
ABSTRACT
Membrane Contact Sites (MCS) are areas of close apposition of organelles that serve as hotspots for crosstalk and direct transport of lipids, proteins and metabolites. Contact sites play an important role in Ca2+ signalling, phospholipid synthesis, and micro autophagy. Initially, altered regulation of vesicular trafficking was regarded as the key mechanism for intracellular pathogen survival. However, emerging studies indicate that pathogens hijack MCS elements – a novel strategy for survival and replication in an intracellular environment. Several pathogens exploit MCS to establish direct contact between organelles and replication inclusion bodies, which are essential for their survival within the cell. By establishing this direct control, pathogens gain access to cytosolic compounds necessary for replication, maintenance, escaping endocytic maturation and circumventing lysosome fusion. MCS components such as VAP A/B, OSBP, and STIM1 are targeted by pathogens through their effectors and secretion systems. In this review, we delve into the mechanisms which operate in the evasion of the host immune system when intracellular pathogens hostage MCS. We explore targeting MCS components as a novel therapeutic approach, modifying molecular pathways and signalling to address the disease’s mechanisms and offer more effective, tailored treatments for affected individuals.
Introduction
Eukaryotic cells exhibit the hallmark of having membrane-bound organelles of multiple kinds with dedicated functions. Through our textbook illustrations, we have had insights that the organelles are compartmentalized into remote entities with their sole mode of communication being either by diffusion of vesicles or through active transport. Nevertheless, research conducted in the past decade has documented a significant shift in this paradigm. Organelles are now categorized not as elusively separate compartments but as dynamic communicating moieties. The structures that facilitate communication between organelles are referred to as membrane contact sites (MCS). MCS are microdomains juxtaposed between membrane-bound organelles and tethered by contact-specific proteins. The functions of MCS encompass – signalling, regulation of organellar membrane dynamics, lipid transport and metabolic channelling [Citation1]. The concept of contact sites was initially expounded upon in 1956 by Bernhard and Rouiller [Citation2]. They had specified observing dense granules between mitochondria and endoplasmic reticulum – “the granules were coated by several layers of complex membranes and helped in maintaining a close topographical relationship between the two organelles in regenerating hepatic cells” [Citation2]. Jones et al. (1972) too had pointed at the physical contacts between endoplasmic reticulum and the replication organelle of Toxoplasma gondii as a prima facie evidence for the role of such contacts in pathogenesis [Citation3]. In Schizosaccharomyces pombe, MCS plays a notable role in morphogenesis by restricting exocytosis in growing cell tips [Citation4]. The deletion of tethered proteins in the S. cerevisiae strain (Δ-super-tether) exhibited a deficiency in ER-PM MCS which resulted in a lethal outcome. However, this lethality could be reversed by introducing an artificial ER-PM staple. This finding underscores the critical importance of ER-PM MCS for the survival of the organism [Citation5]. It was fascinating to observe that in Candida albicans, the causative agent of Candidiasis where the hyphae invade compromised cells, loss of Sac1 (an MCS component) attenuated virulence by adversely affecting biofilm formation, hyphal development and adhesion [Citation6]. These were examples which illustrate the significance of MCS in the regulation of cellular homoeostasis.
The field began to gain momentum when this functional juxtaposition became annotated and the microdomains were termed membrane contact sites. MCS was first annotated as a site of phospholipid biosynthesis between the endoplasmic reticulum and mitochondria [Citation7], following few years as Ca2+ transfer sites [Citation8] and microautophagy hotspots [Citation9]. At present, by virtue of better imaging and biotechniques, we have been able to identify ultramolecular MCS structures, such as tether proteins, spacers and pillars. Among these ultramolecular structures, tether proteins bridge organelles that form MCS. Proteins that reside at contact sites can be further classified as (i) structural proteins (comprise of tethers which clench two organelles together and spacers or pillars which hold the two membranes at a stipulated distance and thus form a scaffold of a contact site. Few examples of tethers include – Vesicle-Associated Membrane Protein (VAMP)-associated protein (VAPs) which interact with multiple biomolecules of opposing membrane [Citation10], Oxysterol-Binding Protein (OSBP)-related protein 5 (ORP5) and ORP8 [Citation11] ( and ) and Mdm1 tethering with Snx13 [Citation12], (ii) functional proteins (proteins that attribute to the functionality of the contact sites, such as transport of ions, lipids or metabolite commerce) e.g. – Lam6 participates in sterol transfer between mitochondria and ER [Citation13], Kv2.1 (a potassium channel) has been recently shown to have a role in tethering mitochondria and ER [Citation14]), (iii) regulatory proteins (control proximity of membrane and the function of protein present in MCS) – p53 alters the redox potential of Ca2+ managing proteins thereby changing mitochondria-ER tethering, phosphorylation of contact proteins alter mitochondria-vacuole tethering [Citation15] (iv) sorter/Recruitment proteins specify the proteome and lipidome at contacts – Phosphofurin Acidic Cluster Sorting protein 2 (PACS-2) coordinates the rationing of contact sites proteins [Citation16]. From the aforementioned MCS categorisation we do not exclusively classify the proteins as bona fide MCS proteins but count them to have ancillary function at the contact sites.
Figure 1. The figure elucidates inter-organellar membrane contact sites (MCS) highlighting the interactions between different organelles within a cell.
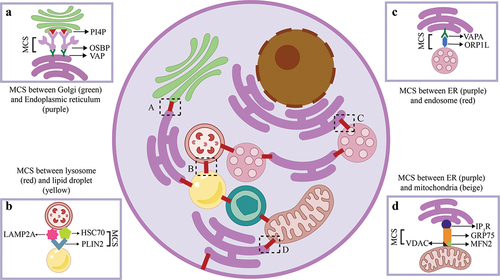
Table 1. The table gives a snippet of MCS categorization alongside their example.
MCS play an important role in the pathogenesis of infectious agents, such as bacteria and viruses. Pathogens hijack the contact sites, thus stalling the normal cellular functions and initiate direct contact between the vacuolar niches [Citation19] and the organelles to gain access to lipids, proteins and ATP that facilitate their growth and maintenance. Bacterial effector proteins rewire host MCS components thereby promoting bacterial pathogenesis followed by impaired cellular processes such as calcium signalling, homoeostasis and increased organellar clearance (e.g. mitochondria) which are hallmarks of pathogen survival and escape from host surveillance machinery [Citation20]. This review elucidates the mechanisms through which pathogens exploit the host’s innate MCS, rewire the existing connection and establish novel connection thereby threading an aggressive infection phenotype within the host followed by how MCS can be targeted for therapy.
In yeast, the MCS tether oxysterol binding homology (Osh) protein Osh3 functions as a sensor for P
() MCS between Golgi and Endoplasmic Reticulum (ER) which includes Phosphatidylinositol 4-phosphate (P14P) and Vesicle Associated Membrane Protein A/B (VAP A/B) with the lipid transfer protein (LTP) being oxysterol binding protein (OSBP) that transports lipids from the ER to Golgi [Citation21]. () MCS between lysosome and lipid droplet (LD) where the proteins involved are lysosome-associated membrane protein type 2A (LAMP2A), heat shock protein (HSC70) and LD-associated protein PLIN2. HSC70 binds to PLIN2 and translocate LD-associated proteins to LAMP2A, which is transported from the lysosomal surface to the lumen for degradation [Citation22]. () MCS flanked by ER and endosome where the tethering proteins involved are VAP and ORP1L4, which aids in cholesterol transfer from ER to endosome. () MCS amid ER and mitochondria that includes proteins tetrameric inositol 1,4,5-triphosphate receptor (IP3R), GRP75, mitofusin2 (MFN2), voltage-dependent anion channel (VDAC). This MCS mediates conduction of Ca2+ from its high concentration in the ER lumen to the mitochondrial matrix [Citation23]. These are few representative examples of MCS proteins however, we speculate that there might be more proteins involved in the formation of the complex machinery and more research needs to be undertaken in deciphering more mysterious proteins involved.
