Strains. Front Microbiol. 2022;13. doi: 10.3389/fmicb.2022.875581.
- Camargo TM, Stipp RN, Alves LA, et al. Novel two-component system of Streptococcus sanguinis affecting functions associated with viability in saliva and biofilm formation. Infect Immun. 2018;86(4). doi: 10.1128/IAI.00942-17
- Agarwal V, Sroka M, Fulde M, et al. Binding of Streptococcus pneumoniae endopeptidase O (PepO) to complement component C1q modulates the complement attack and promotes host cell adherence. J Biol Chem. 2014;289(22):15833–15844. doi: 10.1074/jbc.M113.530212
- Agarwal V, Riesbeck K, Fulde M, et al. Streptococcus pneumoniae: Endopeptidase O (PepO): A novel C1q binding complement inhibitor. Mol Immunol. 2013;56(3):243. doi: 10.1016/j.molimm.2013.05.018
- Honda-Ogawa M, Sumitomo T, Mori Y, et al. Streptococcus pyogenes endopeptidase O contributes to evasion from complement-mediated bacteriolysis via binding to human complement factor C1q. J Biol Chem. 2017;292(10):4244–4254. doi: 10.1074/jbc.M116.749275
- Alves LA, Ganguly T, Én H-C, et al. PepO is a target of the two-component systems VicRK and CovR required for systemic virulence of Streptococcus mutans. Virulence. 2020;11(1):521–536. doi: 10.1080/21505594.2020.1767377
- Lynskey NN, Reglinski M, Calay D, et al. Multi-functional mechanisms of immune evasion by the streptococcal complement inhibitor C5a peptidase. PLOS Pathog. 2017;13(8):e1006493. doi: 10.1371/journal.ppat.1006493
- Rowe HM, Karlsson E, Echlin H, et al. Bacterial Factors Required for Transmission of Streptococcus pneumoniae in Mammalian Hosts. Cell Host Microbe. 2019;25(6):884–891.e6. doi: 10.1016/j.chom.2019.04.012
- Carter R, Wolf J, Van Opijnen T, et al. Genomic analyses of pneumococci from children with sickle cell disease expose host-specific bacterial adaptations and deficits in current interventions. Cell Host Microbe. 2014;15(5):587–599. doi: 10.1016/j.chom.2014.04.005
- Kilian M, Mikkelsen L, Henrichsen J. Taxonomic study of viridans streptococci: Description of Streptococcus gordonii sp. nov. and emended descriptions of Streptococcus sanguis (White and Niven 1946), Streptococcus oralis (Bridge and Sneath 1982), and Streptococcus mitis (Andrewes and Horder 1906). Int J Syst Bacteriol. 1989;39.
- Bishop CJ, Aanensen DM, Jordan GE, et al. Assigning strains to bacterial species via the internet. BMC Biol. 2009;7(1). doi: 10.1186/1741-7007-7-3
- Xu P, Alves JM, Kitten T, et al. Genome of the opportunistic pathogen Streptococcus sanguinis. J Bacteriol. 2007;189(8):3166–3175. doi: 10.1128/JB.01808-06
- Moraes JJ, Stipp RN, Harth-Chu EN, et al. Two-component system VicRK regulates functions associated with establishment of Streptococcus sanguinis in biofilms. Infect Immun. 2014;82(12):4941–4951. doi: 10.1128/IAI.01850-14
- Dunny GM, Lee LN, LeBlanc DJ. Improved electroporation and cloning vector system for gram-positive bacteria. Appl Environ Microbiol. 1991;57(4):1194–1201. doi: 10.1128/aem.57.4.1194-1201.1991
- Alves LA, Nomura R, Mariano FS, et al. CovR regulates Streptococcus mutans susceptibility to complement immunity and survival in blood. Infect Immun. 2016;84(11):3206–3219. doi: 10.1128/IAI.00406-16
- Domenech M, Ramos-Sevillano E, García E, et al. Biofilm formation avoids complement immunity and phagocytosis of Streptococcus pneumoniae. Infect Immun. 2013;81(7):2606–2615. doi: 10.1128/IAI.00491-13
- Yuste J, Botto M, Bottoms SE, et al. Serum amyloid P aids complement-mediated immunity to Streptococcus pneumoniae. PLOS Pathog. 2007;3(9):3. doi: 10.1371/journal.ppat.0030120
- Vilela SFG, Barbosa JO, Rossoni RD, et al. Lactobacillus acidophilus ATCC 4356 inhibits biofilm formation by C. Albicans and attenuates the experimental candidiasis in Galleria mellonella. Virulence. 2015;6(1):29–39. doi: 10.4161/21505594.2014.981486
- Jorjão AL, Oliveira LD, Scorzoni L, et al. From moths to caterpillars: Ideal conditions for Galleria mellonella rearing for in vivo microbiological studies. Virulence. 2018;9(1):383–389. doi: 10.1080/21505594.2017.1397871
- Dijokaite A, Humbert MV, Borkowski E, et al. Establishing an invertebrate Galleria mellonella greater wax moth larval model of Neisseria gonorrhoeae infection. Virulence. 2021;12(1):1900–1920. doi: 10.1080/21505594.2021.1950269
- Lemoine F, Correia D, Lefort V, et al. NGPhylogeny.fr: new generation phylogenetic services for non-specialists. Nucleic Acids Res. 2019;47(W1):W260–W265. doi: 10.1093/nar/gkz303
- Marquart ME. Pathogenicity and virulence of Streptococcus pneumoniae: Cutting to the chase on proteases. Virulence. 2021;12(1):766–787. doi: 10.1080/21505594.2021.1889812
