
ABSTRACT
Nephrotic syndrome (NS) describes a group of kidney disorders in which there is injury to podocyte cells, specialized cells within the kidney's glomerular filtration barrier, allowing proteins to leak into the urine. Three mutations in ARHGDIA, which encodes Rho GDP dissociation inhibitor α (GDIα), have been reported in patients with heritable NS and encode the following amino acid changes: ΔD185, R120X, and G173V. To investigate the impact of these mutations on podocyte function, endogenous GDIα was knocked-down in cultured podocytes by shRNA and then the cells were re-transfected with wild-type or mutant GDIα constructs. Among the 3 prototypical Rho-GTPases, Rac1 was markedly hyperactivated in podocytes with any of the 3 mutant forms of GDIα while the activation of RhoA and Cdc42 was modest and variable. All three mutant GDIα proteins resulted in slow podocyte motility, suggesting that podocytes are sensitive to the relative balance of Rho-GTPase activity. In ΔD185 podocytes, both random and directional movements were impaired and kymograph analysis of the leading edge showed increased protrusion and retraction of leading edge (phase switching). The mutant podocytes also showed impaired actin polymerization, smaller cell size, and increased cellular projections. In the developing kidney, GDIα expression increased as podocytes matured. Conversely, active Rac1 was detected only in immature, but not in mature, podocytes. The results indicate that GDIα has a critical role in suppressing Rac1 activity in mature podocytes, to prevent podocyte injury and nephrotic syndrome.
Nephrotic syndrome (NS) is a class of kidney diseases in which the glomerular filtration barrier is injured, thereby allowing proteins to leak out of the glomerular capillaries into the urine. This leads to a low level of albumin in the serum (hypoalbuminemia) and edema. The kidney's glomerular filtration barrier consists of 3 basic components: fenestrated endothelial cells, acellular glomerular basement membrane, and specialized epithelial cells called podocytes. The podocytes have actin-rich finger-like projections, called foot processes. Foot processes from adjacent podocytes interdigitate, tightly surrounding glomerular capillaries to help retain proteins in the capillaries. Abnormalities in the podocyte actin cytoskeleton can cause nephrotic syndrome.Citation1,2
NS can be either genetic or acquired. In both cases, the primary defect is at the glomerular filtration barrier and in particular, most cases of NS are caused by abnormal morphology and function of podocytes.Citation3,4 NS with an onset within the first 3 months of life is called congenital nephrotic syndrome.Citation5 It is known that the majority of congenital NS are genetic and arise from mutations in NPHS1 (nephrin), NPHS2 (podocin), LAMB2 (laminin), WT1 (Wilms tumor suppressor 1), or PLCE1 (phospholipase C ϵ).Citation6,7 As the age on onset increases, the probability of a genetic cause decreases whereas the probability of an acquired form increases; however, there are some cases in which genetic NS presents during adult life (e.g. ACTN4, TRPC6, INF2).Citation8-10 We and others reported recently that mutations in ARHGDIA, which encodes Rho GDP dissociation inhibitor α (GDIα), can cause congenital or infantile NS.Citation11,12
Rho-family small GTPases (Rho-GTPases) are critical regulators of the actin cytoskeleton and Rac1, Cdc42, and RhoA are the 3 prototypes. GDIα regulates Rho-GTPases by sequestering them in their inactive, GDP-bound state in the cytosol.Citation13 Thus far, 3 GDIα mutations have been identified in patients with NS: ΔD185, R120X, and G173V. ΔD185 is an in-frame deletion, which removes one of the 3 consecutive aspartic acids.Citation11 G173V is a missense mutation causing one amino acid substitution while R120X is a misssense mutation that causes frame-shift and premature truncation.Citation12 Patients with ΔD185 and R120X mutations presented with congenital NS, whereas the patient with the G173V mutation had a later onset (1 – 2.4 years). Previous studies suggested that all the 3 mutations are loss-of-function mutations.Citation11,12 However, studies on the R120X and G173V mutations were performed using human podocytes in which the mutated GDIα was overexpressed, but the endogenous GDIα was still present.Citation12 Furthermore, the ΔD185 GDIα has not yet been studied in podocytes.Citation11 The goal of the current study was to establish the impact of each mutation on podocyte morphology and function by creating podocytes that express mutant GDIαs after the endogenous GDIα has been knocked down.