Pathogens hijacking MCS
Intracellular bacterial hijacking
Intracellular pathogens such as Legionella, Chlamydia and Coxiella have evolved exclusive mechanisms to hijack the normal host cellular processes for their own replication and survival. It was previously known that they hijack the vesicular trafficking system [Citation24,Citation25]. However, recent research has shown that the intracellular pathogens target non-vesicular transport as well for their replication. Despite their distinct life cycles, these entities are connected through their ability to manipulate the conventional MCS and establish a direct interaction between the inclusion (their place of residence) and the endoplasmic reticulum (ER) using effector proteins. In this context, we elucidate the mechanisms by which intracellular bacteria, specifically Legionella, Coxiella, and Chlamydia, exploit and manipulate the host’s MCS.
Coxiella burnetii is a causative agent of Q fever or “query fever,” a zoonosis which was first described in 1937 originating in Australia. C. burnetii is an intracellular gram-negative bacterium that infects an array of organisms ranging from arthropods to humans. During infection, Coxiella takes over host cells by establishing a Coxiella-containing vacuole (CCV), which is indispensable for its replication [Citation26,Citation27]. Coxiella requires abundant cholesterol for both the expansion of CCV and its replication. It has recently been reported that Coxiella hijacks oxysterol-binding related protein 1 L (ORP1L) through the Type 4B secretion system, a protein machinery that like T3SS delivers bacterial effector proteins to the host [Citation28]. Coxiella’s main role in cholesterol-dependent endosomal trafficking is the establishment of MCS between late endosomes/lysosomes and the ER [Citation29]. ORP1L consists of four functional domains, including three N-terminal ankyrin repeats that bind with Rab7,an FFAT motif, a PH domain and a C-terminal OSBP-related domain (ORD) that binds sterols [Citation30–32]. In mammalian cells, ORP1L acts as a cholesterol sensor where the cholesterol content of endosomes induces conformational changes in ORP1L. At low cholesterol level, endosomes form MCS with the ER. However, in Coxiella-infected cells, an enigmatic Coxiella effector protein facilitates the association of ORP1L with CCV, and then the FFAT motif of ORP1L interacts with VAP on the membrane of the ER, eventually forming CCV-ER MCSs () [Citation28,Citation33]. The depletion of ORP1L led to smaller CCV, but does not affect bacterial growth, further research is needed to decipher its role in Coxiella infection. It has been hypothesized that ORP1L is assumed to allocate cholesterol from lysosomes to the ER, independent of the lysosomal cholesterol transporter NPC1, with the cholesterol then being incorporated into lipid droplets. CCV – ER MCS may also participate in the exchange of calcium or other small molecules or manipulate ER stress and apoptosis. However, these are all speculations with no staunch evidences documented yet. Therefore, Coxiella may hijack this cholesterol-sensing signalling to transfer cholesterol to the CCV for replication of Coxiella within host cells [Citation34].
Figure 2. The figure illustrates bacteria-induced membrane contact sites demonstrating how bacteria establish close interactions between host cell organelles to promote their replication.
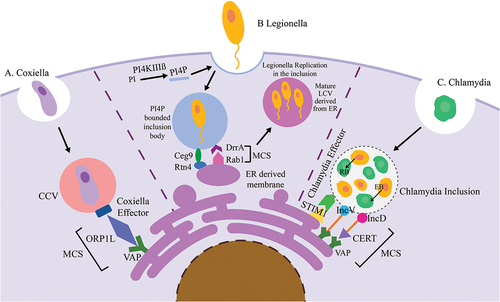
Legionella pneumophila is a Gram-negative, water-borne bacterium that grows in natural biofilms. It is a causative agent of deadly respiratory infection Legionnaires’ disease [Citation35]. Legionella creates a protective vacuole known as Legionella containing vacuole (LCV); which upon internalization circumvents with endo-lysosomal encounter [Citation36]. Legionella hijacks the conventional MCS that occurs between the ER and Golgi and redirects it to the LCV. Following the formation of a new MCS between the LCV and ER, there is transport of essential proteins into the LCV which are required for proper replication of Legionella in its protective niche. Once replication is complete, it exits the cell and infects a new host [Citation37]. Legionella expresses effectors that assist in direct lipid transfer between organelles through MCS. They adhere to a crucial regulatory lipid called phosphatidylinositol phosphate (PIP) [Citation38]. PIP, in a particular organellar membrane recruits PIP-binding proteins, which is attached to the juxtaposed membrane of another organelle, thereby leading to MCS formation. Legionella resides in a nascent phosphatidylinositol 4-monophosphate (PI4P)-rich LCV derived from the plasma membrane (PM) (). Phosphatidylinositol 4-kinase III alpha (PI4KIIIα), is an enzyme which generates PI4P from Phosphatidylinositol (PI) in the PM, from which the LCV is derived [Citation37]. In addition, inside macrophages, Legionella, remodels the PI4P-rich LCV [Citation38,Citation39] by effectors like DrrA (also known as SidM), LidA, LeB and SidD [Citation40,Citation41]. There is also another effector protein Ceg9 that interacts with the ER protein, reticulon 4 (Rtn4). After a brief period of internalization of the bacteria, this MCS between LCV and ER directs tubular ER towards the LCV where they ultimately coalesce thereby mimicking the identity of the host membrane () [Citation42]. The protein DrrA possesses a phosphatidylinositol 4-phosphate (PI4P)-binding domain located at its C-terminal region, which facilitates its attachment to PI4P-enriched Legionella-containing vacuoles (LCVs). Following the recruitment of DrrA onto the Legionella-containing vacuole (LCV), a subsequent activation is initiated through the triggering of Rab1 [Citation43,Citation44]. The binding of Rab1 facilitates the recruitment of endoplasmic reticulum-derived vesicles to the LCV [Citation45]. In yeast, the MCS tether oxysterol binding homology (Osh) protein Osh3 functions as a sensor for PI4P. When the levels of PI4P increase at the PM, Osh3 localises to ER-PM MCS and promotes the activity of the phosphatase enzyme Sac1 [Citation46]. Since overexpression of Sac1 leads to the dissociation of Rab1 from LCV [Citation37]. It is likely that a similar mechanism, potentially involving mammalian ORP homologs, also occurs in Legionella-infected mammalian cells. In addition, the Legionella protein SidD also releases Rab1 from the LCV. Legionella protein SidD performs an analogous function as that of Sac1 in eukaryotes by de AMPylation (dephosphorylation of Adenosine Monophosphate) activity which releases Rab1 from the LCV [Citation47]. Thus, during the process of remodeling, Legionella pneumophila exploits the host’s ER-PM MCS, along with key components such as PI4KIIIα, PI4P, and SidD. This “hijacking mechanism” facilitates the trafficking of vesicles from ER to the Golgi apparatus, ultimately directing them towards the LCV through the recruitment of Rab1. This phenomenon leads to the tethering of ER-derived vesicles with the LCV. Legionella do not form stable ER-LCV MCS; instead, they hijack components of ER-PM MCS to form ER-derived vesicles to promote their survival and replication within the host cell. The Legionella effector protein RaIF helps in recruiting ADP-ribosylation factor (Arf) directly from the Golgi [Citation48]. Arfs are essential for the establishment of secretory vesicles, helping in the engagement of coat proteins or protein complexes that activate lipid modifying enzymes and influence traffic in the secretory pathway. Therefore, RaIF also forms a transient microdomain that directly helps to transport the Arf from Golgi to the LCV [Citation29]. In a recent study conducted by Simone Vormittag and colleagues, it was empirically demonstrated that the components of the MCS are integral to the LCV and that the replication of the bacterium is contingent upon the presence of MCS components, such as VAP or OSBP11 and Sac1. Notably, the absence of these MCS components resulted in a significant reduction in replication efficiency. Hence, it can be inferred that the components of the MCS play a crucial role in the viability and reproduction of Legionella within the host [Citation49].
