
ABSTRACT
African swine fever (ASF) is a rapidly fatal viral haemorrhagic fever in Chinese domestic pigs. Although very high mortality is observed in pig farms after an ASF outbreak, clinically healthy and antibody-positive pigs are found in those farms, and viral detection is rare from these pigs. The ability of pigs to resist ASF viral infection may be modulated by host genetic variations. However, the genetic basis of the resistance of domestic pigs against ASF remains unclear. We generated a comprehensive set of structural variations (SVs) in a Chinese indigenous Xiang pig with ASF-resistant (Xiang-R) and ASF-susceptible (Xiang-S) phenotypes using whole-genome resequencing method. A total of 53,589 nonredundant SVs were identified, with an average of 25,656 SVs per individual in the Xiang pig genome, including insertion, deletion, inversion and duplication variations. The Xiang-R group harboured more SVs than the Xiang-S group. The F-statistics (FST) was carried out to reveal genetic differences between two populations using the resequencing data at each SV locus. We identified 2,414 population-stratified SVs and annotated 1,152 Ensembl genes (including 986 protein-coding genes), in which 1,326 SVs might disturb the structure and expression of the Ensembl genes. Those protein-coding genes were mainly enriched in the Wnt, Hippo, and calcium signalling pathways. Other important pathways associated with the ASF viral infection were also identified, such as the endocytosis, apoptosis, focal adhesion, Fc gamma R-mediated phagocytosis, junction, NOD-like receptor, PI3K-Akt, and c-type lectin receptor signalling pathways. Finally, we identified 135 candidate adaptive genes overlapping 166 SVs that were involved in the virus entry and virus-host cell interactions. The fact that some of population-stratified SVs regions detected as selective sweep signals gave another support for the genetic variations affecting pig resistance against ASF. The research indicates that SVs play an important role in the evolutionary processes of Xiang pig adaptation to ASF infection.
Background
African swine fever (ASF) is a highly fatal and contagious (near to 100%) haemorrhagic disease in domestic and wild pigs. ASF threatens global pig production and food security, and raises widespread and colossal economic losses in most countries in Africa, Asia, and Europe [Citation1]. ASF is caused by an enveloped double-stranded DNA virus belonging to the genus Asfivirus family Asfarviridae [Citation2]. Almost all domestic pigs of all ages and breeds are susceptible to ASFV (ASF virus), which causes varying degrees of clinical symptoms [Citation3]. However, African wild swine, such as bush pigs, red river hogs, and warthogs, can be asymptomatically infected with ASFV and show obvious resistance [Citation4,Citation5]. Increased resistance to ASF has also been reported in many parts of the world, including eastern Zambia [Citation6], Vietnam [Citation7], Estonia [Citation8], the Democratic Republic of Congo [Citation9], and Kenya–Uganda [Citation10]. The clinically healthy and antibody-positive domestic pigs have been found in epidemic areas, with few virus and viral genomes detected [Citation11]. Many experiments have confirmed that after the ASF outbreak, the recovered pigs and their offspring are neither carriers nor relapses of the disease [Citation7,Citation12]. The difference in ASFV resistance is likely related to genetic differences among various pig breeds [Citation11,Citation13,Citation14]. Genetic improvement of host resistance by breeding domestic pigs with natural resistance to ASFV has been proposed as a viable strategy for preventing and controlling ASFV infection [Citation15].
Host gene variations affect susceptibility to pathogenic infections, some of which may be the basis for the main mechanisms leading to variations in disease resistance [Citation16,Citation17]. Interactions between the host and pathogen can shape natural variations by implementing strong selection in the affected population [Citation18]. Many studies have illustrated that the outcomes of infection with Zika virus, HIV, hepatitis virus, Ebola virus, and SARS-CoV-2 are correlated with host genetic background [Citation19–23]. Genomic structural variation (SVs) is an important genetic variation that includes insertions, deletions, segmental duplications, inversions and complex changes of larger than 50 base pairs (bp) to several megabases (Mb) in size [Citation24]. They are also the main source of genetic variation behind phenotypic diversity [Citation25]. Numerous studies on humans and animals have shown that SVs contribute to phenotypic variations in complex diseases and traits [Citation17,Citation25,Citation26]. The identification of structural variations and potential genetic markers is important for a better understanding of disease resistance as well as genomic prediction and genetic improvement using marker-assisted selection technology [Citation27].
African swine fever was firstly reported in the Liaoning province on 3 August 2018, and rapidly spread throughout China. By the end of 2021, many ASF cases occur in 31 provinces and regions of China, which result in morbidity and mortality rates of almost 100% [Citation15]. In this study, ASF-resistant pigs were found in a population of the Xiang pig breed on a pig farm in Guizhou province after an ASF outbreak in August 2019. Pigs with ASFV genome-negative and antibody-positive were found in the blood samples of the surviving pigs. To decipher the natural genetic variations of these pigs’ resistance to ASFV infection, we detected the entire spectrum of SV events using a combination of three programs (Delly [Citation28], Manta [Citation29], and Dysgu [Citation30]) between susceptible and resistant populations to ASF in Xiang pigs (named Xiang-S and Xiang-R groups), respectively. In addition, SV comparisons in Xiang, Yorkshire, and warthog pigs were performed to obtain more innate immune genes and to reveal the genetic basis. Candidate genes significantly associated with ASFV disease resistance were identified. Overall, our findings provide novel insights into the genetic architecture of host innate immunity to ASFV, which would make sense for the elucidation of pig resistance to viral diseases, and would be beneficial for revealing the molecular genetic basis of host resistance to other viral diseases in swine and other mammals in the future.
Results
Genomic structural variant data sets
In the present study, 12 DNA libraries were independently constructed from the genomic DNA samples of Xiang pigs representing the two states of susceptibility and resistance to ASF, and then sequenced using the Illumina HiSeq 2500 platform by paired-end DNA sequencing technology, which generated a raw data of 458.23 Gb totally. We obtained 431.60 Gb of clean reads after quality control by NGSQC Toolkit [Citation31], with an average of 35.97 Gb per individual, representing an average depth of ~ 14.22 fold (). Then, the clean reads were mapped onto the pig reference genome Sscrofa11.1 using BWA software [Citation32]. Consequently, the base-mapping rates for each individual ranged from 95.58% to 97.87%, with an average of 96.73%.
Table 1. Summary of sequencing and mapping statistics.
Four types of classical SVs (DEL, INS, DUP, and INV) with lengths greater than 50 bp were called via these softwares. To remove redundant SVs, we reserved the SVs called by at least two software packages with more than three paired-end reads in each sample. The SVs presented in only one sample were excluded to reduce the probability of false positives. On average, 25,995 SVs per individual were called and merged into a non-redundant dataset of 54,863 SVs. We filtered out 1,274 unreliable SVs with a hard threshold and obtained a total of 53,589 unique SVs. Finally, 25,656 high-confidant SVs per sample on average were identified, ranging from 16,449 to 30,987 (Additional file 1: Table S1; ). A total of 44,235 SVs were mutual between two groups, with 2,279 and 7,075 SVs specific to Xiang-S and Xiang-R groups, respectively (). The general distribution of SVs was similar between groups with predominant types of deletion. The samples from Xiang-S and Xiang-R groups contained an average of 21,361 and 23,659 DELs (89.26% and 86.41%, respectively), 428 and 838 INSs (1.79% and 3.06%, respectively), 966 and 1,351 DUPs (4.04% and 4.93%, respectively), and 1,177 and 1,532 INVs (4.92% and 5.60%, respectively) (). Compared with the results observed previously [Citation33], we identified more deletions than insertions using these three SV callers. The numbers of SVs on chromosome 1 was the highest in both populations, and the lowest on chromosome 18 (Additional file 1: Table S1; ). The SV numbers in a chromosome were related to chromosome length.
Figure 1. The identified SVs in the Xiang-R and Xiang-S groups. (a) Total SV numbers of these 12 individuals in two groups. (b) The venn diagram showing the overlapped SVs between two groups. The Xiang-R group was shown in green colour and Xiang-S group in blue. The intersection of two groups was showed in light green. (c) Types of identified SVs in the Xiang-R and Xiang-S groups. The Xiang-R group was present in green columns and Xiang-S group in blue.