- Wilkening RV, Chang JC, Federle MJ. PepO, a CovRS-controlled endopeptidase, disrupts Streptococcus pyogenes quorum sensing. Mol Microbiol. 2016;99(1):71–87. doi: 10.1111/mmi.13216
- Do H, Kumaraswami M. Structural Mechanisms of Peptide Recognition and Allosteric Modulation of Gene Regulation by the RRNPP Family of Quorum-Sensing Regulators. J Mol Biol. 2016;428(14):2793–2804. doi: 10.1016/j.jmb.2016.05.026
- Yamaguchi M, Terao Y, Ogawa T, et al. Role of Streptococcus sanguinis sortase a in bacterial colonization. Microbes Infect. 2006;8(12–13):2791–2796. doi: 10.1016/j.micinf.2006.08.010
- Martini AM, Moricz BS, Ripperger AK, et al. Association of Novel Streptococcus sanguinis Virulence Factors with Pathogenesis in a Native Valve Infective Endocarditis Model. Front Microbiol. 2020;11:11. doi: 10.3389/fmicb.2020.00010
- Soehnlein O. Multiple roles for neutrophils in atherosclerosis. Circ Res. 2012;110(6):875–888. doi: 10.1161/CIRCRESAHA.111.257535
- Kim H, Conway EM. Platelets and Complement Cross-Talk in Early Atherogenesis. Front Cardiovasc Med. 2019 6;6. doi: 10.3389/fcvm.2019.00131
- Alves LA, Harth-Chu EN, Palma TH, et al. The two-component system VicRK regulates functions associated with Streptococcus mutans resistance to complement immunity. Mol Oral Microbiol. 2017;32(5):419–431. doi: 10.1111/omi.12183
- Harth-Chu E N, Alves L A, Theobaldo J D, et al. PcsB Expression Diversity Influences on Streptococcus mitis Phenotypes Associated With Host Persistence and Virulence. Front Microbiol. 2019;10:2567. doi: 10.3389/fmicb.2019.02567
- Gillespie SH, McWhinney PHM, Patel S, et al. Species of alpha-hemolytic streptococci possessing a C-polysaccharide phosphorylcholine-containing antigen. Infect Immun. 1993;61(7):3076–3077. doi: 10.1128/iai.61.7.3076-3077.1993
- Langeggen H, Berge KE, Johnson E, et al. Human umbilical vein endothelial cells express complement receptor 1 (CD35) and complement receptor 4 (Cd11c/CD18) in vitro. Inflammation. 2002;26(3):103–110. doi: 10.1023/a:1015585530204
- Yin W, Ghebrehiwet B, Weksler B, et al. Regulated complement deposition on the surface of human endothelial cells: Effect of tobacco smoke and shear stress. Thromb Res. 2008;122(2):221–228. doi: 10.1016/j.thromres.2007.11.005
- Parente R, Clark SJ, Inforzato A, et al. Complement factor H in host defense and immune evasion. Cell Mol Life Sci. 2017;74(9):1605–1624. doi: 10.1007/s00018-016-2418-4
- Andrews RN, Caudell DL, Metheny-Barlow LJ, et al. Fibronectin Produced by Cerebral Endothelial and Vascular Smooth Muscle Cells Contributes to Perivascular Extracellular Matrix in Late-Delayed Radiation-Induced Brain Injury. Radiat Res. 2018;190(4):361. doi: 10.1667/RR14961.1
- Agarwal V, Asmat TM, Luo S, et al. Complement regulator factor H mediates a two-step uptake of Streptococcus pneumoniae by human cells. J Biol Chem. 2010;285(30):23486–23495. doi: 10.1074/jbc.M110.142703
- Jokiranta TS, Cheng ZZ, Seeberger H, et al. Binding of complement factor H to endothelial cells is mediated by the carboxy-terminal glycosaminoglycan binding site. Am J Pathol. 2005;167(4):1173–1181. doi: 10.1016/S0002-9440(10)61205-9
- Liu F, Li J, Yan K, et al. Binding of Fibronectin to SsPepO Facilitates the Development of Streptococcus suis Meningitis. J Infect Dis. 2018;217(6):973–982. doi: 10.1093/infdis/jix523
- Baker SP, Nulton TJ, Kittena T, et al. Genomic, phenotypic, and virulence analysis of Streptococcus sanguinis oral and infective-endocarditis isolates. Infect Immun. 2019;87(1). doi: 10.1128/IAI.00703-18
- Tsai CJY, Loh JMS, Proft T. Galleria mellonella infection models for the study of bacterial diseases and for antimicrobial drug testing. Virulence. 2016;3:214–229. doi: 10.1080/21505594.2015.1135289
- Wilkening RV, Federle MJ. Evolutionary Constraints Shaping Streptococcus pyogenes–Host Interactions. Trends Microbiol. 2017;25(7):562–572. doi: 10.1016/j.tim.2017.01.007
- Lemos JA, Lin VK, Nascimento MM, et al. Three gene products govern (p)ppGpp production by Streptococcus mutans. Mol Microbiol. 2007;65(6):1568–1581. doi: 10.1111/j.1365-2958.2007.05897.x
- Aleru O, Barber MF, Coers J. Battlefronts of evolutionary conflict between bacteria and animal hosts. PLOS Pathog. 2020;16(9):e1008797. doi: 10.1371/journal.ppat.1008797
- Cagliani R, Forni D, Filippi G, et al. The mammalian complement system as an epitome of host-pathogen genetic conflicts. Mol Ecol. 2016;25(6):1324–1339. doi: 10.1111/mec.13558