Chlamydia species are obligate intracellular Gram-negative bacterial pathogens that have evolved stratagems to invade, survive and replicate within the host genital, ocular, and pulmonary epithelial surfaces. Chlamydia exhibits a biphasic life cycle that occurs subsequent to its invasion of the host. The bacteria undergo a cycle in which they transition between two distinct forms: an infectious and metabolically inactive form known as the elementary body (EB), and a replicative and metabolically active form called the reticulate body (RB) [Citation50]. Chlamydia establishes its residence within a vacuole that is enclosed by a membrane, commonly referred to as inclusion bodies. Sphingomyelin is an essential component that plays a pivotal role in facilitating Chlamydial growth, promoting bacterial replication, and facilitating inclusion biogenesis. Thus, Chlamydia has evolved a novel mechanism to redirect SM biosynthesis to its replicative niche. It manipulates vesicular trafficking from the Golgi to the PM by hijacking tether protein of the ER- Golgi MCS i.e. ceramide-transfer protein (CERT) [Citation51]. An exclusive MCS forms between Chlamydia inclusions and host ER which establishes an on-site biosynthetic factory of SM at or near the inclusion (). Ceramide gets transported from the ER to inclusion, where it is converted to SM. CERT contains several functional domains including (i) a START domain that catalyzes the intermembrane transfer of ceramide, (ii) two phenylalanine residues [FF] in an Acidic Tract (FFAT) motif that interacts with the ER membrane protein VAP and (iii) a pleckstrin homology (PH) domain, which binds to the Golgi by recognizing PI4P [Citation52]. In ER-Golgi MCS, the FFAT motif of CERT interacts with the ER membrane protein VAP and thus anchors ER to Golgi by binding its PH domain to the Golgi apparatus. START domain transfers ceramide from the ER to sphingomyelin synthase 1 (SMS1) localized in the Golgi. Then Golgi transfers the ceramides to the PM [Citation53]. Like all other bacteria, Chlamydia encodes the type III secretion system (T3SS) which is a 3.5 megadalton syringe like complex protein machinery embedded at the bacterial membrane which helps in delivering bacterial effector proteins into the host for a successful invasion thereby promoting aggressive infection and circumventing anti-bacterial response [Citation54]. In ER-Inclusion MCS, the Chlamydia effector protein IncD gets translocated to the host via the T3SS where interacts with the PH domain of CERT and the ER membrane proteins VAP-A and VAP-B interact with the FFAT motif of CERT [Citation55] ().
The START domain of CERT mediates the transfer of ceramide to sphingomyelin synthase 1 (SMS1) and SMS2 to locally synthesize sphingomyelin [Citation56]. The loss of CERT activity caused a reduction in inclusion size as well as ~ 60% decrease in progeny production [Citation56]. The IncV effector initiates an MCS with ER through its FFAT domain directly with the VAPs through phosphorylation by the host kinase CK2 [Citation57], IncV on deleterious mutation did not abolish an ER-inclusion MCS since the inclusion already forms an MCS with ER through IncD and CERT [Citation58]. Henceforth, synthesis of sphingomyelin by hijacking host CERT protein by Chlamydia promotes its survival and bacterial replication within the host. Recent studies have identified that stromal interaction molecule 1 (STIM1) is a crucial component of ER-PM MCS (Fig 2.2), as well as an ER-localized calcium (Ca2+) sensor. When the ER Ca2+ store is depleted, Ca2+ triggers STIM1 oligomerization and relocates to ER-PM MCS where it binds and activates the calcium channel Orai1, thus promoting Ca2+ signalling in the inclusion [Citation59]. It has been shown that STIM1 mobilise/ER cisternae near phagosome for signalling and opening of phagosomal Ca2+ channels that lead to efficient phagocytosis; however, ablation of STIM1 decreases ER-Phagosome contacts thereby decreasing phagocytosis [Citation60]. Therefore, it is hypothesised that Chlamydia exhibits specific interactions with STIM1, which could potentially lead to the downregulation of STIM1 and facilitate the robust formation of its inclusion body, allowing it to evade the host’s defence mechanisms.
Another intracellular pathogen in addition to the above listed is Mycobacterium tuberculosis that serves as an aetiological agent for tuberculosis, an “age-old” disease that remains as a contemporary threat. It demonstrates a high degree of efficacy in evading the immune system by virtue of parasitizing the macrophages of the host. It tempers the typical progression of phagosome into an acidic hydrolytic compartment and avoids the occurrence of localized immune response of the host [Citation61]. While there is still much unknown about tuberculosis in regards to MCS biology, there is evidence suggesting that MCS components may play a role in the propagating its pathogenesis. In infected cells bacterial cell wall lipids lipoarabinomannan (LAM) and glycerophosphoinositolmannoside (PIM) interact with host protein Rab7 and thus initiate an LD-phagosome contact [Citation62]. After the establishment of the said contact, LDs (lipid droplet) translocate into the phagosome. The complete complex is engulfed by “foamy macrophages” that contain an abundance of fatty acids and LD, which serve as a source of nutrients for bacterial proliferation and propagation [Citation63]. Another novel contact is found between protein pair PE9 (Rv1088)-PE10 (Rv1089) with macrophages in which M.tb reside. This interaction results in subduing an immune response by stimulating the production of type 1 IFN which is known to be associated with suppression of innate immune response [Citation64].
Thus, from the aforementioned hijacking patterns it has been observed that the intracellular bacteria follow similar pattern of hijacking MCS. They decimate the typical MCS residing between the organelles by introducing novel MCS between the organelles and their inclusion bodies. In Legionella it is observed how the pathogen remodels the lipid composition of its LCV and refashions it into an ER-like alcove. Both Chlamydia and Coxiella target the lipid transfer proteins thereby showing similar lipid acquisition mechanisms. A striking similarity among all the
intracellular pathogens are their dependency on the secretion of bacterial effector proteins which, by virtue of molecular mimicry, allow the bacteria to avert cellular components to their inclusion bodies. The association of MCS with bacterial pathogens has heralded a new paradigm of bacterial pathogenesis that promises to open up new inquiries in disease modelling research. The bacteria induced MCS direct lipids, metabolites and proteins from the ER to the inclusion bodies (containing the bacteria) to facilitate their replication. (a) Coxiella Containing Vacuole (CCV) forms an MCS through its effectors (yet to be identified), ORP1L (The Oxysterol-binding Protein Homologue) and VAP. It is assumed that ORP1L helps in allocating cholesterol in the CCV to aid in the replication of Coxiella. (b) Legionella establishes MCS between the endoplasmic reticulum and the Legionella containing vacuole (LCV). Immediately after internalization, a PI4P-rich inclusion surrounds the bacteria, leading to MCS proteins Ceg9 and Rtn4 forming a contact that enjoins the tubular ER to the LCV where they bind to the LCV. This represents a perfect example of molecular mimicry of bacterial effectors with host membrane proteins. Rab1 and DrrA form an MCS that decreases the PI4P content by converting it to PI and thus, accelerating the ER-associated organellar mimicry of LCV so as to evade host immune cells (c) Chlamydia leads a biphasic life with two morphological forms: the infectious extracellular elementary body (EB) and the metabolically active intracellular reticulate body (RB). During septicity, EB binds to host cells and is then requisitioned within the inclusion where the EB gets converted to the RB, thereby ensuring activation of its replication forms. Chlamydia forms three MCS with the inclusion body and the ER: VAP docked on IncD (effector of Chlamydia) with CERT (Ceramide transfer protein), CERT helps to transport Ceramide via MCS from the ER to the inclusion, where it is transformed into Sphingomyelin which is necessary for the bacterial replication. STIM1 (Stromal interaction molecule 1) binds to a Chlamydia effector and forms a novel MCS; since STIM1 is a Ca2+ sensor it has been speculated that it activates calcium channel orai1 promoting Ca2+ signalling in the inclusion. There is a third type of MCS that is formed via IncV and VAP without including CERT, helping in a conclusive and tight interaction between the two juxtaposed organellar membranes.