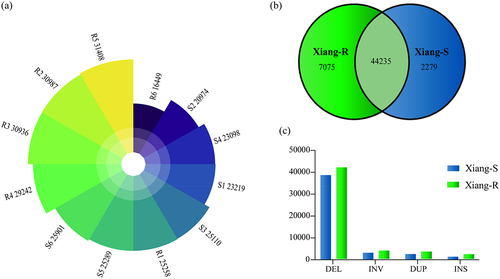
Figure 2. Chromosome distributions of identified SVs in the chromosome of Xiang-R and Xiang-S groups. The Xiang-R group was shown in green column and Xiang-S group in blue. The X axis represented the chromosome position in Mb. The Y axis showed the 19 chromosomes.

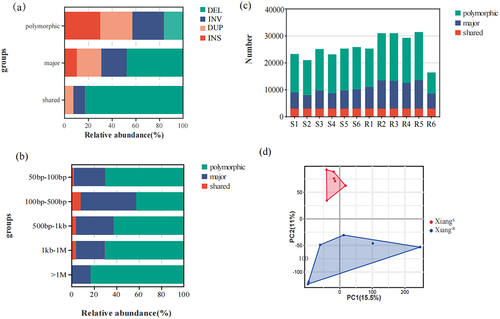
According to the suggestions previously reported [Citation34], these SVs were classified into three categories: shared (identified in all samples), major (identified in ≥ 50% of samples), and polymorphic SVs (identified in >1 sample). Approximately 5.61% (n = 3,005) of the SVs were shared among all samples, 39.24% (n = 21,028) were identified as major variants, and 55.15% (n = 29,556) of them were polymorphic. For each variant type, the proportion of polymorphic SVs was the highest, whereas the shared SVs did not contain INS (). Among the three types of polymorphic, major, and shared SVs, SVs with a length of 100–500 bp accounted for the highest proportion, up to 40.44%, 66.68%, and 74.64%, respectively, whereas the segments with a length of more than 1 Mb were polymorphic SVs (529, 83.31%) (). The Xiang-R5 sample contained the most SVs, whereas the Xiang-R6 sample had the least SVs ().
Figure 3. Distribution of the identified SVs in the Xiang-R and Xiang-S groups. (a) The frequencies of the four SV types of deletion (DEL), inversion (INV), duplication (DUP), and insertion (INS), including shared (identified in all samples), major (identified in ≥ 50% of samples), polymorphic (identified in > 1 sample). (b) Proportions for SVs discovered with different base pair sizes in length. (c) The number of SVs for each discovery category was shown per sample. (d) Principal component analyses for all of the 12 pig individuals in Xiang-R and Xiang-S groups. Different colours represented different populations.
The connected length of all the SVs was 596.18 Mb and 819.33 Mb for Xiang-S and Xiang-R samples, respectively, representing approximately 23.85% and 32.79% of the pig reference genome, where the average SV lengths of DELs were 105.30 Mb (17.66%) and 151.50 Mb (18.49%), respectively. Both of INVs (339.28 and 451.91 Mb, 56.91% and 55.16%) and DUPs (151.56 and 215.84 Mb, 25.42% and 26.34%) contributed much more to the total SV lengths due to their considerably longer sequence lengths.
We performed a principal component analysis (PCA) on the SV pattern dataset (). The PC1 could not be used to differentiate between the two populations. PC2 reflected the biological differences between Xiang-S and Xiang-R groups.
Population-stratified SVs
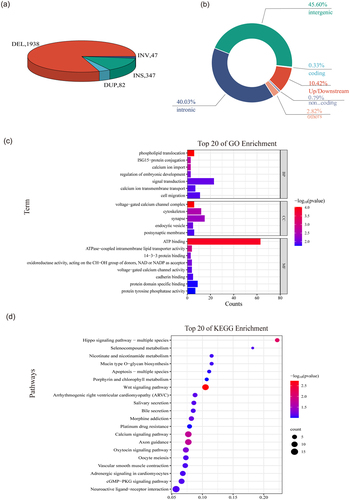
To identify SVs that differentiated between the Xiang-S and Xiang-R populations, the FST values between the Xiang-S and Xiang-R samples were calculated using the NGS data at each locus. A total of 2,414 population-stratified SVs (FST ≥0.25) were identified, including 1,938 DELs, 347 INSs, 82 DUPs and 47 INVs, respectively (). It showed that 234 population-stratified SVs had a length > 1 kb, and the largest event was a ~ 3,721 kb INV in gene NAP1L3 on chromosome X. Most of the population-stratified SVs were located in intergenic regions or introns, 251 SVs in the upstream or downstream of genes, 6 SVs in 5’- or 3’-flanking regions, 22 DUPs leading to the amplification of 20 protein coding genes, 1 pseudogene and 1 lincRNA transcripts (Additional file 2: Table S2; ). The top candidate variants with the maximum FST (FST >0.8) were 5 deletions located in the intron of DLG2 (293 bp), PRKCH (223 bp) and ENSSSCG00000047762 (lncRNA, 92 bp), in the upstream of ENSSSCG00000063149 (lncRNA, 190 bp) or the downstream of LOC100523732 (185 bp) and in the intergenic region between ENSSSCG00000043148 and ENSSSCG00000050489 (58 bp) genes.
Figure 4. The population-stratified SVs identified in the two groups. (a) Pie chart of the population-stratified SVs. Each slice of the pie represented one type of SV. (b) A summary of location classification of the population-stratified SVs. (c) GO analysis of the population-stratified SVs. (d) Top 20 of KEGG enrichment of the population-stratified SVs.
The annotation showed that among those highly differentiated SVs, 1,326 SVs affected 1,152 Ensembl genes, including eight pseudogenes, 158 noncoding RNA genes, and 986 protein genes. Most of those genes (n = 1010, 87.67%) harboured a single population-stratified SV, whereas fewer (n = 142, 12.33%) contained two or more population-stratified SVs. Notably, DLG2 contained eight DELs with 67–303 bp in introns 3, 5, 6, 19, and 20. We further investigated the effects of population-stratified SVs on host genes using a Variant Effect Predictor (VEP). Twelve deletions that were predicted to have a loss-of-function resulted in high impacts on genes, including missing stop codon, transcript ablation, transcript amplification, and frame shift variants in host genes. Twenty DUPs were characterized as whole-gene duplications, which generally caused copy gain of an entire protein-coding gene.
Gene annotation and KEGG analysis of the population-stratified SVs and candidate adaptive genes
To gain insights into the biological functions of population-stratified SVs harbouring genes and the mechanisms underlying the relationship between SVs and disease resistance, we performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses (Additional file 3: Table S3) using the KOBAS 3.0 and WEB-based GEne SeT AnaLysis Toolkit. The Gene Ontology analyses revealed 103 significant GO terms (p < 0.05), which were mainly comprised of the voltage-gated calcium channel complex, phospholipid translocation, calcium ion import, cytoskeleton, endocytic vesicle, cadherin binding, focal adhesion, cytokine production, positive regulation of B cell activation, CD40 signalling pathway, microtubule minus-end, and synaptic membrane adhesion (). KEGG enrichment analysis indicated that genes overlapping with population-stratified SVs were mainly enriched in the Wnt signalling pathway, axon guidance, hippo signalling pathway, calcium signalling pathway, and oxytocin signalling pathway (p < 0.05, ).
Additionally, we found that many genes harboured by population-stratified SVs were involved in the KEGG pathways of apoptosis, ECM–receptor interaction, Fc gamma R-mediated phagocytosis, Th1 and Th2 cell differentiation, c-type lectin receptor signalling pathway, endocytosis, NOD-like receptor signalling pathway, and immune, bacterial and viral infections, etc.
We then focused on the genes involved in biological processes and pathways of immunity, apoptosis, endocytosis, and bacterial and viral infection. We identified 166 candidate SVs with 0.25 ~ 1.0 FST values that overlapped with 135 genes (Additional file 4: Table S4), including 141 DELs, 18 INSs, 4 DUPs and 3 INVs. The lengths of these SVs ranged from 50 to 7,464 bp with an average length of 411 bp, except for two DUPs with larger fragments. There were 144 SVs in introns, 2 in exonic region, 13 in upstream of genes, 4 in downstream of genes, 2 in 3’-UTR, and 1 DUP resulting in transcript amplification.
The patterns of repetitive sequences were analysed for the candidate SVs and found that 87.95% of the breakpoints of SVs overlapped with repetitive elements, including SINEs (53.61%), SINEs+LINEs (4.82%), tandem repeats (12.05%), LINEs (13.25%), SINE+LINE+LTR (1.81%), LTRs (1.81%), LTR+SINE (0.6%), and no repetitive sequences (12.05%). The SINEs derived from tRNA named Pre0_SS (86.52%), PRE1 family (8.99%), and from other SINEs (4.49%, SINE1B_SS, MIR3). LINEs originated from the L1 and L2 families. LTR originated from the ERV1 and ERVL-MaLR families.
