
ABSTRACT
Norovirus (NV) infection causes acute gastroenteritis in children and adults. Upon infection with NV, specific CD8+ T cells, which play an important role in anti-infective immunity, are activated in the host. Owing to the NV’s wide genotypic variability, it is challenging to develop vaccines with cross-protective abilities against infection. To aid effective vaccine development, we examined specific CD8+ T-cell responses towards viral-structural protein (VP) epitopes, which enable binding to host susceptibility receptors. We isolated peripheral blood mononuclear cells from 196 participants to screen and identify predominant core peptides towards NV main and small envelope proteins using ex vivo and in vitro intracellular cytokine staining assays. Human leukocyte antigen (HLA) restriction characteristics were detected using next-generation sequencing. Three conservative immunodominant VP-derived CD8+ T-cell epitopes, VP294–102 (TDAARGAIN), VP2153–161 (RGPSNKSSN), and VP1141–148 (FPHIIVDV), were identified and restrictively presented by HLA-Cw * 0102, HLA-Cw * 0702, and HLA-A *1101 alleles, separately. Our findings provide useful insights into the development of future vaccines and treatments for NV infection.
Introduction
Norovirus (NV) infection, which is generally more prevalent in winter and spring, is the leading cause of sporadic cases and global outbreaks of acute gastroenteritis [Citation1,Citation2]. It is a highly contagious and transmissible disease, accounting for approximately 35,000 deaths annually worldwide [Citation3,Citation4]. According to the World Health Organization (WHO), NV vaccine development should be considered a priority [Citation5]. Vaccine development has encountered considerable challenges because of the genotypic diversity and rapid evolution of NV. NV is genetically diverse and can be classified into at least 10 genogroups (GI – GX). Of these, genomes GI, GII, GIV, GVII, and GIX are present in humans. These 10 genogroups can be further divided into 49 capsid genotypes. More than 30 NV genotypes that can infect humans have been identified, of which GII.4 is the most common worldwide and is responsible for 70% of NV outbreaks. As GII.4 NVs are antigenically diverse, frequent changes in the major antigenic epitopes have led to the emergence of new GII.4 variants, which can potentially escape from the immunogenic responses developed against the previous variants [Citation6]. The GII.4 was the most prevalent genotype in human, and also has caused six pandemics since 1995. Subsequently, the novel lineage virus containing the GII.P16 polymerase and pandemic GII.4 Sydney 2012 capsid has been recently detected around the world since Oct. 2014 or earlier. The novel lineage virus containing polymerase substitutions, which was suggested that may have induced in more transmissible virus [Citation7].
Among the NV vaccine candidates undergoing clinical trials, TAK-214 developed by Takeda Pharmaceuticals International AG [Citation8] is the most studied one. It contains two types of NV-VLPs from GI.1 and three GII.4 variants and confers cross-genotype protection. Another NV vaccine candidate in clinical studies is a recombinant VP1-based bivalent vaccine (VXA-NVV-104) [Citation9] developed by Vaxart Pharmaceutical Inc. based on its proprietary oral tablet technology with recombinant adenovirus-based vectors. It is designed to be administered orally to elicit a mucosal immune response locally within the intestine to prevent NV infection. Although currently several NV vaccines are being tested in preclinical and clinical trials, there are no licenced NV vaccines available. The development of an effective NV vaccine has proven to be difficult owing to the genotype diversity and rapid genetic variation of NV. Recent findings highlight the need to look beyond antibodies for strategies to obtain better and more persistent protection owing to the poor neutralization of antibodies against variant virus strains [Citation10]. Importantly, recent studies have shown that CD8+ T-cell responses are not substantially affected by mutant variants [Citation11]. To establish an effective and safe NV vaccine with appropriate antigen composition, it is vital to identify specific epitopes that are highly conserved among different NV genotypes [Citation12], which may serve as an effective solution to induce sufficiently broad protection against NV. Thus, focusing on T-cell epitopes when designing next-generation vaccines has the potential to provide prolonged protection in the face of emerging variants.
As an endogenous pathogen, NV mainly triggers a specific CD8+ T-cell response to scavenge the virus [Citation13–15]. The capsid protein (VP) of NV contains an immunogenic core component that binds to host receptors and may mediate the infection process, and its amino acid-related immunogenicity and receptor binding sites influence the antiviral effect of host-specific immunity. Therefore, studies targeting VP-specific responses are important for designing vaccines. Recent studies investigating NV-specific T-cell responses have focused on the number and intensity of responding cells; however, antigen-specific (peptide) differences, response profile characteristics, and the emergence of gastrointestinal diseases following NV infection are underreported [Citation16–18]. In this study, we comprehensively analysed immunodominant CD8+ T-cell response profiles associated with the NV VP with the aim to identify candidate antigenic molecules from previously infected individuals. It will provide a theoretical foundation for NV vaccine research in the future.
Materials and methods
Participants and blood samples
This study was approved by the Human Ethics Review Committee of the Eighth Affiliated Hospital of Sun Yat-sen University (approval number: 2020-058-01), and the experiments were conducted in accordance with the World Medical Association Declaration of Helsinki. Informed consent was waived owing to the retrospective nature of the study. Sample collection was performed randomly, and subsequent data analysis revealed no bias in age or sex between the groups. Samples were collected according to the Sex and Gender Equity in Research (SAGER) guidelines. Participants aged between 12 and 89 years were recruited from the Physical Examination Center and the Department of Gastroenterology at the Eighth Affiliated Hospital of Sun Yat-sen University.