(+) RNA viruses raiding MCS
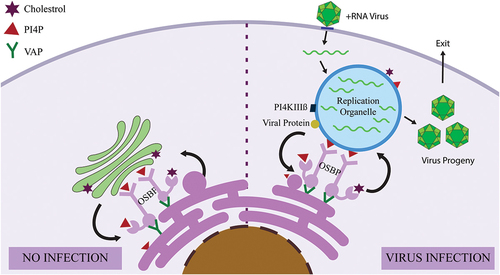
Positive sense (+) RNA viruses mostly enteroviruses are often linked to an array of pathological conditions like asthma, hepatitis, poliomyelitis, diabetes and meningitis. A fundamental step in enteroviral life cycle is replication of their positive sense RNA which occurs within specialized membranous replication organelles that have distinct lipid composition to facilitate robust virus replication, called replicating organelles (ROs) () [Citation65]. These infecting viruses have been observed to induce membrane contact sites that help in transporting lipids to viral membranes and orchestrate the lipid composition for the favourable formation and operation of these distinct compartments () [Citation66]. The RO spawned by the virus maintains a robust interaction with organelles and the cytosol to obtain the proteins, lipids, and essential metabolites required for viral replication. Since the virus prompted MCS comprises both viral and host factors, they are known as vMCSs. Many picornaviruses require an essential multifunctional protein Acyl coenzyme A binding domain 3 (ACBD3) for viral infection. This protein interacts with viral 3A proteins at membrane contact sites aiding in efficient lipid transfer to the ROs through vMCS. The three key players in viral replication are ACBD3, PI4KIIIA/B (gene symbol PI4K A/B)-host factors that synthesize PI4P essential for virus replication and OSBP that localizes at the MCS. Under typical physiological conditions, ACBD3 [Citation67] recruits PI4KB into the Golgi and Trans Golgi Network (TGN) networks so that it elevates the enzymatic activity of PI4KB to produce PI4P locally [Citation68]. In virus-compromised physiology, neutral lipid synthesis is stopped and the ACBD3 protein is hijacked, leading to the recruitment of PI4KB to the ROs to synthesize PI4P [Citation67]. It serves the simple function of (i) helping to gather components required for genome replication and RO formation, and (ii) docking site of viral RNA polymerase, for example, 3Dpol [Citation69,Citation70]. Under uninfected conditions, OSBP delivers to the integral ER membrane protein, VAP-A/B through the FFAT motif followed by binding to PI4P and Arf1 in trans-Golgi, thus bringing the two organelles to close proximity, forming an MCS (). OSBP helps to transport cholesterol directly through MCS without forming any vesicles to Golgi from the ER against the concentration gradient (). Transport requires ATP generated by the opposite flux of PI4P from Golgi to the ER, where it is hydrolysed by Sac1 phosphatase [Citation71,Citation72]. In infected conditions, ROs are formed that prompt OSBP recruitment to juxtapose the Golgi and ER membranes to form a novel MCS for cholesterol transport (PI4P antiport) followed by importing it to the RO by seizing the PIFKIIIB – PI4P-OSBP pathway to support viral replication [Citation73–75] (). Cholesterol is crucial for virus replication since it is the main structural component of RO [Citation76]. They help to dock and gather viral replication proteins by stimulating viral enzymatic reactions followed by the generation of high curvature membrane compartments so that the virus can evade the host’s innate immune defences [Citation77]. Once cholesterol aids in viral replication, the viral progeny escapes from the RO, thereby lysing the entire cell.
Figure 3. MCS between virus-induced replication organelles and cellular organelles.
On the contrary Dengue virus which is also a +ve ssRNA virus shows a replication organelle independent mechanism of viral replication and perpetuation. The hijacking machinery initiates with Triacylglycerol breakdown (lipolysis) of fatty acids stored in lipid droplets (LD) [Citation78]. The viral nonstructural protein NS4A crosstalks with LD dwelling Ancient Ubiquitous protein 1 and thus forms a LD-autophagosome contact to initiate lipophagy [Citation79]. This episodic lipolysis followed by lipophagy releases fatty acids in the cytoplasm which is used as energy source for aberrant viral replication. A recent proteomics study reported that upon human cytomegalovirus infection there is a global 2 to 5-fold increase in MCS proteins. In contrary influenza downregulates by 2 fold and Human coronavirus OC43 and Human Sarcoma Virus-1 cause moderate changes [Citation80]. Although they are not +ve ssRNA viruses but there are evidences that indeed there is a global alteration of abundance of MCS proteins on viral infection.
+RNA viruses hijack the pre-existing MCS between the endoplasmic reticulum (ER) and the Golgi apparatus and direct it to the replication organelle (RO). (a) The MCS components are VAP, OSBP, PI4P and cholesterol. (b) Virus replication requires cholesterol acquisition. The energy against the concentration gradient is provided by the counter-transport of PI4P from the inclusion to the ER where it is finally converted to PI
The parallels drawn between both the bacterial and viral raid and evasion of the host immune system is analogous to each other. Much akin to a medieval monarch who upon entering their newly conquered domain disbands the pre-existing norms of the land to establish their control, the pathogens (both bacteria and viruses) too, upon entering cell, impair the existing cellular communication channels to introduce their own network of communication extensively throughout the cell via the novel MCS routes. Both microbes establish their inclusion bodies and through their secretion systems present their microbial effectors which aid in further biomolecule acquisition into the inclusions. The strategy that both classes of pathogens adopt for avoiding the host immune system is molecular mimicry and activation of inflammasome signalling, which again are comparable to each other. Thus, it can be concluded by reiterating that both intracellular bacteria and virus show corresponding feats in terms of their association “hijacking” pattern, and evasion concerning MCS.
MCS aiding in evasion of host immune surveillance
We have come across definite mechanisms that intracellular pathogens have adopted to sidestep detection by the host immune system. Legionella forms a replicative niche that shares antigenic determinants between parasite and host that eventually evades recognition by the host’s immune system. This concept of imitation is widely known as molecular mimicry [Citation66]. Legionella effector Ra1F contains the Sec7 domain whose eukaryotic host counterpart is Arf GEF (confirmed by structural analysis) [Citation81]. Ra1F activates and helps to recruit Arf1 to the LCV, thus facilitating replication [Citation82]. Legionella also recruits host ER vesicles to adhere to its LCV, thus imitating ER so that it can evade immune recognition. Chlamydia hijacked MCS participate as stimulator of interferon genes (STING)-dependent pathogen sensing [Citation83] that induces the expression of type 1 interferons (IFNs). IFNs help to remove pathogen; however, Chlamydia infection activates an intracellular peptidoglycan binding molecule – nucleotide-binding oligomerization domain containing 1 (NOD1) due to which Chlamydial inclusion has access to the host cytosol [Citation84]. Intriguingly, an increase in the acquisition or utilization of host lipids in inclusion leads to the activation of the inflammasome [Citation85] that promotes infection and evades host’s cytosolic detection of Chlamydial components. +RNA viruses have evolved extensive measures to mitigate the mechanism of innate sensing, snoozing immune alarm. Viral proteins impede the production of interferons (IFNs) and cytokines from infected cells, thus supporting viral replication. During infection, dynamin-like protein 1 (Drp1) [Citation86] and mitofusin 1 (Mfn1) [Citation87] are drained by viral proteins leading to MCS disruption and mitochondrial membrane remodelling. This feat is another example of molecular mimicry in which the cytosolic pattern recognition receptors (PRRs) cannot sense foreign RNA. Recently, it has been documented that the loss of Drp1 coronavirus-infected cells resulted in the formation of elongated mitochondria with elevated viral replication and inhibition of immune activation [Citation87].