Results
The ΔD185 and R120X mutant proteins abolish the interaction between RhoGDIα and the Rho-GTPases
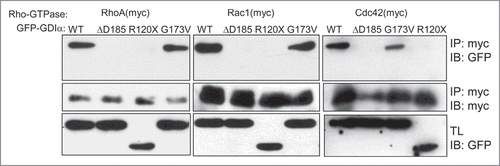
Since all 3 GDIα mutations have been reported to be loss-of-function mutations,Citation11,12 we determined whether the mutant forms of GDIα proteins can bind to their downstream Rho-GTPases. HEK293T cells were transiently transfected with one of the myc-tagged Rho-GTPases (either RhoA, Rac1, or Cdc42) and either a wild-type (WT) or one of the mutated forms of GDIα tagged with green fluorescent protein (GFP). Rho-GTPases were immunoprecipitated by anti-myc antibody and GDIα was detected by anti-GFP antibody (). The WT GDIα co-immunoprecipitated with the all 3 Rho-GTPases, whereas the ΔD185 GDIα and R120X GDIα did not, suggesting that these mutations impair the interaction between the Rho-GTPases and GDIα. Note that the R120X mutation results in a premature truncation that gives rise to a smaller protein, as seen on the Western Blot (). In contrast, the G173V GDIα co-immunoprecipitated with all 3 Rho-GTPases. Thus, the ΔD185 GDIα and R120X GDIα have impaired interaction with Rho-GTPases while the G173V GDIα appears to interact with the Rho-GTPases.
Figure 1. The ΔD185 and R120X mutant proteins abolish the interaction between RhoGDIα and the Rho-GTPases. HEK293T cells were transiently transfected with a myc-tagged Rho-GTPase and GFP-GDIα (either WT or one of the mutated forms). Cell lysates were immunoprecipitated using anti-myc antibody. Both the precipitates and the total lysates (TL) were immunoblotted for GFP (GDIα). WT GDIα and G173V GDIα co-immunoprecipitated with all 3 Rho-GTPases, but the ΔD185 GDIα and R120X GDIα did not.
Establishment of GDIα knockdown and mutant GDIα mouse podocytes
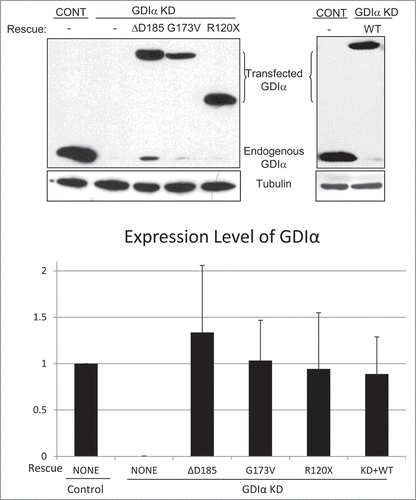
To study the consequence of each GDIα mutant protein in podocytes, we next created podocyte lines that express mutant GDIαs. First, the endogenous GDIα was stably knocked down in mouse podocytes using an shRNA (GDIα KD). The knockdown was efficient with minimal expression of GDIα protein detected by immunoblot (). Control mouse podocytes were also created using control shRNA that did not affect GDIα levels. GDIα KD cells were then stably transfected with WT or one of the mutant forms of GDIα tagged with GFP. The shRNA used for knockdown targets the 3′ UTR of the GDIα mRNA that is missing in the GFP-GDIα constructs; thus these constructs are resistant to the shRNA. Expression of the GFP-GDIα was confirmed by immunoblot for each line (). Thus, 3 cell lines were created in which the endogenous WT GDIα was replaced by GFP-tagged mutant forms of GDIα (ΔD185 GDIα, G173V GDIα, and R120X GDIα podocytes) or of WT GDIα (WT GDIα-GFP).