Among 196 participants, 40 were negative for both the NV-Ig G antibody and nucleic acid and 156 were positive for the NV-Ig G antibody but negative for nucleic acid. A serum NV VP-IgG antibody ELISA (DEIA2068S, CD Creative Diagnostics, Shirley, NY, USA) and viral nucleic acid real-time qPCR (YZB/LDQS003–2013, Hubei Langde Medical Technology Co., Ltd., Hubei, China) were used to identify whether individuals were infected. A flowchart of sample collection and general features of the study population are provided in Fig. S1 and Table S4. Blood samples were collected from all participants. Peripheral blood mononuclear cells (PBMCs) were isolated using a Ficoll-Hypaque (LTS1077; TBD Science, Tianjin, China) density gradient centrifuge as per the manufacturer’s instructions and stored in liquid nitrogen until further analysis. After separation, the lowest layers of nucleated and red blood cells were collected and stored at −20°C for HLA typing. Next-generation sequencing (NGS) was conducted at Bofurui Medical Laboratory Co., Ltd. (Guangzhou, China).
Synthetic peptides and antibodies
A series of 131 18 mer peptides covering the NV main and small envelope proteins, VP1 (540 aa) and VP2 (268 aa), respectively, with a total length of 808 aa, was synthesized at China Peptide Co., Ltd. (Shanghai, China). Our study focused on the new genotype viral strain GII.P16-GII.4 Sydney 2012. Sequence information was deposited in the National Center for Biotechnology Information (NCBI) https://www.ncbi.nlm.nih.gov/nuccore/NC_039477.1/. A total of 131 18mer peptides were first divided into two VP1-POOLs and VP2-POOLs, each of which contained 88 and 43 peptides, respectively. Then, VP1-POOL and VP2-POOL contained nine VP1 sub-peptide pools and four VP2 sub-peptide pools, respectively. For detailed peptide pool information, referred to Table.S1. NV VP1/VP2 overlapping 18mers with 6-aa shifts, 13mers with 2-aa shifts, and 10mers, 9mers, and 8mers with a 1-aa shift were synthesized as cleaved pin peptides. All peptides had > 90% purity. Details regarding the peptides are provided in Table S1 and S2. The peptides were dissolved in dimethylsulfoxide, DMSO (Sigma-Aldrich, St. Louis, MO, USA) and stored at − 80°C.
Anti-hCD3 (317336; PerCp-Cy5.5), anti-hCD8 (300912; allophycocyanin; APC), anti-hCD4 (300506; fluorescein isothiocyanate; FITC), anti-hIFN-γ (502510; phycoerythrin; PE), and anti-HLA-C were purchased from BioLegend (373302, San Diego, CA, USA). Anti-HLA-B and anti-HLA-A were purchased from Novus Biologicals (NBP2–45000, Centennial, CO, USA) and Santa Cruz Biotechnology (sc -365,485, Dallas, TX, USA), respectively.
Ex vivo IFN-γ identification using intracellular cytokine staining (ICS)
PBMCs were seeded into 96-well plates at 5 × 105 cells/well and stimulated with 1 µmol/L NV VP1/VP2 peptide pools or 18mer (or even shorter) peptides in RF-10 at 37°C in the presence of monensin (554724, BD Biosciences, San Jose, CA, USA) ex vivo for 5 h. The immunodominant response peptides were screened and identified using ex vivo ICS. The results were visualized as a heat-map. Data are representative of three independent experiments. RF-10 contained RPMI 1640 supplemented with 10% foetal bovine serum, 2-mercaptoethanol (5 × 10−5 mol/L), antibiotics (penicillin 100 U/mL, streptomycin 100 µg/mL), and l-glutamine (2 mmol/L). All reagents were purchased from Gibco (Carlsbad, CA, USA).
Cells were harvested and stained with anti-hCD3-PerCP-Cy5.5 (OKT3, BioLegend), anti-hCD4-FITC (RPA-T4, BioLegend), and anti-hCD8-APC (HIT8α, BioLegend) primary antibodies at 1:200 at 4°C for 30 min, followed by washing, fixing, and staining with the anti-hIFN-γ-PE (4S.B3, BioLegend) antibody at 1:100 at 4°C for 30 min. Fluorescence-activated cell sorting (FACS) data were acquired using an LSRFortessa Flow Cytometer (BD Biosciences) and analysed using FlowJo (Tree Star, Ashland, OR, USA). Responses were considered positive if the results were at least three times the mean of those of the negative control wells [Citation19]. Results are representative of three independent experiments. A flowchart of the gating strategy for NV VP-specific CD4+ and CD8+ T cells in PBMCs is shown in Fig. S2.
Immunodominant peptide-specific T-cell bulk culture
First, 1 × 106 PBMCs were resuspended in 200 µL of RF-10 and pulsed with 1 µmol/L of an immunodominant peptide pool (consisting of the identified dominant 18mer peptides). PBMCs were cultivated in a cell incubator at 37°C for 1 h, during which they were vortexed twice to ensure a sufficient load of dominant peptides from autologous antigen-presenting cells. PBMCs were washed twice with phosphate-buffered saline (pH 7.2, Gibco), followed by peptide pulsing and incubation with the remaining 9 × 106 PBMCs (in RF-10 containing 25 IU/mL recombinant human IL-2 [PeproTech, Rocky Hill, NJ, USA]), for 10 consecutive days in 48-well culture plates. On the fifth day, 50% of the medium was replaced with RF-10 containing 50 IU/mL recombinant human IL-2. Cell growth was closely monitored, and the cell culture medium was changed as needed until detection. After 10 days, the cultured cells were seeded in 96-well plates at 5 × 105 cells/well and stimulated with RF-10 with 13mer, 10mer, 9mer, or 8mer peptides separately at 1 µmol/L in the presence of monensin at 37°C for 5 h. ICS was then performed as described above. Data are representative of three independent experiments.