MCS in therapeutics
Since it has been observed how pathogens (namely bacteria and viruses) exploit pre-existing MCS components and direct them to their site of proliferation, namely, the replication organelle, the machinery can be inhibited or blocked in such a way that they are not accessible to the pathogen’s replicative machinery. We speculate that targeting the altered MCS sites in infected cells can aid in reducing the onslaught of rampant infection progression. In the case of cellular infection by a virus, the entire machinery of OSBP is redirected towards the RO from the ER. Consequently, by inhibiting the functioning of OSBP within the infected cell, we can effectively deprive the virus of crucial biomolecules necessary for its replication. This interference with infection propagation holds potential for impeding the spread of the virus. Literature has documented instances where down-regulation of CERT causes cancer cell sensitization to cytotoxic drugs [Citation88]. Targeting OSBP improves the anticancer activity of OSW-1 (Orsaponin), a naturally occurring compound that has been proven to be a potent anticancer drug through in vitro studies [Citation89]. Viral infections can be combated by targeting ORPs with small molecular antagonist since controlling organeller lipid composition by OSBPs/ORps is critical for many pathogenic viruses. As there are documented evidences of the MCS components being viable targets in anticancer therapy, we can therefore speculate that they are crucial for therapeutic intervention against infectious pathogens as well. Given below () is a table of inhibitors of the various MCS proteins. It must be considered that during the development of therapeutics care must be taken such that only the MCS components that are hijacked by the pathogens are inhibited and not the entire machinery which might lead to a complete collapse of the cell. essential for dug targeting such that the MCS formed between the host organelle and the inclusion bodies can be successfully targeted and obliterated such that the pathogens cannot evade the immune system.
Table 2. The table summarizes all inhibitors that act on specialized MCS components that might be essential for dug targeting such that the MCS formed between the host organelle and the inclusion bodies can be successfully targeted and obliterated such that the pathogens cannot evade the immune system.
Conclusion and future directions
In summary, this review delves into an anthology of mechanism adopted by pathogens that hijack the typical MCS and form novel connection that aids them in their replication. We also discuss the probable compounds that might be useful in the development of therapeutics that target the hijacked MCS. We have discussed about MCS components and their mode of exploitation by both bacteria and viruses (especially +RNA) and how they evade the immune system, thereby being detrimental to the host cell. We have further listed few inhibitors that might be useful in therapeutics by targeting the MCS proteins. If the association between the organelles and the pathogen’s inclusion body can be hampered then the pathogen will not get access to essential biomolecules for their replication thereby perishing. We have delved into the plausible causes of dodging immune surveillance by virtue of MCS and hope the dearth in this research narrows with time. While, we have summarised the recent trend in this expanding field a plethora of novel and exhilarating questions remain unanswered e.g. – Are there more contact site junctions that have not been described yet? Are there more molecular tethers, pillars, and spacers that play a role in pathogenesis? There have been various instances were effector proteins of pathogens mediate host pathogen interaction, however they have not been staunchly classified as MCS proteins. Additionally, we believe that many proteins other than what we have listed above might mimic and function like that of MCS proteins. For example, there is a PE_PPE family of proteins mostly found to be cell wall-associated or secreted in culture supernatants of M.tb. These proteins function to evade host immune to promote pathogenicity and also suppress the host immune response [Citation98]. Even though the authors have not explicitly stated that the proteins in question are interact with MCS proteins, it is reasonable to hypothesise that they, along with numerous other proteins that facilitate host-pathogen interactions, could potentially mimic the components of the MCS to interact with it. Is there any other function of MCS other than directing lipids and Ca2+ in inclusions? The existing body of literature primarily focuses on the transportation of Ca2+ ions and lipids. However, it is our conjecture that in addition to these molecules, energy molecules such as ATP and NAD are also directly transported, rather than being translocated via vesicles. What is the mechanism of PI4K recruitment by 3A? [Citation99]Are there other ORPs that help the +RNA virus replication? How does lipid exchange occur at MCS, which do not have lipid transport proteins? How is the regulation of MCS numbers affected when the body is hijacked by pathogens? [Citation100] Does MCS play a role in retroviral replication? How does the loss of MCS from their original sites during pathogen invasion affect the communication and functioning of the organelles from which they have been hijacked? These questions are of an open-ended nature, necessitating further investigation in order to provide comprehensive answers. It is highly probable that this field will experience significant growth and will garner attention from various academic disciplines. It is expected that the aforementioned inquiries will be addressed in the future, leading to the exploration of new avenues for research and actualization.
Author contributions
PP and BT wrote the manuscript. Both authors corrected and approved the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
No data sets were analysed in the manuscript and thus availability is not applicable.
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Prinz WA, Toulmay A, Balla T. The functional universe of membrane contact sites. Nat Rev Mol Cell Biol. 2020;21(1):7–13. doi: 10.1038/s41580-019-0180-9
- Bernhard W, Rouiller C. Microbodies and the problem of mitochondrial regeneration in liver cells. J Biophys Biochem Cytol. 1956;2(4 Suppl):355–360. doi: 10.1083/jcb.2.4.355
- Jones TC, Yeh S, Hirsch JG. The interaction between Toxoplasma gondii and mammalian cells. I. Mechanism of entry and intracellular fate of the parasite. J Exp Med. 1972;136(5):1157–1172. doi: 10.1084/jem.136.5.1157
- Ng AYE, Ng AQE, Zhang D. ER-PM contacts restrict exocytic sites for polarized morphogenesis. Curr Biol. 2018;28(1):146–153 e5. doi: 10.1016/j.cub.2017.11.055
- Quon E, Sere YY, Chauhan N, et al. Endoplasmic reticulum-plasma membrane contact sites integrate sterol and phospholipid regulation. PLoS Biol. 2018;16(5):e2003864. doi: 10.1371/journal.pbio.2003864
- Kadosh D, Lopez-Ribot JL. Candida albicans: adapting to succeed. Cell Host Microbe. 2013;14(5):483–485. doi: 10.1016/j.chom.2013.10.016
- Vance JE. Phospholipid synthesis in a membrane fraction associated with mitochondria. J Biol Chem. 1990;265(13):7248–7256. doi: 10.1016/S0021-9258(19)39106-9
- Rizzuto R, Pinton P, Carrington W, et al. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 1998Jun 12;280(5370):1763–1766. doi: 10.1126/science.280.5370.1763. PubMed PMID: 9624056.
- Roberts P, Moshitch-Moshkovitz S, Kvam E, et al. Piecemeal microautophagy of nucleus in Saccharomyces cerevisiae. Mol Biol Cell. 2003Jan;14(1):129–141. doi: 10.1091/mbc.e02-08-0483. PubMed PMID: 12529432; PubMed Central PMCID: PMCPMC140233.
- Murphy SE, Levine TP. VAP, a versatile access point for the endoplasmic reticulum: review and analysis of FFAT-like motifs in the VAPome. Biochim Biophys Acta. 2016;1861(8 Pt B):952–961. doi: 10.1016/j.bbalip.2016.02.009
- Chung J, Torta F, Masai K, et al. INTRACELLULAR TRANSPORT. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science. 2015Jul 24;349(6246):428–432. doi: 10.1126/science.aab1370. PubMed PMID: 26206935; PubMed Central PMCID: PMCPMC4638224.