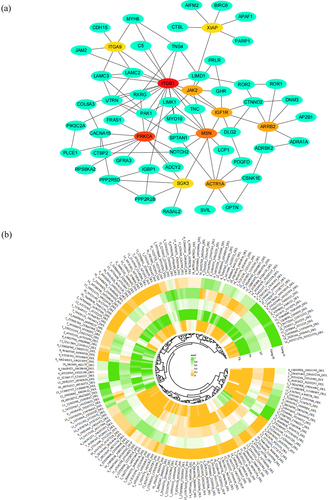
Furthermore, the interactions between the candidate genes were interpreted that 111 of the 135 genes could form a protein–protein interaction network (). Of those, 13 candidate genes were identified as hub genes connected with 6 to 22 genes based on the network relationships of 135 genes using the STRING online platform (https://cn.string-db.org/, Version: 12.0).
Figure 5. The 166 candidate SVs refered to viral infections. (a) Protein–protein interaction network analysis of the candidate genes related to immune pathways. Top 10 hub genes were colored in red, orange and yellow, and the others colored in green. (b) Heat map of the 166 candidate SVs based on the variation frequency in all the groups (Xiang-R, Xiang-S, Xiang pigs (Xiang-R and Xiang-S), Yorkshire, and Phacochoerus africanus).
Comparison of the candidate SVs to genomic sequences of Yorkshire pig and warthog (phacochoerus africanus)
To compare the genetic differentiation of the candidate SVs between Xiang pigs and other pig breeds, we aggregated the whole-genome data of Yorkshire (n = 12) and warthog (n = 12) pigs from the NCBI database. And then the FST was calculated between samples from Xiang-R and Yorkshire or warthog pigs using the 166 SV loci. The SVs from two datasets were detected and genotyped using the same protocol described in the Methods section. Groups of Xiang-R and the warthogs showed more variations than that of Xiang-R and Yorkshire pig compares (). The results are presented in Table S5 (Additional file 5).
It was observed that 41.57% (n = 69) of these SV loci presented significantly higher differentiation (FST = 0.250–0.885) for Xiang-R and Yorkshire comparison, and 53.61% (n = 89) showed significantly higher differentiation between Xiang-R and Warthog's samples (FST = 0.25–1.0). PCA showed that Xiang-R, Xiang-S, and Yorkshire pigs were clustered by PC1, and warthogs were clustered on the other side, whereas the Xiang and Yorkshire pigs were significantly separated by PC2 (Additional file17: Figure S1a). In addition, the branches of the phylogenetic tree (Additional file17: Figure S1b) were consistent with the PCA results and were divided into three populations displaying genetically distinct clusters. Furthermore, population admixture analysis (Additional file17: Figure S1c) distinguished the three populations as expected based on their origins at K = 3.
Validation of 28 structural variations in pig populations
Total of 28 structural variations located in the exonic, intronic and intergenic regions were randomly selected using PCR method to confirm the efficiency of our approach and authenticity of the SV calling results (Additional file 6: Table S6). Clear PCR products of the expected sizes were obtained (Additional file 17: Figure S2) and the sequences of alleles W and V of each SV were confirmed by Sanger sequencing, and the variation regions were confirmed by comparing both sequences. All 28 SVs were verified to be truly existed, implying that our detection method was reliable and effective. However, the exact terminal location of the SV and the genotype frequency varied to some extent, which may be related to the combination of SV regions.
Discussion
ASF is a viral haemorrhagic fever in domestic pigs that leads to almost 100% mortality [Citation35,Citation36]. The outbreak of ASF in China has led to severe losses in the pig industry since August 2018 in China [Citation37]. ASFV infects domestic and wild pigs and ticks. Although ASF is normally associated with very high case fatality rates, a proportion of infected domestic pigs would recover from the infection and survive, showing an increased resistance to the pathogenic effects of the disease, with a high proportion of healthy pigs having antibodies against the ASF virus [Citation36,Citation38]. However, the genetic basis of the resistance of domestic pigs to ASF is complex and remains incompletely understood. Therefore, it is important to determine the genetic basis of ASF resistance or susceptibility in pigs to provide vital clues for ASF prevention. In this study, we analysed the complete spectrum of SVs in ASF-resistant and ASF-susceptible populations of Xiang pigs, a Chinese indigenous miniature pig breed, using high-throughput sequencing. We identified population-stratified SVs between ASF-susceptible and ASF-resistant samples, genes, and enriched pathways, and explored the genetic basis for the resistance of the Xiang pig breed to ASF.
We obtained the reliable SV datasets and identified 25,656 high-confidence SVs on average. Nearly 45% were identified as shared or major variants and 55.15% as polymorphic variants. Near to 88% of the SV breakpoints overlapped with repetitive elements, most of which were interspersed repeats (56.8%) and tandem repeats (21.3%). Consistent with previous studies [Citation39,Citation40], the profiles of SVs length distribution displayed obvious peaks at 300 bp, corresponding to SINEs, and at 6 kb, corresponding to LINEs (Additional file17: Figure S3). Although the general distribution of SVs was similar in both groups, ASF-resistant pigs harboured more SVs than the ASF-susceptible pigs. We performed PCA on the SV pattern dataset. PC2 reflected the biological differences between the Xiang-S and Xiang-R groups. And then the SVs were genotyped and calculated the F-statistics (FST) between the Xiang-S and Xiang-R samples using the NGS data at each locus. A total of 2,414 population-stratified SVs (FST >0.25) were identified in samples from the two groups. After annotattion, 1,326 SVs overlapped with or near 1,152 Ensembl genes, including 986 protein-coding genes, 8 pseudogenes, and 158 noncoding RNA genes.
It has been reported that ASFV infection activates several signalling pathways and regulates the expression of related genes, such as EGFR, PI3K-Akt, cytokine – cytokine receptor interaction, c-type lectin receptor, TNF, IL-17, Jak-STAT, toll-like receptor, NF-κB, apoptosis, Th1 and Th2 cell differentiation, NOD-like receptor, Fc epsilon RI, Hippo, and chemokine signalling pathways [Citation41]. In our study, KEGG enrichment analysis showed that the genes with population-stratified SVs were mainly enriched in the wnt, Hippo, calcium, axon guidance, and oxytocin signalling pathways. But more than that, the other important pathways associated with the ASFV infection were also found in our study (Additional file 3: Table S3), such as the endocytosis (ko04144; P-value = 0.157), cGMP-PKG (ko04022; P-value = 0.077), apoptosis (ko04210; P-value = 0.298), ECM–receptor interaction (ko04512; P-value = 0.101), focal adhesion (ko04510; P-value = 0.135), Fc gamma R-mediated phagocytosis (ko04666; P-value = 0.322), NOD-like receptor (ko04621; P-value = 0.628), C-type lectin receptor (ko04625; P-value = 0.638), PI3K-Akt (ko04151; P-value = 0.554), Th17 cell differentiation (ko04659; P-value = 0.645), Th1 and Th2 cell differentiation (ko04658; P-value = 0.542) signalling pathways. The enriched genes were mapped to GO terms, such as calcium, phospholipid, cytoskeleton, endocytic vesicle, cadherin binding, adhesion, cytokine, CD40, B cell activation, microtubule, and cell junction. These results indicated that the structural variations in the genome may be associated with resistance to ASFV infection in the surviving Xiang pig populations. We then focused on investigating the genes involved in viral entry and virus–host cell interactions, such as immunity, apoptosis, endocytosis, and viral infection. Finally, we identified 135 genes that overlapped with 166 candidate SVs (FST = 0.25–1.0).