HLA restriction and HLA-I antibody-blocking assays
HLA alleles in immunodominant responders were identified using NGS. Each participant’s response to a single dominant epitope was analysed, and the HLA-I allele restriction profile was generated. The HLA-I restriction characteristics presenting the same epitope were confirmed. This method, combined with antibody-blocking assays, allowed further characterization of HLA-I-restricted genotypes.
In the antibody-blocking assay, 5 × 105 cells/well were incubated with 2 µL of anti-HLA-A, 3 µL of anti-HLA-B, and 2 µL of anti-HLA-C antibodies separately for 30 min, followed by the addition of 1 µM/L dominant peptide and monensin. T-cell activation was measured via ICS. Results are representative of three independent experiments.
Bioinformatics analysis
The protein sequences used to identify the immunodominant core short peptide were assessed using the NCBI NV database and BLAST online, and the genotypes of NV strains were identified to determine the similarities among them. The search criteria were as follows: the dominant peptides were first introduced into the sequence frame, and then, the comparison peptide was selected to target all the NV strains contained in the database without being restricted to a particular genotype. The NCBI database was used to compare protein sequences to map immunodominant peptides and determine the frequency of mutations in the NV sequences of different genotypes.
Statistical analysis
Statistical analysis was performed using Prism version 9.0 (GraphPad Software, San Diego, CA, USA). Pearson’s chi-square or Fisher’s exact tests were used to compare T-cell response rates of categorical variables. Continuous variables and T-cell response frequencies are presented as means ± SD. One-way analysis of variance was used to compare differences among three groups, whereas an unpaired t-test was used to analyse differences between two groups; when the variance differed, the unpaired t-test with Welch’s correction was applied. Statistical significance was set at p values <0.05.
Results
NV VP-specific responses of CD8+ T cells were stronger than those of CD4+ T cells in previously infected individuals
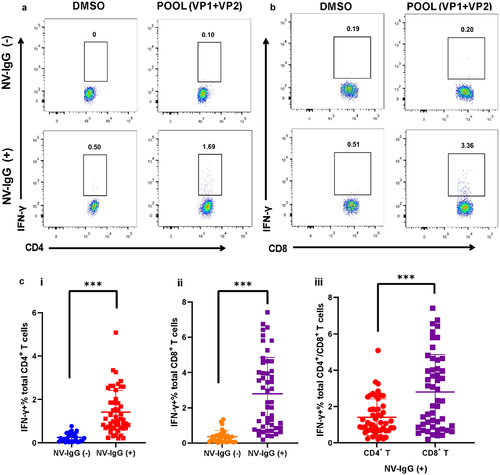
To assess NV VP (VP1+VP2)-specific CD4+ and CD8+ T-cell responses of individuals in the NV-IgG (−) and NV-IgG (+) groups, PBMCs were isolated and stimulated ex vivo using specific VP peptide pools. Random sample collection was performed, and subsequent data analysis revealed no bias in age or sex between the groups. Sample collection was performed in accordance with the SAGER guidelines. The stimulation pool of 18mer peptides encompassed the full-length NV VP1 and VP2 proteins. Following stimulation, VP-specific CD4+/CD8+ T-cell responses in NV-infected individuals were detected using ICS ex vivo.
IFN-γ-producing CD4+/CD8+ T cells were not detected in NV-IgG (−) participants under identical stimulation conditions (). The detection frequencies of both VP-specific CD4+ and CD8+ T cells were substantially higher in the NV-IgG (+) group than in the NV-IgG (−) group (). The frequency of VP-specific CD8+ T-cell responses in the NV-IgG (+) group was markedly higher than that of CD4+ T cells (), suggesting that although both VP-specific CD4+/CD8+ T cells are involved in host immunity following NV infection, the CD8+ T-cell response was higher than that of CD4+ T cells. Therefore, we focused on NV-specific CD8+ T-cell responses in individuals previously infected with NV.
Figure 1. NV VP-specific CD4+ and CD8+ T-cell responses in individuals of the NV-IgG (−) and NV-IgG (+) groups. Representative FACS plots of NV VP-specific CD4+ T-cell responses (a) and CD8+ T-cell responses (b) of individuals in the NV-IgG (−) and NV-IgG (+) groups. (c) Percentages of CD4+ T cells (i) and CD8+ T cells (ii) in PBMCs from NV-IgG (−) (n = 30) and NV-IgG (+) (n = 50) individuals. (iii) Percentages of CD4+ and CD8+ T cells in PBMCs from NV-IgG (+) individuals (n = 50). Data are presented as means ± SD; ns: no significant difference; ***p < 0.001.