- Henne WM, Zhu L, Balogi Z, et al. Mdm1/Snx13 is a novel ER-endolysosomal interorganelle tethering protein. J Cell Bio. 2015 Aug 17;210(4):541–551. doi: 10.1083/jcb.201503088. PubMed PMID: 26283797; PubMed Central PMCID: PMCPMC4539980.
- Gatta AT, Wong LH, Sere YY, et al. A new family of StART domain proteins at membrane contact sites has a role in ER-PM sterol transport. Elife. 2015 May 22;4. doi: 10.7554/eLife.07253. PubMed PMID: 26001273; PubMed Central PMCID: PMCPMC4463742.
- Johnson B, Leek AN, Solé L, et al. Kv2 potassium channels form endoplasmic reticulum/plasma membrane junctions via interaction with VAPA and VAPB. Proc Natl Acad Sci U S A. 2018;115(31):E7331–E7340. doi: 10.1073/pnas.1805757115
- Scorrano L, De Matteis MA, Emr S, et al. Coming together to define membrane contact sites. Nat Commun. 2019;10(1):1287. doi: 10.1038/s41467-019-09253-3
- Raturi A, Simmen T. Where the endoplasmic reticulum and the mitochondrion tie the knot: the mitochondria-associated membrane (MAM). Biochim Biophys Acta. Biochim Biophys Acta, Mol Cell Res. 2013;1833(1):213–224. doi: 10.1016/j.bbamcr.2012.04.013
- Giordano F, Saheki Y, Idevall-Hagren O, et al. PI(4,5)P(2)-dependent and Ca(2+)-regulated ER-PM interactions mediated by the extended synaptotagmins. Cell. 2013Jun 20;153(7):1494–1509. doi: 10.1016/j.cell.2013.05.026. PubMed PMID: 23791178; PubMed Central PMCID: PMCPMC3716012.
- Raiborg C, Wenzel E, Pedersen N, et al. Phosphoinositides in membrane contact sites. Biochem Soc Trans. 2016;44(2):425–430. doi: 10.1042/BST20150190
- Kumar Y, Valdivia RH. Leading a sheltered life: intracellular pathogens and maintenance of vacuolar compartments. Cell Host Microbe. 2009;5(6):593–601. doi: 10.1016/j.chom.2009.05.014
- Jiang C, Huang X, Yao J, et al. The role of membrane contact sites at the bacteria-host interface. Crit Rev Microbiol. 2022;48(3):270–282. doi: 10.1080/1040841X.2021.1961678
- Peretti D, Dahan N, Shimoni E, et al. Coordinated lipid transfer between the endoplasmic reticulum and the Golgi complex requires the VAP proteins and is essential for Golgi-mediated transport. Mol Biol Cell. 2008;19(9):3871–3884. doi: 10.1091/mbc.e08-05-0498
- Liao PC, Yang EJ, Borgman T, et al. Touch and go: membrane contact sites between lipid droplets and other organelles. Front Cell Dev Biol. 2022;10:852021. doi: 10.3389/fcell.2022.852021
- Basso V, Marchesan E, Ziviani E. A trio has turned into a quartet: DJ-1 interacts with the IP3R-Grp75-VDAC complex to control ER-mitochondria interaction. Cell Calcium. 2020;87:102186. doi: 10.1016/j.ceca.2020.102186
- Asrat S, de Jesús DA, Hempstead AD, et al. Bacterial pathogen manipulation of host membrane trafficking. Annu Rev Cell Dev Biol. 2014;30(1):79–109. doi: 10.1146/annurev-cellbio-100913-013439
- Ge J, Shao F. Manipulation of host vesicular trafficking and innate immune defence by Legionella Dot/Icm effectors. Cell Microbiol. 2011;13(12):1870–1880. doi: 10.1111/j.1462-5822.2011.01710.x
- Heinzen RA, Scidmore MA, Rockey DD, et al. Differential interaction with endocytic and exocytic pathways distinguish parasitophorous vacuoles of Coxiella burnetii and Chlamydia trachomatis. Infect Immun. 1996Mar;64(3):796–809. doi: 10.1128/iai.64.3.796-809.1996. PubMed PMID: 8641784; PubMed Central PMCID: PMCPMC173840.
- Carey KL, Newton HJ, Luhrmann A, et al. The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PLOS Pathog. 2011May;7(5):e1002056. doi: 10.1371/journal.ppat.1002056. PubMed PMID: 21637816; PubMed Central PMCID: PMCPMC3102713
- Justis AV, Hansen B, Beare PA, et al. Interactions between the Coxiella burnetii parasitophorous vacuole and the endoplasmic reticulum involve the host protein ORP1L. Cell Microbiol. 2017Jan;19(1):e12637. doi: 10.1111/cmi.12637. PubMed PMID: 27345457; PubMed Central PMCID: PMCPMC5177503.
- Jackson CL, Casanova JE. Turning on ARF: the Sec7 family of guanine-nucleotide-exchange factors. Trends Cell Biol. 2000;10(2):60–67. doi: 10.1016/S0962-8924(99)01699-2
- Loewen CJ. A conserved ER targeting motif in three families of lipid binding proteins and in Opi1p binds VAP. EMBO J. 2003;22(9):2025–2035. doi: 10.1093/emboj/cdg201
- Im YJ, Raychaudhuri S, Prinz WA, et al. Structural mechanism for sterol sensing and transport by OSBP-related proteins. Nature. 2005;437(7055):154–158. doi: 10.1038/nature03923
- Loewen CJ, Levine TP. A highly conserved binding site in vesicle-associated membrane protein-associated protein (VAP) for the FFAT motif of lipid-binding proteins. J Biol Chem. 2005;280(14):14097–14104. doi: 10.1074/jbc.M500147200
- Rocha N, Kuijl C, van der Kant R, et al. Cholesterol sensor ORP1L contacts the ER protein VAP to control Rab7-RILP-p150 glued and late endosome positioning. J Cell Bio. 2009Jun 29;185(7):1209–1225. doi: 10.1083/jcb.200811005. PubMed PMID: 19564404; PubMed Central PMCID: PMCPMC2712958.
- Stanhope R, Derre I. Making contact: VAP targeting by intracellular pathogens. contact (Thousand Oaks). 2018;1. doi: 10.1177/2515256418775512
- Eisenreich W, Heuner K. The life stage-specific pathometabolism of Legionella pneumophila. FEBS Lett. 2016;590(21):3868–3886. doi: 10.1002/1873-3468.12326
- Swanson MS, Isberg RR. Association of Legionella pneumophila with the macrophage endoplasmic reticulum. Infect Immun. 1995;63(9):3609–3620. doi: 10.1128/iai.63.9.3609-3620.1995
- Hubber A, Arasaki K, Nakatsu F, et al. The machinery at endoplasmic reticulum-plasma membrane contact sites contributes to spatial regulation of multiple Legionella effector proteins. PLOS Pathog. 2014Jul;10(7):e1004222. doi: 10.1371/journal.ppat.1004222. PubMed PMID: 24992562; PubMed Central PMCID: PMCPMC4081824.
- Hubber A, Roy CR. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu Rev Cell Dev Biol. 2010;26(1):261–283. doi: 10.1146/annurev-cellbio-100109-104034
- Robinson CG, Roy CR. Attachment and fusion of endoplasmic reticulum with vacuoles containing Legionella pneumophila. Cell Microbiol. 2006;8(5):793–805. doi: 10.1111/j.1462-5822.2005.00666.x
- Murata T, Delprato A, Ingmundson A, et al. The Legionella pneumophila effector protein DrrA is a Rab1 guanine nucleotide-exchange factor. Nat Cell Biol. 2006;8(9):971–977. doi: 10.1038/ncb1463
- Machner MP, Isberg RR. A bifunctional bacterial protein links GDI displacement to Rab1 activation. Science. 2007;318(5852):974–977. doi: 10.1126/science.1149121
- Haenssler E, Ramabhadran V, Murphy CS, et al. Endoplasmic reticulum tubule protein reticulon 4 Associates with the Legionella pneumophila vacuole and with translocated substrate Ceg9. Infect Immun. 2015Sep;83(9):3479–3489. doi: 10.1128/IAI.00507-15. PubMed PMID: 26099580; PubMed Central PMCID: PMCPMC4534651.