Infection to host cells by a virus is a complex and diverse process related to the biochemical and molecular interactions between viruses and hosts [Citation42]. ASFV predominantly infects porcine monocyte macrophages and replicates in the cell cytoplasm [Citation43]. Several studies have demonstrated that ASFV invades through several models of cell entry, including receptor-dependent mechanism, clathrin- and dynamin-mediated endocytosis, and constitutive micropinocytosis [Citation44–46]. A recent study demonstrated that ASFV uptake was significantly affected in a dose-dependent manner by macropinocytosis inhibitors such as EIPA (5-N-Ethyl-N-isopropyl) amiloride, which blocks vacuolar Na+/H+ antiporters, IPA-3 (3-indolepropionic acid), an inhibitor of p21-activated kinase 1 (Pak1); and cytochalasin D, an inhibitor of actin polymerization [Citation45]. ASFV uptake is also strongly decreased by clathrin inhibitors (chlorpromazine and pitstop2) and the dynamin inhibitors (dynasore, DYN). ASFV infection is also strongly inhibited by both classes of endocytosis inhibitors [Citation47]. In the present study, we identified 26 population-stratified SVs that overlapped with 20 protein genes involving in clathrin- and dynamin-mediated endocytosis and Fc gamma R-mediated phagocytosis.
Among them, 11 SVs were the Xiang-R enriched deletions with SINEs and/or LINEs in intron regions, including 275 bp deletion (Pre0-SS) in intron 10 of RAB11FIP4 gene, 281 bp deletion (Pre0-SS) in intron 3 of ASAP1 gene, 322 bp deletion (Pre0-SS) in intron 2 of GRK3 gene, 308 bp deletion (Pre0-SS and L1–2) in intron 14 of DNM3 gene, 482 bp deletion (Pre0 and L1) in intron 2 of SH3KBP1 gene, 287 bp deletion (Pre0-SS and L1–2) in intron 6 of RAB10 gene, 340 bp deletion (Pre0-SS and L2A) in intron 18 of AP2B1 gene, 260 bp deletion (Pre0-SS) in intron 2 of PAK1 gene, 285 bp and 263 bp deletion (Pre0-SS) in intron 10 and intron 25 of KIF5C gene, and 67 bp deletion (SINE MIR3) in intron 6 of USP8 gene. The three SINE (Pre0-SS) deletions were downstream of SNX12 (337 bp) and upstream of EHD4 (266 bp) and LIMK1 (276 bp). These genes play important roles in clathrin-dependent and dynamin-mediated endocytosis (AP2B1, PAK1, DNM3, KIF5C, and GRK3) and Fc gamma R-mediated phagocytosis (PAK1 and LIMK1). The presence or absence of SINEs or LINEs may cause the variations in the regulatory regions and introns of the genes. Previous studies have revealed that SINE or LINE insertions can supply regulatory sequences to adjacent genes at a new integration site, and thus have a potential influence on gene regulation. They can block the transcription of some protein-coding genes by binding to Pol II and significantly repress the promoter activity of genes in cells [Citation48] causing exon skipping, a frame shift, a downstream premature termination codon, and possibly a truncated protein product [Citation49]. Furthermore, they may play a role in the post-transcriptional regulation of gene expression. It has been found that SINEs in 3’-untranslated regions (3’-UTRs) regulate gene expression by Staufen-mediated mRNA decay in human and mouse myoblasts [Citation50]. In our study, the presence/absence of SINE or LINE might act as regulators that increase or decrease the expression of genes related to clathrin-dependent and dynamin-mediated endocytosis and phagocytosis in mononuclear-phagocytic cells, thus reducing ASFV uptake and infection/lesions in Xiang pigs. For example, the 308 bp insertion in intron 14 of DNM3, 340 bp insertion in intron 18 of AP2B1, 260 bp insertion in intron 2 of PAK1, and 322 bp insertion in intron 2 of GRK3, 337 bp insertion downstream of SNX12, and 281 bp insertion in intron 8 of PRKCA also comprised Pre0-SS SINEs derived from tRNA. These SINEs contained an internal RNA promoter. Pol III within the 5’ terminus, a polyadenylation signal (AATAAA), and A-rich tail at the 3’ terminus (Additional file17: Figure S4). The RNAs of SINEs transcribed by pol III would block the transcription of DNM3, AP2B1, PAK1, GRK3, and SNX12 by binding to Pol II and repressing promoter activity. In addition, SINE can be transcribed as part of gene introns by RNA polymerase II.
Signalling pathways mediated by G-protein-coupled receptor (GPCR) are important targets for viruses to rewire cells during infection [Citation33]. It has been found that a great deal of viruses can activate and/or require GPCR-mediated signalling pathways for viral infection. GPCR is a seven-transmembrane-spanning receptor that transmits signals from extracellular stimuli into intracellular signals and performs crucial effects on cellular functions [Citation51]. When the GPCR is activated by its ligand such as a chemokine, or a pathogen, the GPCR recruits intracellular effectors, such as heterotrimeric G proteins, GPCR-kinases, or β-arrestins, and binds to the intracellular loops of the receptor [Citation52]. GPCRs phosphorylated by GRKs drive the receptors to recruit β-arrestins, which are the GPCR-associated scaffolding proteins involved in endocytosis. The binding of β-arrestins brings out further recruitment for the endocytic proteins, including clathrin and adaptor protein 2 (AP2). Finally, these effects result in clathrin-mediated endocytosis and GPCR receptor recycling [Citation53]. The porcine repetitive element 1 (PRE1) is the most abundant SINEs in the genome. A previous report revealed that the proximal PRE1 in HORMAD1, TCEA3 and HACD3 genes had tissue-specific enhancer and promoter functions [Citation54]. In our study, a 215 bp deletion 1,447 bp downstream of ARRB2 comprised PRE1b. PRE1b is the proximal SINE in ARRB2 gene. Sequence analysis of PRE1b showed that this SINE contained many cis-regulation elements, including the GT1-motif, SP1 element, CAAT-box, and dOCT elements. The proximal PRE1b in ARRB2 probably has enhancer and promoter functions. In general, the effects of SINEs on gene expression were eliminated by the deletion of the SINEs. Therefore, the deletion of PRE1b at the proximal end of ARRB2 leads to the loss of enhancer or promoter functions, thus downregulating the expression of ARRB2 gene. At the same time, the gene expression would decrease because of the Pre0 SINE insertion in intron 2 of GRK3 gene. The decrease of GRK3 and β-arrestin 2 might lead to the interruption of GPCR-mediated pathway for viral infections in Xiang pig population.
Several macrophage receptors, such as CD163, CD45, Fc-receptors, and MHCII, have been considered to play a role in viral infection. Nevertheless, specific receptors for ASFV are yet to be identified, suggesting that other receptors or entry mechanisms might be essential for viral infection [Citation45]. Insulin-like growth factor-1 receptor (IGF1R), also known as CD221 or JTK13, is a receptor tyrosine kinase (RTK) to the insulin receptor (IR) family. It has been reported that IGF1R is an entry receptor for the respiratory syncytial virus (RSV) [Citation55]. In our study, it was observed that a 61 bp deletion and a 62 bp insertion in intron 1 of IGF1R gene also were Xiang-R-enriched deletions and insertions and did not overlap with any transposon elements. The analysis of sequences indicated that the two SVs contained several conserved cis-elements for RNA binding proteins (RBPs), such as SFPQ (GGAGUUU), PTBP1 (UUCUUCU), QKI (ACUAAGG), RBM5 (UAAGGUA), HNRNPA1 (TAGGGG), TARDBP (UGUAUGCG), SRSF1 (GGAAGUGC), NONO (AGGGU), IGF2BP1 (UGGCACCCCUG), and PCBP1 (GCAUCCC) (Additional file17: Figure S5). RBPs are key regulators that can directly bind to specific target RNAs via consensus sequences, and control RNA splicing and metabolism [Citation56]. The consensus RNA sequences of the RBPs are called cis-elements, which determine the specificity of the targets and functions of RBPs. RNA splicing is strictly regulated in various tissues and developmental stages. Abnormal regulation of RNA splicing can be caused by mutations in cis-elements that eliminate the utilization of correct splice sites. In particularly, long introns are prone to intron retention and alternative splicing, with concomitant premature termination [Citation57]. The 61 bp deletion and 62 bp insertion in intron 1 of IGF1R gene led to a lack or gain of these cis-elements of the RBPs such as SFPQ, SF1, SRSF1, and NONO. Because intron 1 of IGF1R gene is a longer intron (51,725 bp), the two SVs would probably cause abnormal splicing of IGF1R mRNA such as intron retention and alternative splicing. Thus, the mutation of IGF1R might lead to the inhibition of endocytosis and ASF viral infections triggered by IGF1R in the Xiang pig population.