Identification of immunodominant CD8+ T-cell response to NV VP-POOL
VP1 and VP2 play important roles in maintaining the natural viral structure and host susceptibility to NV. To analyse differences in VP1-/VP2-specific CD8+ T-cell responses, PBMCs were isolated using the Ficoll-Hypaque gradient and then stimulated by VP1-POOL and VP2-POOL separately to determine the production of the dominant responding peptide VP-POOL. At the beginning of peptide screening, we considered the use of the negative control peptide, OVA323–339 (ISQAVHAAHAEINEAGR), for screening (Fig. S3). The peptide POOL composition is detailed in Table S1A and S1B. Significantly higher IFN-γ secretion was detected after VP1-/VP2-POOL stimulation compared with that after DMSO simulation. However, the response frequency of the VP1-POOL was slightly higher than that of the VP2-POOL but not significantly ().
Figure 2. Ex vivo identification of NV VP1 and VP2 dominant peptide pools and epitope-specific CD8+ T-cell responses in NV-infected individuals. (a) Production of IFN-γ measured using ICS after 5 h of ex vivo stimulation of PBMCs with VP peptide pools (1 µM/L). Results are representative of three independent experiments. (i) Comparison analysis of the percentage of CD8+ T cells stimulated with VP1-POOL or VP2-POOL. Screening of dominant sub-VP1-POOL (ii) and sub-VP2-POOL (iii). ***p < .001. (b) The relative strength of VP1-POOL3-specific CD8+ T-cell response in individuals previously infected with NV. The immunodominant hierarchies of 33 individuals responding to VP1-POOL3 shown in were compiled as a “heat-map.” for each individual, the strongest response was considered the dominant response (DR) and is shown in dark red. All the other responses were evaluated by their relative strength against the DR. The responses were considered non-specific when their strengths were not higher than 40% of their corresponding DR; these are marked in white. The responses with strengths between 41% and 99% of the DR are assigned a specific colour in the heat-map. The percentage of DR peptides in different individuals is counted and shown on the right of the graph. It is presented as a histogram demonstrating the percentages of dominant peptides in VP1-POOL3. (c) The relative strength of VP2-POOL2-specific CD8+ T-cell response in individuals previously infected with NV. The immunodominant hierarchies of 31 individuals responding to VP2-POOL2 shown in were compiled as a “heat-map.” the detailed description is similar to the one in B. (d) The relative strength of VP2-POOL3-specific CD8+ T-cell response in individuals previously infected with NV. The immunodominant hierarchies of 30 individuals responding to VP2-POOL3 shown in were compiled as a “heat-map.” the detailed description is similar to the one in B.
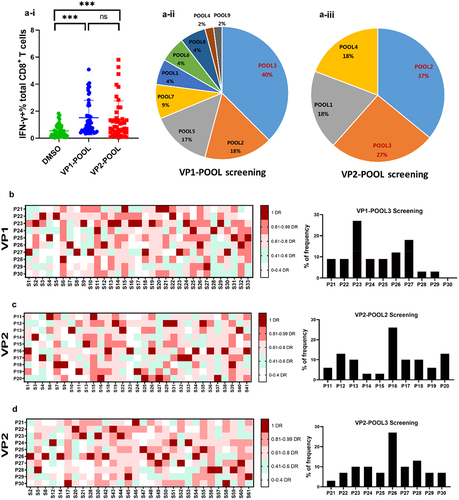
We investigated VP1-/VP2-specific CD8+ T-cell responses separately. After identification of the dominant VP-POOL, we further measured the dominant sub-peptide pool of VP1/VP2-POOL. We grouped 10 consecutive peptides into a sub-peptide pool and obtained nine VP1 and four VP2 sub-peptide pools (Table S1A, B). By systematically exploring characteristics of potential VP-specific CD8+ T-cell responses, we identified that VP1-POOL3, VP2-POOL2, and VP2-POOL3 were the dominant response sub-peptide pools, comprising 40% (18/45), 37% (17/45), and 27% (12/45) of the VP1 and VP2 peptide pools, respectively (). Thus, we identified three dominant response VP-POOLs.
Ex vivo and in vitro validation of dominant epitopes with VP1-P23, VP2-P16, and VP2-P26
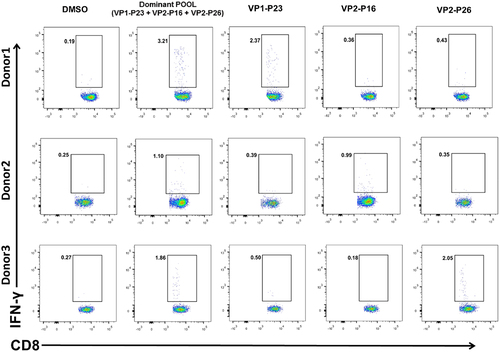
We screened dominant peptides ex vivo. First, 33 individuals responding to VP1-POOL3 were screened from participants previously infected with NV who were responsive to VP1-POOL, and we found nine responders to VP1-P23 (27%) (). Similarly, the VP2-POOL2 and VP2-POOL3 immunodominant peptides were screened individually. In total, 31 and 30 participants responding to VP2-POOL2 and VP2-POOL3, respectively, were selected among the VP2-POOL responders. There were eight responders to VP2-P16 and another eight responders to VP2-P26 among the VP2-POOL2 and VP2-POOL3 responders, with frequencies of approximately 26% and 27%, respectively (). A total of 61 responders were involved in dominant peptide identification. The numbers of individuals previously infected with NV who responded to both VP1-POOL3 and VP2-POOL2 or VP1-POOL3 and VP2-POOL3 were 23 and 10, respectively (Fig. S1).