- Zhu Y, Hu L, Zhou Y, et al. Structural mechanism of host Rab1 activation by the bifunctional Legionella type IV effector SidM/DrrA. Proc Natl Acad Sci U S A. 2010;107(10):4699–4704. doi: 10.1073/pnas.0914231107
- Brombacher E, Urwyler S, Ragaz C, et al. Rab1 guanine nucleotide exchange factor SidM is a major phosphatidylinositol 4-phosphate-binding effector protein of Legionella pneumophila. J Biol Chem. 2009;284(8):4846–4856. doi: 10.1074/jbc.M807505200
- Machner MP, Isberg RR. Targeting of host rab GTPase function by the intravacuolar pathogen Legionella pneumophila. Dev Cell. 2006;11(1):47–56. doi: 10.1016/j.devcel.2006.05.013
- Stefan CJ, Manford AG, Baird D, et al. Osh proteins regulate phosphoinositide metabolism at ER-plasma membrane contact sites. Cell. 2011Feb 4;144(3):389–401. doi: 10.1016/j.cell.2010.12.034. PubMed PMID: 21295699.
- Tan Y, Luo ZQ. Legionella pneumophila SidD is a deAmpylase that modifies Rab1. Nature. 2011;475(7357):506–509. doi: 10.1038/nature10307
- Amor JC, Swails J, Zhu X, et al. The structure of RalF, an ADP-ribosylation factor guanine nucleotide exchange factor from Legionella pneumophila, reveals the presence of a cap over the active site. J Biol Chem. 2005;280(2):1392–1400. doi: 10.1074/jbc.M410820200
- Vormittag S, Husler D, Haneburger I, et al. Legionella- and host-driven lipid flux at LCV-ER membrane contact sites promotes vacuole remodeling. EMBO Rep. 2023Mar 6;24(3):e56007. doi: 10.15252/embr.202256007. PubMed PMID: 36588479; PubMed Central PMCID: PMCPMC9986823.
- Grieshaber S, Grieshaber N, Yang,H, et al. Impact of active metabolism on Chlamydia trachomatis elementary body transcript profile and infectivity. J Bacteriol. 2018Jul 15;200(14). doi: 10.1128/JB.00065-18. PubMed PMID: 29735758; PubMed Central PMCID: PMCPMC6018357.
- Hackstadt T, Scidmore MA, Rockey DD. Lipid metabolism in Chlamydia trachomatis-infected cells: directed trafficking of Golgi-derived sphingolipids to the chlamydial inclusion. Proc Natl Acad Sci U S A. 1995;92(11):4877–4881. doi: 10.1073/pnas.92.11.4877
- Hanada K, Kumagai K, Tomishige N, et al. CERT and intracellular trafficking of ceramide. Biochim Biophys Acta. 2007;1771(6):644–653. doi: 10.1016/j.bbalip.2007.01.009
- Hanada K, Kumagai K, Yasuda S, et al. Molecular machinery for non-vesicular trafficking of ceramide. Nature. 2003;426(6968):803–809. doi: 10.1038/nature02188
- Radics J, Konigsmaier L, Marlovits TC. Structure of a pathogenic type 3 secretion system in action. Nat Struct Mol Biol. 2014;21(1):82–87. doi: 10.1038/nsmb.2722
- Derre I, Swiss R, Agaisse H. The lipid transfer protein CERT interacts with the Chlamydia inclusion protein IncD and participates to ER-Chlamydia inclusion membrane contact sites. PLOS Pathog. 2011;7(6):e1002092. doi: 10.1371/journal.ppat.1002092
- Elwell CA, Jiang S, Kim JH, et al. Chlamydia trachomatis co-opts GBF1 and CERT to acquire host sphingomyelin for distinct roles during intracellular development. PLOS Pathog. 2011Sep;7(9):e1002198. doi: 10.1371/journal.ppat.1002198. PubMed PMID: 21909260; PubMed Central PMCID: PMCPMC3164637.
- Ende RJ, Murray RL, D’Spain SK, et al. Phosphoregulation accommodates type III secretion and assembly of a tether of ER-Chlamydia inclusion membrane contact sites. Elife. 2022;11:11. doi: 10.7554/eLife.74535
- Stanhope R, Flora E, Bayne C, et al. IncV, a FFAT motif-containing Chlamydia protein, tethers the endoplasmic reticulum to the pathogen-containing vacuole. Proc Natl Acad Sci USA. 2017 Nov 7;114(45):12039–12044. doi: 10.1073/pnas.1709060114. PubMed PMID: 29078338; PubMed Central PMCID: PMCPMC5692559.
- Agaisse H, Derre I, Trebak M. STIM1 is a novel component of ER-Chlamydia trachomatis inclusion membrane contact sites. PLoS One. 2015;10(4):e0125671. doi: 10.1371/journal.pone.0125671
- Nunes P, Cornut D, Bochet V, et al. STIM1 juxtaposes ER to phagosomes, generating Ca(2)(+) hotspots that boost phagocytosis. Curr Biol. 2012Nov 6;22(21):1990–1997. doi: 10.1016/j.cub.2012.08.049. PubMed PMID: 23041196.
- Russell DG. Mycobacterium tuberculosis: here today, and here tomorrow. Nat Rev Mol Cell Biol. 2001;2(8):569–577. doi: 10.1038/35085034
- Roque NR, Lage SL, Navarro R, et al. Rab7 controls lipid droplet-phagosome association during mycobacterial infection. Biochim Biophys Acta, Mol Cell Biol Lipids. 2020;1865(8):158703. doi: 10.1016/j.bbalip.2020.158703
- Peyron P, Vaubourgeix J, Poquet Y, et al. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLOS Pathog. 2008Nov;4(11):e1000204. doi: 10.1371/journal.ppat.1000204. PubMed PMID: 19002241; PubMed Central PMCID: PMCPMC2575403.
- Tiwari B, Ramakrishnan UM, Raghunand TR. The Mycobacterium tuberculosis protein pair PE9 (Rv1088)-PE10 (Rv1089) forms heterodimers and induces macrophage apoptosis through Toll-like receptor 4. Cell Microbiol. 2015;17(11):1653–1669. doi: 10.1111/cmi.12462
- Creasey EA, Isberg RR. Maintenance of vacuole integrity by bacterial pathogens. Curr Opin Microbiol. 2014;17:46–52. doi: 10.1016/j.mib.2013.11.005
- Nagy PD, Strating JR, van Kuppeveld FJ. Building viral replication organelles: close encounters of the membrane types. PLOS Pathog. 2016;12(10):e1005912. doi: 10.1371/journal.ppat.1005912
- Nchoutmboube JA, Viktorova EG, Scott AJ, et al. Increased long chain acyl-coa synthetase activity and fatty acid import is linked to membrane synthesis for development of picornavirus replication organelles. PLOS Pathog. 2013;9(6):e1003401. doi: 10.1371/journal.ppat.1003401. PubMed PMID: 23762027; PubMed Central PMCID: PMCPMC3675155.
- Schneider F. [Enzyme biochemistry]. Acta Histochem Suppl. 1975;14:19–31.
- Hsu NY, Ilnytska O, Belov G, et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell. 2010May 28;141(5):799–811. doi: 10.1016/j.cell.2010.03.050. PubMed PMID: 20510927; PubMed Central PMCID: PMCPMC2982146.