Furthermore, the cytoskeleton, focal adhesions and cell–cell junctions play essential roles in viral infections [Citation42]. The cytoskeleton participates in virus surfing before entering the cells and provides forces for viral entry and virus uncoating. Focal adhesions can sense extracellular mechanical signals and also influence viral infections in multiple ways. The junctions among cells can perceive the forces from the extracellular and intracellular environments and may be used by viruses to assist their infection [Citation58]. Actin-cytoskeleton rearrangement, activation of the EGFR and PI3K-Akt signalling pathways can promote a macropinocytosis-mediated endocytic processes for ASFV entry [Citation59]. Inhibition of PI3K and other key regulators of micropinocytosis leads to a remarkable decrease in ASFV entry and infection. In our study, it was found 47 genes with 62 population-stratified SVs related to the cytoskeleton, adhesions, and junctions (Additional file 7: Table S7). In particular, 10 SVs in 9 genes (DLG2, PTPN14, TNS4, FRMPD1, PCLO, GRM1, JAK2, CDH15, ITGA9) were highly differentiated in these two groups of Xiang pigs (FST = 0.5–1.0), indicating that these SVs also played a role in the process of Xiang pig’s adaptation to the ASFV infection. The cytoskeleton, focal adhesion, and junctions are regulated by the Wnt and Ras signalling pathways. We also found that 26 SVs in the introns of 18 genes involved in the Wnt and Ras signalling pathways were population stratified between the two populations.
After ASFV infects pig macrophages, the hosts first induce an innate immune response and then produce an adaptive immune response. However, to survive and produce progeny viruses, ASFV encodes various proteins and uses various mechanisms to evade the host immune response, such as inhibition of the cGAS/STING pathway, expression regulation of cytokines and chemokines, inhibition of apoptosis and autophagy, inhibition of the adaptive immune response, regulation of host inflammation, interferon response, and activation of specific target genes [Citation60]. In present study, 48 SVs were identified to be overlapped with 44 genes, which related to the immune response, NOD-like receptor, apoptosis, autophagy, inflammation, TNF, and NF-κB signalling pathways. Of those, 32 SVs contained transposable elements (TEs) including SINEs, LINEs, or LTRs. Compared with the TEs insertion frequency in the Xiang-S group, the majority of TEs in Xiang-R group were SINEs, LINEs or LTRs. Reports on mammalian lineages have shown that lineage-specific TEs have vital effects on innate immune functions through different mechanisms. TEs and virus-derived proteins have been repeatedly selected as immune proteins that typically limit viruses through dominant-negative activity in animals [Citation61–63]. The non-coding transcripts derived from TEs readily form double-stranded RNAs or DNAs with immunostimulatory effects [Citation64,Citation65]. TEs can also regulate the expression of IFN-inducible genes by acting as regulatory elements [Citation66–68]. Recent reports have indicated that some transposons such as SINEs and LINEs can be activated and upregulated during infection with DNA viruses [Citation69]. Transcription of endogenous retrotransposon elements can induce an IFN response in the infected cells [Citation70]. Our results showed that the Xiang-R population presented higher interferon levels (Additional file17: Figure S6) and antiviral ability compared to the Xiang-S population due to the presence of TEs activated during ASFV infection and the induced interferon response, which might reduce the susceptibility to African swine fever virus.
Compared with a previous report on host gene expression patterns in pigs infected with non-lethal ASFV using RNA-Seq technology [Citation71], 11 candidate genes in our study were also upregulated in ASFV-infected pigs, including GHR upregulated in the liver, JAK2 upregulated in peripheral blood mononuclear cells (PBMC), and ANTXR1, CTBP2, EPB41L4B, FARP1, FRMPD4, GPC4, MYO10, PRINKLE2, and THSD4 upregulated in the submandibular lymph nodes (SMLN). TE elements were present in all variants except for SVs in MYO10, suggesting that the presence of SVs in these genes might be the reasons for their differential expression after ASF infection in pigs.
Selective sweep analysis based on FST and θπ ratio could screen many small selective regions. To detect the genome-wide selection signatures, we used whole genome re-sequencing data to calculate the pairwise FST and θπ in 100 kb sliding windows with a step length of 10 kb across the chromosomes between the Xiang-R and Xiang-S groups. And the 11,957 windows (top 5 %) were selected for FST and θπ ratio calculation (Additional file 8-10: Table S8–10). The overlapping regions detected by both approaches (threshold, 5 %) were defined as key selection signatures. Finally, 1023 outlier windows were identified as the regions of selective sweeps with θπ ratio > 1.12 and FST >0.12, which contained 535 genes (Additional file 17: Figure S7 and Additional file 11: Table S11), including 271 known protein genes, and 251 novel genes. Notably, 45.7% of the selected regions were located on chromosomes 1, 13, and 14. Gene MECOM on chromosome 13 was subjected to a relatively strong selection pressure (Additional file 17: Figure S8). After comparing these selected genes with our population-stratified genes between the Xiang-R and Xiang-S groups, it was found that 29 genes overlapping with 42 SVs referred to these selective regions, which involved three candidate genes (GHR, PCLO, and HTR7) (Additional file 12: Table S12). Furthermore, functional enrichment analysis revealed that 30 selected genes were significantly enriched in pathways involved in viral infection, immune regulation, and antigen processing and presentation (Additional file 17: Figure S9 and Additional file 13: Table S13). The fact that some of the regions detected using selective sweep analysis overlapped with population-stratified SVs strengthens the possible involvement of genes that may affect the phenotypic traits of pig disease resistance.
Conclusions
Our study reveals that SVs play a crucial role in the evolutionary adaption of Xiang pigs to ASFV infections. The whole-genome SV dataset, consisting of 53,589 high-confidence SVs, provides a significant number of previously unresolved common variants in a surviving pig population infected with ASFV. In addition, we identified 135 protein-coding adaptive genes affected by 166 population-stratified SVs between the two groups of ASF-susceptible and -resistant pigs, revealing new evidence of adaptation to ASFV infection and improving our understanding of the genetic basis for the resistance of the Xiang pig breed to ASFV. Our research provides meaningful information regarding pig genomic variation, which could be used to uncover the relationship between genetic variation and disease resistance traits in pigs.
Methods
Animal and sample collection
In this study, the ASF-resistant pigs were found in Xiang pig breed and its cross on an ASF outbreak pig farm in Guizhou province after 6 August 2019. No ASFV genome-positive and 32 antibody-positive pigs were found in blood samples from the 34 survived pigs. To decipher the natural genetic variation in resistance to ASFV infection in pigs, six pigs resistant to ASF (4 Xiang pigs, 2 Xiang × Duroc × Berkshires) from the Xiang pig farm were bought to investigate the natural resistance to African swine fever. According to the guidance on measures to prevent and eradicate ASF in China, the pigs were assembled in a local quarantine facility after an outbreak of ASF occurred. The pig blood samples were tested by qPCR and ELISA and monitored by local veterinary authorities for three months. All six pigs were seropositive and negative on qPCR detection for ASFV genome without developing notable clinical signs after the outbreak. Information on the selected pigs was shown in Table S14 (Additional file 14: Table S14). Meanwhile, 6 Xiang pigs on this pig farm were sampled as susceptible individuals before the onset of the ASF, and their deaths were confirmed after the outbreak of the disease. These pigs were designed to be populations resistant to ASFV (Xiang-R) and susceptible to ASFV (Xiang-S), respectively.
In addition, another 24 samples from Yorkshire pigs (n = 12) and warthogs (n = 12) with Illumina reads were collected from the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject) as outer groups under the accession numbers of PRJNA550237 and PRJNA691462. Information on all the samples used in this study was presented in Table S15 (Additional file 15: Table S15).
Serological tests
Fresh peripheral blood was routinely sampled from the front cavity vein of each pig using an 18-gauge needle into heparin-plus vacuum blood collection tube (5 mL) and EDTA.Na2-plus vacuum blood collection tube (5 mL), and serum was obtained by centrifugation (1500 g, 4°C) for 30 min and refrigerated for later use. The African swine fever virus and immunoglobulin G antibodies against ASF virus were detected in serum samples by real-time PCR and an indirect enzyme-linked immunosorbent assay (ELISA) using the African swine fever virus nucleic acid real-time PCR rapid test kit and African swine fever antibody ELISA test kit (Wuhan Keqian Biology Co., Ltd.) according to the manufacturer’s instructions.
Besides that, the cytokines of IFN-α, IFN-β and IFN-γ were detected by porcine ELISA Kits (Solarbio, Beijing) and then analysed by a microplate reader. The experimental steps strictly followed the manufacturer’s recommendations.