Donor 1 showed obvious CD8+ T-cell-specific secretion of IFN-γ after 5 h of stimulation with VP1-P23; similar results were observed with Donor 2 and 3 after stimulation with VP2-P16 and VP2-P26, respectively (). These three representative donors were randomly selected from the 61 responders. Thus, we confirmed in vitro that the VP-immunodominant 18mer peptides in previously infected individuals were VP1-P23, VP2-P16, and VP2-P26.
Figure 3. Specific CD8+ T-cell responses to the dominant peptides, identified through in vitro assays. PBMCs were treated with dominant peptides at 37°C for 1 h followed by bulk in vitro culture of specific CD8+ T cells for 10 d. Results are representative of three independent experiments. Representative FACS plots of dominant responses are presented.
Characterization of immunodominant VP1-P23-specific CD8+ T-cell responses
The most common core sequence was identified by adopting a set of 13mer overlapping peptides covering the initially detected VP1-P23 sequence. Five 13mer peptides were synthesized (Table S2a). The core 13mer sequence was screened and titrated. For Donor 1, immunodominant CD8+ T cells responding to the VP1133–150 (VP1-P23) 18mer peptide () recognized two 13mer peptides, VP1137–149 and VP1139–151 (). VP1137–149 stimulated the highest CD8+ T-cell response, indicating that it was the most potent core sequence (). A set of 10mer, 9mer, and 8mer peptides was synthesized based on the 13mer peptide derived from VP1137–149. Successive screening of these peptides showed that the core peptide was VP1141–148 (), which was the minimal epitope of VP1-P23. We then confirmed the restricting HLA genotype characteristics of VP1141–148 using an HLA-I antibody-blocking assay. The anti-HLA-A antibody efficiently blocked CD8+ T-cell activation through the peptide VP1141–148, but anti-HLA-B and anti-HLA-C antibodies were unavailable ().
Figure 4. Detailed characterization of the immunodominant VP1-P23-specific CD8+ T-cell responses. (a) (i) 13mer overlapping peptides within the 18mer VP1133–150 (VP1-P23 responsive Donor 1) were screened. (ii) Comparative dominant 13mer peptides were titrated and identified. (iii) Ten-mer overlapping peptides within the 13mer VP1137–149 were screened. (iv) Nine-mer and 8mer peptides within the 10mer VP1140–149 were screened. (v) An HLA-I antibody-blocking assay was used to identify the restricted characteristics of VP1141–148. Results are representative of three independent experiments. (b) Other representative VP1-P23 responder core peptides were identified from 13mer, 10mer, 9mer, and 8mer peptides in sequence. (c) The sequences of HLA-A alleles in VP1141–148 responders were evaluated using NGS.
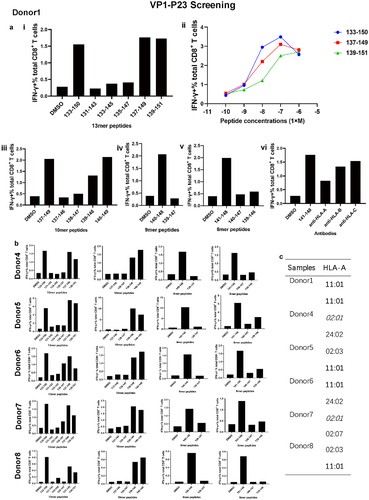
Similarly, the VP1-P23 core peptide was identified in five other responding donors (). VP1137–149 was the dominant epitope of the 13 mer peptide from Donor 4 to 8. The CD8+ T-cell-restricted characteristic was HLA-A. Thereafter, we performed HLA-A genotyping of the alleles of six donors to identify a possible common HLA-A-restricting molecule that binds the VP1141–148 epitope. Four donors carried the HLA-A *1101 allele, and Donor 1 carried the HLA-A *1101 homozygous alleles. As both Donor 4 and Donor 7 harboured HLA-A *0201 alleles, we speculated that the dominant VP1141–148 epitope was presented by both HLA-A *1101 and HLA-A *0201 alleles ().
Confirmation of immunodominant CD8+ T-cell response characteristics of VP2-P16 and VP2-P26
A similar strategy was applied to confirm the features of the VP2-P16 and VP2-P26 epitopes. In Donor 2, the immunodominant CD8+ T cells responded to VP2-P16 (). The dominant 13mer was identified as VP294–106 based on the 18mer peptide VP291–108 (). VP294–102 was confirmed as the main 9mer epitope (). The synthetic peptide sequences are presented in Table S2b. The HLA-I-restricted characteristic of VP294–102 was HLA-Cw (). The other six donors who responded to the dominant VP294–102 epitope were identified (), and all seven donors carried HLA-Cw * 0102 alleles. Donor 9 and Donor 11 carried HLA-Cw * 0102 homozygous alleles (). We inferred that the dominant epitope VP294–102 was carried by HLA-Cw * 0102 alleles.