- De Matteis MA, Wilson C, D’Angelo G. Phosphatidylinositol-4-phosphate: the Golgi and beyond. BioEssays. 2013;35(7):612–622. doi: 10.1002/bies.201200180
- Kentala H, Weber-Boyvat M, Olkkonen VM. OSBP-Related protein family: mediators of lipid transport and signaling at membrane contact sites. Int Rev Cell Mol Biol. 2016;321:299–340.
- Mesmin B, Bigay J, Moser von Filseck J, et al. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell. 2013 Nov 7; 155(4):830–843. doi: 10.1016/j.cell.2013.09.056. PubMed PMID: 24209621.
- Strating JR, van der Linden L, Albulescu L, et al. Itraconazole inhibits enterovirus replication by targeting the oxysterol-binding protein. Cell Rep. 2015 Feb 3;10(4):600–615. doi: 10.1016/j.celrep.2014.12.054. PubMed PMID: 25640182; PubMed Central PMCID: PMCPMC4383725.
- Arita M, Kojima H, Nagano T, et al. Phosphatidylinositol 4-kinase III beta is a target of enviroxime-like compounds for antipoliovirus activity. J Virol. 2011;85(5):2364–2372. doi: 10.1128/JVI.02249-10
- Roulin PS, Lotzerich M, Torta F, et al. Rhinovirus uses a phosphatidylinositol 4-phosphate/cholesterol counter-current for the formation of replication compartments at the ER-Golgi interface. Cell Host Microbe. 2014 Nov 12;16(5):677–690. doi: 10.1016/j.chom.2014.10.003. PubMed PMID: 25525797.
- Stoeck IK, Lee JY, Tabata K, et al. Hepatitis C virus replication depends on endosomal cholesterol homeostasis. J Virol. 2018Jan 1;92(1). doi: 10.1128/JVI.01196-17. PubMed PMID: 29046459; PubMed Central PMCID: PMCPMC5730777.
- Altan-Bonnet N. Lipid tales of viral replication and transmission. Trends Cell Biol. 2017;27(3):201–213. doi: 10.1016/j.tcb.2016.09.011
- Heaton NS, Randall G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host Microbe. 2010;8(5):422–432. doi: 10.1016/j.chom.2010.10.006
- Zhang J, Lan Y, Li MY, et al. Flaviviruses exploit the lipid droplet protein AUP1 to trigger lipophagy and drive virus production. Cell Host Microbe. 2018Jun 13;23(6):819–831 e5. doi: 10.1016/j.chom.2018.05.005. PubMed PMID: 29902443.
- Cook KC, Tsopurashvili E, Needham JM, et al. Restructured membrane contacts rewire organelles for human cytomegalovirus infection. Nat Commun. 2022;13(1):4720. doi: 10.1038/s41467-022-32488-6
- Pipaliya SV, Schlacht A, Klinger CM, et al. Ancient complement and lineage-specific evolution of the Sec7 ARF GEF proteins in eukaryotes. Mol Biol Cell. 2019;30(15):1846–1863. doi: 10.1091/mbc.E19-01-0073
- Nagai H, Kagan JC, Zhu X, et al. A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science. 2002;295(5555):679–682. doi: 10.1126/science.1067025
- Barker JR, Koestler BJ, Carpenter VK, et al. STING-dependent recognition of cyclic di-AMP mediates type I interferon responses during Chlamydia trachomatis infection. MBio. 2013Apr 30;4(3):e00018–13. doi: 10.1128/mBio.00018-13. PubMed PMID: 23631912; PubMed Central PMCID: PMCPMC3663186.
- Pham OH, Lee B, Labuda J et al. NOD1/NOD2 and RIP2 regulate endoplasmic reticulum Stress-induced Inflammation during Chlamydia infection. MBio. 2020;11(3). doi: 10.1128/mBio.00979-20
- Itoh R, Murakami I, Chou B, et al. Chlamydia pneumoniae harness host NLRP3 inflammasome-mediated caspase-1 activation for optimal intracellular growth in murine macrophages. Biochem Biophys Res Commun. 2014;452(3):689–694. doi: 10.1016/j.bbrc.2014.08.128
- Chen S, Liu S, Wang J et al. TBK1-mediated DRP1 targeting confers nucleic acid sensing to Reprogram mitochondrial dynamics and physiology. Mol Cell. 2020;80(5):810–827 e7. doi: 10.1016/j.molcel.2020.10.018
- Castanier C, Garcin D, Vazquez A et al. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 2010;11(2):133–138. doi: 10.1038/embor.2009.258
- Swanton C, Marani M, Pardo O et al. Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs. Cancer Cell. 2007;11(6):498–512. doi: 10.1016/j.ccr.2007.04.011
- Zhou Y, Garcia-Prieto C, Carney DA, et al. OSW-1: a natural compound with potent anticancer activity and a novel mechanism of action. J Natl Cancer Inst. 2005;97(23):1781–1785. doi: 10.1093/jnci/dji404
- Albulescu L, Bigay J, Biswas B, et al. Uncovering oxysterol-binding protein (OSBP) as a target of the anti-enteroviral compound TTP-8307. Antiviral Res. 2017;140:37–44. doi: 10.1016/j.antiviral.2017.01.008
- Albulescu L, Strating JRPM, Thibaut HJ, et al. Broad-range inhibition of enterovirus replication by OSW-1, a natural compound targeting OSBP. Antiviral Res. 2015;117:110–114. doi: 10.1016/j.antiviral.2015.02.013
- Arita M, Kojima H, Nagano T, et al. Oxysterol-binding protein family I is the target of minor enviroxime-like compounds. J Virol. 2013;87(8):4252–4260. doi: 10.1128/JVI.03546-12
- Kuang XL, Liu Y, Chang Y, et al. Inhibition of store-operated calcium entry by sub-lethal levels of proteasome inhibition is associated with STIM1/STIM2 degradation. Cell Calcium. 2016;59(4):172–180. doi: 10.1016/j.ceca.2016.01.007
- Petit A, Knabe L, Khelloufi K, et al. Bronchial epithelial calcium metabolism impairment in smokers and chronic obstructive pulmonary disease. Decreased ORAI3 signaling. Am J Respir Cell Mol Biol. 2019;61(4):501–511. doi: 10.1165/rcmb.2018-0228OC
- Wen L, Voronina S, Javed MA Inhibitors of ORAI1 prevent cytosolic calcium-associated Injury of Human Pancreatic Acinar cells and acute pancreatitis in 3 mouse models. Gastroenterology. 2015;149(2):481–92 e7. doi: 10.1053/j.gastro.2015.04.015
- Bandet CL, Hajduch E. CERT-Dependent ceramide transport, a crucial process in cells. Diabetes Clin Res. 2021;3(2):40–45.
- Hanada K, Sakai S, Kumagai K. et al. Natural ligand-mimetic and nonmimetic inhibitors of the ceramide transport protein CERT. Int J Mol Sci. 2022;23(4). doi: 10.3390/ijms23042098
- Tiwari BM, Kannan N, Vemu L, et al. The Mycobacterium tuberculosis PE proteins Rv0285 and Rv1386 modulate innate immunity and mediate bacillary survival in macrophages. PLoS One. 2012;7(12):e51686. doi: 10.1371/journal.pone.0051686
- Altan-Bonnet N, Balla T. Phosphatidylinositol 4-kinases: hostages harnessed to build panviral replication platforms. Trends Biochem Sci. 2012;37(7):293–302. doi: 10.1016/j.tibs.2012.03.004
- Nath VR, Mishra S, Basak B et al. Extended synaptotagmin regulates membrane contact site structure and lipid transfer function in vivo. EMBO Rep. 2020;21(9):e50264. doi: 10.15252/embr.202050264