Whole-genome re-sequencing and data processing
Genomic DNA was extracted from the peripheral blood leukocytes using a SQ Blood DNA Kit (OMEGA, USA) and fragmented into about 500 bp in size using an ultrasonic disruptor. Twelve DNA libraries were constructed using the Illumina DNA Prep library kit (Illumina, Australia) according to the manufacturer’s instructions and sequenced on an Illumina Hiseq 2500 platform with pair-end reads of 150 bp by BGI-Shenzhen, China. The whole-genome sequencing data were available from the NCBI (Information available at http://ftp.ebi.ac.uk/pub/databases/nextgen/).
The NGSQC Toolkit was used to filter adapters and low-quality reads from the raw sequencing data. Pig genome sequence of Sscrofa 11.1 was downloaded from Ensembl (http://ftp.ensembl.org/pub/release-90/fasta/sus_scrofa/dna/) as reference. The yielded clean reads were then mapped and aligned to the primary assembly of pig reference genome (Sscrofa 11.1) using BWA with the following parameters: “bwa mem -M -R.” The realignment data in SAM format files were converted to BAM format and then sorted using Samtools (v1.10) [Citation72].
SV calling and merging
To improve SV detection accuracy, three software programs were applied with default parameters for SV calling: Delly (v0.7.8) [Citation28], Manta (v1.6.0) [Citation29], and Dysgu [Citation30]. As TRA (translocation) with ambiguous location information, we only detected structural variations of inversion (INV), deletion (DEL), insertion (INS), and tandem duplication (DUP). Subsequently, all the SVs that displayed the same SV types were merged with the overlapping length of more than 50 bp and detected by at least two tools using SURVIVOR (v1.0.6) [Citation73] via the following parameters: “SURVIVOR merge files 1000 2 1 1 0 50.” To reduce the numbers of false-positive SVs, we filtered the SVs from only one sample. Data from chromosome Y were excluded to eliminate sex interference. Based on the pipeline suggestion in the previously studied [Citation24], a 2 Mb threshold was set for DELs and INSs, and 5 Mb for INVs and DUPs detection. Larger length of SVs were then discarded according to the thresholds.
Annotation and enrichment analysis of the population stratified SVs
The distribution of different genomic features was evaluated using the Variant Effect Predictor at the Ensembl website (http://www.ensembl.org/Tools/VEP) to track the impacts of variations on gene structure. To provide insight into the functional enrichment of the genes affected by the population-stratified SVs, GO and KEGG pathway analyses were performed using the KOBAS-intelligence (http://kobas.cbi.pku.edu.cn/) and the WEB-based GEne SeT AnaLysis Toolkit (https://www.webgestalt.org/).
Screening of repetitive elements in candidate SVs
To better evaluate the patterns of repetitive elements in candidate SVs, transposable element regions for the genome reference Sscrofa11.1 were downloaded from the UCSC Table browser (http://hgdownload.soe.ucsc.edu/goldenPath/susScr11/database/rmsk.txt.gz). The candidate SVs were then compared with the transposable elements via bedtools-intersect program [Citation74].
F-statistics (FST) of the detected SVs
The frequencies of the reference and alternate alleles were calculated at each locus within each defined population. To identify SVs contribution to population differentiation, the F-statistics (FST) was evaluated according to the previous studies [Citation75,Citation76].
Population structure analyses
To better understand the genetic relationships among the pig populations, we performed PCA, neighbour-joining (NJ) tree, and ADMIXTURE analysis. Vcftools (V0.1.16) [Citation77] and PLINK (V1.9) [Citation78] were used for data processing. PCA was performed using the R platform. A phylogenetic tree was established using the neighbour-joining (NJ) algorithm on the R platform and displayed with the interactive tree of life (iTOL) (V6.0). Population ancestry was estimated using the ADMIXTURE [Citation79] software with K set from 2 to 4, and the results were plotted using our local R scripts.
SNP calling and selective analysis
The Genome Analysis Toolkit (GATK) [Citation80] was applied to mark duplicated sequences of the aligned BAM files and call variants with default parameters. The INDELs in the VCF file were excluded and high-quality SNPs were identified using the following filtering criteria: QUAL > 30, OD > 5.0, S < 60.0, MQ > 40.0, MQRankSum > −12.5, ReadPosRankSum > −8.0, according to Wang et al. [Citation81]. The SNPs were functionally annotated using ANNOVAR [Citation82] software. In order to identify the candidate regions under positive selection in Xiang-R group, we calculated the fixation statistics (FST) and population nucleotide diversity ratio (θπ) according to the procedures described by Li et al. [Citation83].
PCR validation
Total of 28 predicted SVs were randomly selected for verification by pairwise PCR. Primers were designed from the peripheral regions outside of SV breakpoints (Additional file 16: Table S16). The amplicons from the PCR-positive samples were then sequenced using Sanger technology by Sangon Biotech (Shanghai) Co., Ltd. And the expected genotype was directly counted and manually checked based on the SV type, SV size, and genome position.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conceptualization, X.R, J.W.; data curation, F.Q., X.C.; investigation, S.L., X.N., S.H.; writing-the draft, F.Q.; writing-review and editing, X.R., J.W. All authors have read and approved the final manuscript and agree to be personally accountable for author’s own contributions.
Abbreviations
| ASF | = | African swine fever |
| ASFV | = | African swine fever virus |
| SV | = | structural variation |
| Xiang-R | = | ASF-resistant Xiang pig |
| Xiang-S | = | ASF-susceptible Xiang pig |
| FST | = | F-statistics |
| Kb | = | kilobases |
| Mb | = | megabases |
| ELISA | = | enzyme-linked immunosorbent assay |
| INV | = | inversion |
| DEL | = | deletion |
| INS | = | insertion |
| DUP | = | tandem dulpication |
| TRA | = | translocation |
| GO | = | gene ontology |
| KEGG | = | Kyoto Encyclopedia of Genes and Genomes |
| PCA | = | principal component analysis |
| VEP | = | Variant Effect Predictor |
| EIPA | = | 5-N-Ethyl-N-isopropyl |
| IPA-3 | = | 3-indolepropionic acid |
| PAK1 | = | p21- activated kinase 1 |
| DYN | = | dynasore |
| GPCR | = | G-protein coupled receptor |
| AP2 | = | adaptor protein 2 |
| PRE1 | = | porcine repetitive element 1 |
| IGF1R | = | insulin-like growth factor-1 receptor |
| RTK | = | receptor tyrosine kinase |
| IR | = | insulin receptor |
| RSV | = | respiratory syncytial virus |
| RBP | = | RNA binding protein |
| TE | = | transposable element. |
Availability of data and materials
All 12 samples of Yorkshire and 12 samples of warthogs under accession numbers PRJNA550237 and PRJNA691462 were downloaded from the NCBI BioProject database (Additional file 15:Table S15). The sequence data of Xiang pigs used in this study could be obtained from the NCBI Sequence Read Archive (SRA) database under accession number PRJNA1057488.
Ethics approval and consent to participate
All animal procedures have been approved from the Animal Care and Use Committee of Guizhou University (EAE-GZU-2019-E032) and were conducted in accordance with the National Research Council’s “Guidelines for the Care and Use of Experimental Animals” in China.
Supplemental Material
Download Zip (5.1 MB)Acknowledgements
We thank the National Natural Science Foundation of China (32360810, 31960641, and 31672390) for funding, along with the Ministry of Science and Technology of the People’s Republic of China (863 Program) (2013AA102503) and Department of Agriculture and Rural Affairs of Guizhou Province (HQDPPI-2022). We thank the Dashandi pig breeding farm, and Congjiang County, Guizhou province, for providing samples.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental data
Supplemental data for this article can be accessed online at https://doi.org/10.1080/21505594.2024.2382762.