Figure 5. Confirmation of immunodominant VP2-P16- and VP2-P26-specific CD8+ T-cell response characteristics. (a) (i) 13mer overlapping peptides within the 18mer VP291–108 (VP2-P16 responder Donor 2) were screened. (ii) Nine-mer overlapping peptides within the 13mer VP294–106 were screened. (iii) an HLA-I antibody-blocking assay was used to identify the restricted characteristics of VP294–102. (iv) Other representative VP2-P16 responder core 9mer peptides were identified. Results are representative of three independent experiments. (v) The sequences of HLA-Cw alleles in VP294–102 responders were evaluated using NGS. (b) (i) 13mer overlapping peptides within the 18mer VP2151–168 (VP2-P26 responder Donor 3) were screened. (ii) Nine-mer overlapping peptides within the 13mer VP1151–163 were screened. (iii) an HLA-I antibody-blocking assay was used to identify the restricted characteristics of VP2153–161. (iv) Other representative VP2-P26 responder core 9mer peptides were identified. Results are representative of three independent experiments. (v) The sequences of HLA-Cw alleles in VP2153–161 responders were evaluated using NGS.
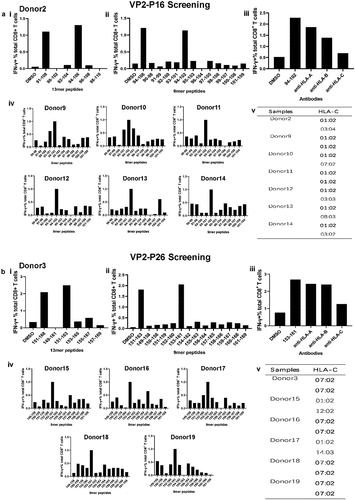
In Donor 3, VP2151–168 18mer peptide-specific immunodominant CD8+ T cells recognized one 13mer peptide, VP2151–163 (). The 9 mer peptide VP2153–161 (Table S2C) was screened and identified (). The VP2153–161 epitope was HLA-Cw restricted (). Five donors in addition to Donor 3 were responsive to VP2153–161 (). Four donors, including Donor 3, carried HLA-Cw * 0702 homozygous alleles (). Both Donor 15 and Donor 17 carried HLA-Cw * 0102 alleles. These findings confirmed that immunodominant responses to VP2153–161 were restricted to HLA-Cw * 0702. Nonetheless, the VP2153–161 epitope may be carried by HLA-Cw * 0102.
BLAST comparison analysis revealed that the three dominant epitopes showed high sequence similarity in epidemic strains and other genotypes of NV strains in several regions (Table S3).
Discussion
Our study demonstrated that NV-specific CD4+ and CD8+ T-cell responses of individuals previously infected with NV who had NV-IgG (+) were higher than those of NV-IgG (−) participants, and CD8+ T-cell responses in previously infected individuals with NV-IgG (+) were higher than CD4+ T-cell responses. Subsequently, we focused on the impact of the NV-specific CD8+ T-cell response against NV infection. We comprehensively profiled the immunodominant CD8+ T-cell responses to capsid proteins and detected candidate antigenic molecules, thereby providing a theoretical basis for studies on vaccines against NV infection. Specific T-cell responses target a particular antigen component. The NV nucleocapsid proteins, consisting of VP1 and VP2, bind to human susceptible receptor histo-blood group antigens and constitute the first step in viral infection of the host [Citation20].
The characteristics of immunogenicity and neutralizing antibody epitopes of VP1 are an essential basis for vaccine design. VP2 as a component of the norovirus nucleocapsid, it played an important role in stabilizing the structure of virus particles, affecting the nuclear localization of VP1 and cooperating with viral particles to bind to the host susceptibility receptor, so it is important to maintain persistent infection [Citation21,Citation22]. Considering the combination of GII.P16 polymerase function and the pandemic GII.4 capsids structure may result in highly transmissible virus in the future [Citation7], we initially synthesized overlapping peptides encompassing the complete NV VP1 and VP2 proteins, based on NV-GII.P16-GII.4 Sydney 2012.
Dominant peptides derived from peptide pool responders were detected. VP1-P23, VP2-P16, and VP2-P26 were the immunodominant 18mer epitopes. As the 18mer peptide was not the optimal epitope recognized by CD8+ T cells, we used truncated peptides to identify core epitopes [Citation23,Citation24]. Successful cultivation of dominant epitope-specific CD8+ T cells in vitro [Citation25] allowed us to determine that the core epitopes of VP1-P23, VP2-P16, and VP2-P26 were VP1141–148, VP294–102, and VP2153–161, respectively. All three dominant core epitopes were 8mer or 9mer, which are the smallest antigenic epitopes recognized by specific CD8+ T cells [Citation23,Citation24,Citation26].
Although VP2-specific CD8+ T-cell responses remain elusive, VP1-dominant epitopes have been reported. Malm et al. [Citation16] identified a similar VP1-P23-specific CD8+ T cell and first reported an 18mer of NV VP1 in combination with a 10mer epitope, as predicted by the Immune Epitope Database. Stepwise screening of Donor 4 and Donor 7 revealed that a previously reported 10mer peptide “TMFPHIIVDV” [Citation17] was VP1139–148 and that its corresponding core epitope was VP1141–148. A highlight of our epitope identification method was the use of an amino acid-truncated approach, which is more accurate and reliable than the predictive approaches that are typically used.