Additional information
Funding
References
- Ravilov RK, Rizvanov AA, Mingaleev DN, et al. Viral vector vaccines against ASF: problems and Prospectives. Front Vet Sci. 2022;9:830244. doi: 10.3389/fvets.2022.830244
- Diaz C, Salat J, Brinek Kolarova D, et al. Examination of immunogenic properties of recombinant antigens based on p22 protein from African swine fever virus. J Vet Res. 2022;66(3):297–17. doi: 10.2478/jvetres-2022-0043
- Arias M, de la Torre A, Dixon L, et al. Approaches and perspectives for development of African swine fever virus vaccines. Vaccines (Basel). 2017;5(4):35. doi: 10.3390/vaccines5040035
- Lee C, Day J, Goodman SM, et al. Genetic origins and diversity of bushpigs from Madagascar (potamochoerus larvatus, family suidae). Sci Rep. 2020;10(1):20629. doi: 10.1038/s41598-020-77279-5
- Roger F, Ratovonjato J, Vola P, et al. Ornithodoros porcinus ticks, bushpigs, and African swine fever in Madagascar. Exp Appl Acarol. 2001;25(3):263–269. doi: 10.1023/a:1010687502145
- Wilkinson PJ, Pegram RG, Perry BD, et al. The distribution of African swine fever virus isolated from ornithodoros moubata in Zambia. Epidemiol Infect. 1988;101(3):547–564. doi: 10.1017/s0950268800029423
- Oh T, Nguyen TM, Ngo TTN, et al. Long-term follow-up of convalescent pigs and their offspring after an outbreak of acute African swine fever in Vietnam. Transbound Emerg Dis. 2021;68(6):3194–3199. doi: 10.1111/tbed.14276
- Nurmoja I, Petrov A, Breidenstein C, et al. Biological characterization of African swine fever virus genotype II strains from north-eastern Estonia in European wild boar. Transbound Emerg Dis. 2017;64(6):2034–2041. doi: 10.1111/tbed.12614
- Patrick BN, Machuka EM, Githae D, et al. Evidence for the presence of African swine fever virus in apparently healthy pigs in South-Kivu Province of the democratic Republic of Congo. Vet Microbiol. 2020;240:108521. doi: 10.1016/j.vetmic.2019.108521
- Abworo EO, Onzere C, Oluoch Amimo J, et al. Detection of African swine fever virus in the tissues of asymptomatic pigs in smallholder farming systems along the kenya-uganda border: implications for transmission in endemic areas and ASF surveillance in East Africa. J Gen Virol. 2017;98(7):1806–1814. doi: 10.1099/jgv.0.000848
- Netherton CL, Connell S, Benfield CTO, et al. The genetics of life and death: virus-host interactions underpinning resistance to African swine fever, a viral hemorrhagic disease. Front Genet. 2019;10:402. doi: 10.3389/fgene.2019.00402
- Stahl K, Sternberg-Lewerin S, Blome S, et al. Lack of evidence for long term carriers of African swine fever virus - a systematic review. Virus Res. 2019;272:197725. doi: 10.1016/j.virusres.2019.197725
- Arias M, Jurado C, Gallardo C, et al. Gaps in African swine fever: analysis and priorities. Transbound Emerg Dis. 2018;65(Suppl 1):235–247. doi: 10.1111/tbed.12695
- Palgrave CJ, Gilmour L, Lowden CS, et al. Species-specific variation in RELA underlies differences in NF-kappaB activity: a potential role in African swine fever pathogenesis. J Virol. 2011;85(12):6008–6014. doi: 10.1128/JVI.00331-11
- Liu Y, Zhang X, Qi W, et al. Prevention and control strategies of african swine fever and progress on pig farm repopulation in China. Viruses. 2021;13(12):2552. doi: 10.3390/v13122552
- Elbers JP, Brown MB, Taylor SS. Identifying genome-wide immune gene variation underlying infectious disease in wildlife populations - a next generation sequencing approach in the gopher tortoise. BMC Genomics. 2018;19(1):64. doi: 10.1186/s12864-018-4452-0
- Bai H, Sun Y, Liu N, et al. Genome-wide detection of CNVs associated with beak deformity in chickens using high-density 600K SNP arrays. Anim Genet. 2018;49(3):226–236. doi: 10.1111/age.12652
- Sterken MG, van Sluijs L, Wang YA, et al. Punctuated loci on chromosome IV determine natural variation in orsay virus susceptibility of caenorhabditis elegans strains bristol N2 and hawaiian CB4856. J Virol. 2021;95(12). doi: 10.1128/JVI.02430-20
- Heim MH, Bochud PY, George J. Host - hepatitis C viral interactions: the role of genetics. J Hepatol. 2016;65(1 Suppl):S22–S32. doi: 10.1016/j.jhep.2016.07.037
- Rasmussen AL, Okumura A, Ferris MT, et al. Host genetic diversity enables Ebola hemorrhagic fever pathogenesis and resistance. Science. 2014;346(6212):987–991. doi: 10.1126/science.1259595
- Dean M, Carrington M, Winkler C, et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia growth and development study, multicenter AIDS cohort study, multicenter hemophilia cohort study, San Francisco City cohort, ALIVE study. Science. 1996;273(5283):1856–1862. doi: 10.1126/science.273.5283.1856
- Yun SI, Song BH, Frank JC, et al. Functional genomics and immunologic tools: the impact of viral and host genetic variations on the outcome of zika virus infection. Viruses. 2018;10(8):422. doi: 10.3390/v10080422
- Hou Y, Zhao J, Martin W, et al. New insights into genetic susceptibility of COVID-19: an ACE2 and TMPRSS2 polymorphism analysis. BMC Med. 2020;18(1). doi: 10.1186/s12916-020-01673-z
- Wu Z, Jiang Z, Li T, et al. Structural variants in the Chinese population and their impact on phenotypes, diseases and population adaptation. Nat Commun. 2021;12(1):6501. doi: 10.1038/s41467-021-26856-x
- Weischenfeldt J, Symmons O, Spitz F, et al. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat Rev Genet. 2013;14(2):125–138. doi: 10.1038/nrg3373
- Wu Y, Fan H, Jing S, et al. A genome-wide scan for copy number variations using high-density single nucleotide polymorphism array in simmental cattle. Anim Genet. 2015;46(3):289–298. doi: 10.1111/age.12288
- Liu C, Ran X, Yu C, et al. Whole-genome analysis of structural variations between Xiang pigs with larger litter sizes and those with smaller litter sizes. Genomics. 2019;111(3):310–319. doi: 10.1016/j.ygeno.2018.02.005
- Rausch T, Zichner T, Schlattl A, et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28(18):i333–i9. doi: 10.1093/bioinformatics/bts378
- Chen X, Schulz-Trieglaff O, Shaw R, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32(8):1220–1222. doi: 10.1093/bioinformatics/btv710
- Cleal K, Baird DM. Dysgu: efficient structural variant calling using short or long reads. Nucleic Acids Res. 2022;50(9):e53. doi: 10.1093/nar/gkac039
- Patel RK, Jain M, Liu Z. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLOS ONE. 2012;7(2):e30619. doi: 10.1371/journal.pone.0030619
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324
- Liu C, Ran X, Wang J, et al. Detection of genomic structural variations in Guizhou indigenous pigs and the comparison with other breeds. PLOS ONE. 2018;13(3):e0194282. doi: 10.1371/journal.pone.0194282
- Quan C, Li Y, Liu X, et al. Characterization of structural variation in tibetans reveals new evidence of high-altitude adaptation and introgression. Genome Biol. 2021;22(1):159. doi: 10.1186/s13059-021-02382-3
- Muangkram Y, Sukmak M, Wajjwalku W. Phylogeographic analysis of African swine fever virus based on the p72 gene sequence. Genet Mol Res. 2015;14(2):4566–4574. doi: 10.4238/2015.May.4.15
- Eble PL, Hagenaars TJ, Weesendorp E, et al. Transmission of African swine fever virus via carrier (survivor) pigs does occur. Vet Microbiol. 2019;237:108345. doi: 10.1016/j.vetmic.2019.06.018
- Sun E, Huang L, Zhang X, et al. Genotype I African swine fever viruses emerged in domestic pigs in China and caused chronic infection. Emerg Microbes Infect. 2021;10(1):2183–2193. doi: 10.1080/22221751.2021.1999779
- Mulumba-Mfumu LK, Goatley LC, Saegerman C, et al. Immunization of African indigenous pigs with attenuated genotype I African swine fever virus OURT88/3 induces protection against challenge with virulent strains of genotype I. Transbound Emerg Dis. 2016;63(5):e323–7. doi: 10.1111/tbed.12303