HLA-I molecules strictly restrict antigen recognition of specific CD8+ T cells to only dominant peptides bound to MHC-I [Citation24,Citation26]. Clarification of the HLA-restricted characteristics of dominant responses indicated that the VP1141–148, VP294–102, and VP2153–161 epitopes were primarily presented by HLA-A *1101, HLA-Cw * 0102, and HLA-Cw * 0702, respectively, whereas VP1141–148 and VP2153–161 were also restricted to HLA-A *0201 and HLA-Cw * 0102, respectively. We identified that the core epitope VP1141–148 from Donor 4 and Donor 7 had the HLA-A *0201 alleles in common. Additionally, the dominant 10mer peptide and its restricted characteristics were identical to those previously reported [Citation17]. Regarding VP2153–161 responders, both Donor 15 and Donor 17 carried HLA-Cw * 0102 alleles, suggesting that dominant epitopes exhibited the characteristics of HLA-I pan-restriction. Pan-restricted epitopes have also been reported in cytomegalovirus, Mycobacterium tuberculosis, and hepatitis C virus studies [Citation27–29]. An investigation of epitope-specific cytomegalovirus pp65 cytotoxic T cells demonstrated that certain epitope-induced T-cell responses were only presented by specific HLA alleles, whereas other epitope-stimulated T-cell responses were presented by several different HLA alleles [Citation28]. Dominant epitope-specific T-cell responses may eliminate local viral colonization and suppress symptom manifestation, thus indicating their potential application in adoptive immunotherapy for individuals with chronic NV infection [Citation30].
In this study, we explored the characteristics of immunodominant CD8+ T-cell response profiles in capsid proteins from cases of previous NV infection. Detection of the candidate antigenic molecules from conservative CD8+ T-cell epitopes can provide a theoretical basis for future studies on vaccines against NV infection. However, this study has some limitations that need to be addressed in future studies. For one thing, NV is a common enteric pathogen that primarily induces gastrointestinal symptoms, so it is important to pay attention to clarify the changes in tissue-specific T-cell responses localized in the intestinal mucosa. For another, although the secretion of IFN-gamma was detected after CD8+ T-cell activation, focusing on the secretion of other cytokines such as different types of IFNs, tumour necrosis factors, perforin, and granzymes can facilitate further understanding of their effects.
Authors’ contributions
Study concept and design: Taojun He, Bin Li, Chao Wu;
Funding acquisition: Taojun He, Chao Wu;
Acquisition of data: Yilin Deng, Fang Zhang, Jin Zhang, Luhong Zhu;
Analysis and interpretation: Qinjin Wang, Jie Ning, Hui Wu, Hanmei Yuan;
Draft the manuscript and preliminary revise: Taojun He, Bin Li;
Study supervision: Bin Li, Chao Wu;
Final approval: Taojun He, Yilin Deng, Fang Zhang, Jin Zhang, Luhong Zhu, Qinjin Wang, Jie Ning, Hui Wu, Hanmei Yuan, Bin Li, Chao Wu.
Supplemental Material
Download MS Word (694.4 KB)Acknowledgements
We acknowledge all members of the Laboratory Medicine Department for their helpful assistance. We would also like to thank Editage (www.editage.cn) for English language editing.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The Data generated during the current study is available at repository name “Characteristics of Norovirus Capsid Protein-Specific CD8+ T-Cell Responses in Previously Infected Individuals” at https://doi.org/10.7910/DVN/6K7AFF.
Supplemental data
Supplemental data for this article can be accessed online at https://doi.org/10.1080/21505594.2024.2360133
Additional information
Funding
References
- Ahmed SM, Hall AJ, Robinson AE, et al. Global prevalence of norovirus in cases of gastroenteritis: a systematic review and meta-analysis. Lancet Infect Dis. 2014 Aug;14(8):725–13. doi: 10.1016/S1473-3099(14)70767-4
- Ahmed SM, Lopman BA, Levy K, et al. A systematic review and meta-analysis of the global seasonality of norovirus. PLoS One. 2013;8(10):e75922. doi: 10.1371/journal.pone.0075922
- Ge L, Chen X, Liu J, et al. Genomic and biological characterization of a pandemic norovirus variant GII.4 Sydney 2012. Vir Gen. 2020 Apr;56(2):174–181. doi: 10.1007/s11262-019-01729-0
- Ozaki K, Matsushima Y, Nagasawa K, et al. Molecular evolution of the protease region in norovirus genogroup II. Front Microbiol. 2019;10:2991. doi: 10.3389/fmicb.2019.02991
- Esposito S, Principi N. Norovirus vaccine: priorities for future research and development. Front Immunol. 2020;11:1383. doi: 10.3389/fimmu.2020.01383
- Parra G. Emergence of norovirus strains: A tale of two genes. Virus Evol. 2019;5(2):vez048. doi: 10.1093/ve/vez048
- Ruis C, Roy S, Brown JR, et al. The emerging GII.P16-GII.4 Sydney 2012 norovirus lineage is circulating worldwide, arose by late-2014 and contains polymerase changes that may increase virus transmission. PLoS One. 2017;12(6):e0179572. doi: 10.1371/journal.pone.0179572
- Baehner F, Bogaerts H, Goodwin R. Vaccines against norovirus: state of the art trials in children and adults. Clin Microbiol Infect. 2016;22:S136–S139. doi: 10.1016/j.cmi.2015.12.023
- Kim L, Liebowitz D, Lin K, et al. Safety and immunogenicity of an oral tablet norovirus vaccine, a phase I randomized, placebo-controlled trial. JCI Insight. 2018;3(13). doi: 10.1172/jci.insight.121077
- Wu K, Werner A, Koch M, et al. Serum neutralizing activity elicited by mRNA-1273 vaccine. N Engl J Med. 2021;384(15):1468–1470. doi: 10.1056/NEJMc2102179
- Tarke A, Sidney J, Methot N, et al. Negligible impact of SARS-CoV-2 variants on CD4 (+) and CD8 (+) T cell reactivity in COVID-19 exposed donors and vaccinees. bioRxiv [Preprint]. 2021 Mar 1. doi: 10.1101/2021.02.27.433180.
- Ford-Siltz LA, Tohma K, Parra GI. Understanding the relationship between norovirus diversity and immunity. Gut Microbes. 2021 Jan-Dec;13(1):1–13. doi: 10.1080/19490976.2021.1900994
- Tomov VT, Palko O, Lau CW, et al. Differentiation and protective capacity of virus-specific CD8(+) T cells suggest murine norovirus persistence in an immune-privileged enteric niche. Immunity. 2017 Oct 17;47(4):723–738.e5. doi: 10.1016/j.immuni.2017.09.017
- He R, Hou S, Liu C, et al. Erratum: Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature. 2016 Oct 19;540(7633):470. doi: 10.1038/nature20107
- Wu C, Zanker D, Valkenburg S, et al. Systematic identification of immunodominant CD8+ T-cell responses to influenza a virus in HLA-A2 individuals. Proc Natl Acad Sci USA. 2011 May 31;108(22):9178–9183. doi: 10.1073/pnas.1105624108
- Malm M, Tamminen K, Vesikari T, et al. Norovirus-specific memory T cell responses in adult human donors. Front Microbiol. 2016;7:1570. doi: 10.3389/fmicb.2016.01570
- Malm M, Vesikari T, Blazevic V. Identification of a first human Norovirus CD8(+) T Cell epitope restricted to HLA-A(*)0201 Allele. Front Immunol. 2018;9:2782. doi: 10.3389/fimmu.2018.02782
- Malm M, Tamminen K, Vesikari T, et al. Type-specific and cross-reactive antibodies and T cell responses in norovirus VLP immunized mice are targeted both to conserved and variable domains of capsid VP1 protein. Mol Immunol. 2016 Oct;78:27–37. doi: 10.1016/j.molimm.2016.08.009
- Peng Y, Mentzer AJ, Liu G, et al. Broad and strong memory CD4(+) and CD8(+) T cells induced by SARS-CoV-2 in UK convalescent individuals following COVID-19. Nat Immunol. 2020 Nov;21(11):1336–1345. doi: 10.1038/s41590-020-0782-6
- Zhang M, Zhang B, Chen R, et al. Human Norovirus induces aquaporin 1 production by Activating NF-κB signaling pathway. Viruses. 2022 Apr 18;14(4):842. doi: 10.3390/v14040842
- Lin Y, Fengling L, Lianzhu W, et al. Function of VP2 protein in the stability of the secondary structure of virus-like particles of genogroup II norovirus at different pH levels: function of VP2 protein in the stability of NoV VLPs. J Microbiol. 2014 Nov;52(11):970–975. doi: 10.1007/s12275-014-4323-6
- Liu Z, Zhang M, Shen Z, et al. The coordinating role of the human norovirus minor capsid protein VP2 is essential to functional change and nuclear localization of the major capsid protein VP1. Arch Virol. 2019 Apr;164(4):1173–1180. doi: 10.1007/s00705-019-04192-2
- Pishesha N, Harmand TJ, Ploegh HL. A guide to antigen processing and presentation. Nat Rev Immunol. 2022 Apr 13;22(12):751–764. doi: 10.1038/s41577-022-00707-2
- Sadegh-Nasseri S, Kim A. Selection of immunodominant epitopes during antigen processing is hierarchical. Mol Immunol. 2019 Sep;113:115–119. doi: 10.1016/j.molimm.2018.08.011
- Huang M, Zhang J, Chen W. FACS isolation of low percentage human antigen-specific CD8(+) T cells based on activation-induced CD3 and CD8 downregulation. J Immunol Methods. 2019 Sep;472:35–43. doi: 10.1016/j.jim.2019.06.013
- Nguyen AT, Szeto C, Gras S. The pockets guide to HLA class I molecules. Biochem Soc Trans. 2021 Nov 1;49(5):2319–2331. doi: 10.1042/BST20210410
- Axelsson-Robertson R, Weichold F, Sizemore D, et al. Extensive major histocompatibility complex class I binding promiscuity for Mycobacterium tuberculosis TB10.4 peptides and immune dominance of human leucocyte antigen (HLA)-B*0702 and HLA-B*0801 alleles in TB10.4 CD8 T-cell responses. Immunology. 2010 Apr;129(4):496–505. doi: 10.1111/j.1365-2567.2009.03201.x
- Hasan AN, Doubrovina E, Sottile R, et al. Dominant epitopes presented by prevalent HLA alleles permit wide use of banked CMVpp65 T cells in adoptive therapy. Blood Adv. 2022 Aug 23;6(16):4859–4872. doi: 10.1182/bloodadvances.2022007005
- Molero-Abraham M, Lafuente EM, Flower DR, et al. Selection of conserved epitopes from hepatitis C virus for pan-populational stimulation of T-cell responses. Clin Dev Immunol. 2013;2013:601943. doi: 10.1155/2013/601943
- Hanajiri R, Sani GM, Saunders D, et al. Generation of Norovirus-specific T cells from human donors with extensive cross-reactivity to variant sequences: implications for immunotherapy. J Infect Dis. 2020 Feb 3;221(4):578–588. doi: 10.1093/infdis/jiz491