- Beyter D, Ingimundardottir H, Oddsson A, et al. Long-read sequencing of 3,622 icelanders provides insight into the role of structural variants in human diseases and other traits. Nat Genet. 2021;53(6):779–786. doi: 10.1038/s41588-021-00865-4
- De Coster W, De Rijk P, De Roeck A, et al. Structural variants identified by oxford nanopore PromethION sequencing of the human genome. Genome Res. 2019;29(7):1178–1187. doi: 10.1101/gr.244939.118
- Li Z, Chen W, Li X, et al. Transcriptome profiling in swine macrophages infected with African swine fever virus (ASFV) uncovers the complex and close relationship with host. Pathogens. 2022;11(12):1411. doi: 10.3390/pathogens11121411
- Liu W, Tang D, Xu XX, et al. How physical factors coordinate virus infection: a perspective from mechanobiology. Front Bioeng Biotechnol. 2021;9:764516. doi: 10.3389/fbioe.2021.764516
- Dixon LK, Islam M, Nash R, et al. African swine fever virus evasion of host defences. Virus Res. 2019;266:25–33. doi: 10.1016/j.virusres.2019.04.002
- Alonso C, Galindo I, Cuesta-Geijo MA, et al. African swine fever virus-cell interactions: from virus entry to cell survival. Virus Res. 2013;173(1):42–57. doi: 10.1016/j.virusres.2012.12.006
- Sanchez EG, Perez-Nunez D, Revilla Y. Mechanisms of entry and endosomal pathway of African swine fever virus. Vaccines (Basel). 2017;5(4):42. doi: 10.3390/vaccines5040042
- Zheng X, Nie S, Feng WH. Regulation of antiviral immune response by African swine fever virus (ASFV). Virol Sin. 2022;37(2):157–167. doi: 10.1016/j.virs.2022.03.006
- Hernaez B, Guerra M, Salas ML, et al. African swine fever virus undergoes outer envelope disruption, capsid disassembly and inner envelope fusion before core release from multivesicular endosomes. PloS Pathog. 2016;12(4):e1005595. doi: 10.1371/journal.ppat.1005595
- Wang Q, Nowak CM, Korde A, et al. Compromised RNA polymerase III complex assembly leads to local alterations of intergenic RNA polymerase II transcription in Saccharomyces cerevisiae. BMC Biol. 2014;12(1):89. doi: 10.1186/s12915-014-0089-x
- Downs LM, Mellersh CS, Wade C. An intronic SINE insertion in FAM161A that causes exon-skipping is associated with progressive retinal atrophy in Tibetan spaniels and Tibetan terriers. PLoS One. 2014;9(4):e93990. doi: 10.1371/journal.pone.0093990
- Maquat LE. Short interspersed nuclear element (SINE)-mediated post-transcriptional effects on human and mouse gene expression: SINE-UP for active duty. Philos Trans R Soc Lond B Biol Sci. 2020;375(1795):20190344. doi: 10.1098/rstb.2019.0344
- Jean-Charles PY, Kaur S, Shenoy SK. G protein-coupled receptor signaling through beta-arrestin-dependent mechanisms. J Cardiovasc Pharmacol. 2017;70(3):142–158. doi: 10.1097/FJC.0000000000000482
- Gurevich VV, Gurevich EV. GPCR signaling regulation: the role of GRKs and arrestins. Front Pharmacol. 2019;10:125. doi: 10.3389/fphar.2019.00125
- Pierce KL, Lefkowitz RJ. Classical and new roles of beta-arrestins in the regulation of G-protein-coupled receptors. Nat Rev Neurosci. 2001;2(10):727–733. doi: 10.1038/35094577
- Zheng M, Guo T, Yang B, et al. Origin, evolution, and tissue-specific functions of the porcine repetitive element 1. Genet Sel Evol. 2022;54(1):54. doi: 10.1186/s12711-022-00745-3
- Griffiths CD, Bilawchuk LM, McDonough JE, et al. IGF1R is an entry receptor for respiratory syncytial virus. Nature. 2020;583(7817):615–619. doi: 10.1038/s41586-020-2369-7
- Chen Y, Qin H, Zheng L. Research progress on RNA-binding proteins in breast cancer. Front Oncol. 2022;12:974523. doi: 10.3389/fonc.2022.974523
- Stagsted LVW, O’Leary ET, Ebbesen KK, et al. The RNA-binding protein SFPQ preserves long-intron splicing and regulates circRNA biogenesis in mammals. Elife. 2021;10. doi: 10.7554/eLife.63088
- Yap AS, Duszyc K, Viasnoff V. Mechanosensing and mechanotransduction at cell–cell junctions. Cold Spring Harb Perspect Biol. 2018;10(8):a028761. doi: 10.1101/cshperspect.a028761
- Zhang K, Li S, Liu S, et al. Spatiotemporally orchestrated interactions between viral and cellular proteins involved in the entry of African swine fever virus. Viruses. 2021;13(12):2495. doi: 10.3390/v13122495
- Wang Y, Kang W, Yang W, et al. Structure of African swine fever virus and associated molecular mechanisms underlying infection and immunosuppression: a review. Front Immunol. 2021;12:715582. doi: 10.3389/fimmu.2021.715582
- Blanco-Melo D, Gifford RJ, Bieniasz PD. Co-option of an endogenous retrovirus envelope for host defense in hominid ancestors. Elife. 2017;6. doi: 10.7554/eLife.22519
- Mura M, Murcia P, Caporale M, et al. Late viral interference induced by transdominant gag of an endogenous retrovirus. Proc Natl Acad Sci U S A. 2004;101(30):11117–11122. doi: 10.1073/pnas.0402877101
- Young GR, Yap MW, Michaux JR, et al. Evolutionary journey of the retroviral restriction gene Fv1. Proc Natl Acad Sci USA. 2018;115(40):10130–10135. doi: 10.1073/pnas.1808516115
- Chiappinelli KB, Strissel PL, Desrichard A, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162(5):974–986. doi: 10.1016/j.cell.2015.07.011
- Schmidt N, Domingues P, Golebiowski F, et al. An influenza virus-triggered SUMO switch orchestrates co-opted endogenous retroviruses to stimulate host antiviral immunity. Proc Natl Acad Sci USA. 2019;116(35):17399–17408. doi: 10.1073/pnas.1907031116
- Kelly CJ, Cg C-M, Chuong EB. Ruminant-specific retrotransposons shape regulatory evolution of bovine immunity. Genome Res. 2022;32(8):1474–1486. doi: 10.1101/gr.276241.121
- Chuong EB, Elde NC, Feschotte C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science. 2016;351(6277):1083–1087. doi: 10.1126/science.aad5497
- Bogdan L, Barreiro L, Bourque G. Transposable elements have contributed human regulatory regions that are activated upon bacterial infection. Philos Trans R Soc Lond B Biol Sci. 2020;375(1795):20190332. doi: 10.1098/rstb.2019.0332
- Macchietto MG, Langlois RA, Shen SS. Virus-induced transposable element expression up-regulation in human and mouse host cells. Life Sci Alliance. 2020;3(2):e201900536. doi: 10.26508/lsa.201900536
- De Cecco M, Ito T, Petrashen AP, et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature. 2019;566(7742):73–78. doi: 10.1038/s41586-018-0784-9
- Feng W, Zhou L, Du H, et al. Transcriptome analysis reveals gene expression changes of pigs infected with non-lethal African swine fever virus. Genet Mol Biol. 2023;46(3):e20230037. doi: 10.1590/1678-4685-GMB-2023-0037
- Li H, Handsaker B, Wysoker A, et al. The sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352
- Jeffares DC, Jolly C, Hoti M, et al. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat Commun. 2017;8(1):14061. doi: 10.1038/ncomms14061
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842. doi: 10.1093/bioinformatics/btq033
- Poptsova M, Banerjee S, Gokcumen O, et al. Impact of constitutional copy number variants on biological pathway evolution. BMC Evol Biol. 2013;13(1):19. doi: 10.1186/1471-2148-13-19
- Hu Y, Xia H, Li M, et al. Comparative analyses of copy number variations between Bos taurus and bos indicus. BMC Genomics. 2020;21(1):682. doi: 10.1186/s12864-020-07097-6
- Danecek P, Auton A, Abecasis G, et al. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–2158. doi: 10.1093/bioinformatics/btr330
- Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795
- Falush D, Wirth T, Linz B, et al. Traces of human migrations in Helicobacter pylori populations. Science. 2003;299(5612):1582–1585. doi: 10.1126/science.1080857
- McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110
- Wang X, Ran X, Niu X, et al. Whole-genome sequence analysis reveals selection signatures for important economic traits in Xiang pigs. Sci Rep. 2022;12(1):11823. doi: 10.1038/s41598-022-14686-w
- Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603
- Li M, Tian S, Jin L, et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat Genet. 2013;45(12):1431–1438. doi: 10.1038/ng.2811