Figure 2. Establishment of GDIα knockdown and GDIα mutant mouse podocyte lines. Mouse podocytes were transduced with control shRNA (control) or GDIα shRNA (GDIα KD) and selected by puromycin. Total lysates were immunoblotted for GDIα, showing effective GDIα KD. The GDIα KD cells were then stably transfected with GFP-tagged WT GDIα (right) or GFP-tagged mutant GDIα (left). The cells with GDIα re-expression were selected with puromycin and G418, lysed and immunoblotted for GDIα. The blot shows effective replacement of endogenous GDIα with GFP-tagged WT or mutant GDIα. The image shown is representative of at least 3 experiments.
Rac1 remains hyperactivated in presence of mutant GDIα proteins
GDIα binds to the Rho-GTPases and maintains them in their inactive states.Citation13 To determine whether the GDIα mutations affect this function, we next quantified basal Rho-GTPase activity in the mouse podocyte lines established above using standard pull-down assays for the active Rho-GTPases. Most strikingly, Rac1 activity was markedly increased in GDIα KD podocytes compared to that in control podocytes (). This hyperactivation was not attenuated by expression of any of the mutant GDIαs. In contrast, the WT GDIα-GFP podocytes showed much less Rac1 activity compared with GDIα KD podocytes. Minimal Rac1 hyperactivation was observed in the WT GDIα-GFP podocytes that was nonetheless significantly higher compared to control podocytes (). This may be attributed to the lower expression level of the transfected WT GDIα-GFP compared to that of the endogenous WT GDIα ().
Figure 3. Rac1 remains hyperactivated in presence of mutant GDIα proteins. Active Rac1, RhoA, and Cdc42 were pulled down and immunoblotted in cell lines as shown (A). Bands were quantified by densitometry and normalized to tubulin (as shown in ) and to the control cell line (B). Rac1 activity was consistently increased in the GDIα KD and mutant GDIα cells compared to control cells. This hyperactivity was markedly diminished in cells rescued with WT GDIα, but nonetheless remained significantly higher than control. RhoA and Cdc42 activity were also increased in the GDIα KD cells, but they were decreased to control levels in the cells rescued with WT GDIα. There was a general increase in RhoA and Cdc42 activity in the cells expressing the mutant GDIαs; however, due to great variability, only the RhoA hyperactivity in the ΔD185 GDIα was statistically significant, as compared with control cells. The blots shown are representative of at least 3 experiments. *p < 0.05 vs control, n = 3–6.
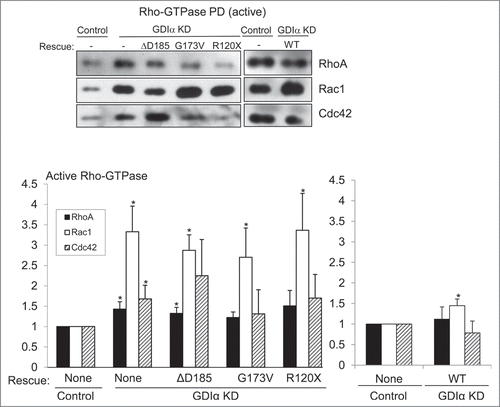
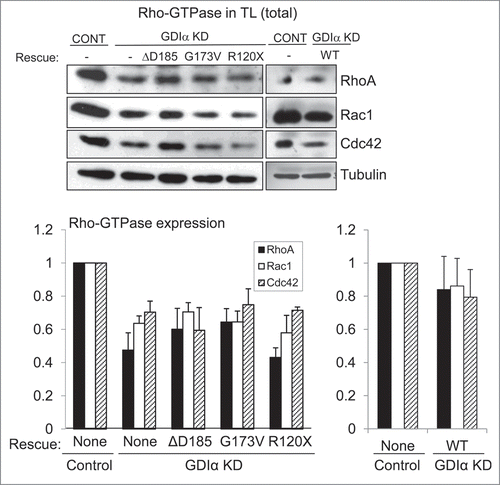
Figure 4. Mutant GDIα proteins do not protect the Rho-GTPases from proteosomal degradation. Western analysis of protein lysates from each mouse podocyte cell line were immunoblotted for Rac1, RhoA, and Cdc42. Total levels of Rho-GTPases were lower in the GDIα KD and the mutant GDIα cells. The blot shown above is representative of at least 3 experiments. Re-expression of the WT GDIα restored Rho-GTPase levels, similar to those in the control levels. Equal loading was confirmed by tubulin. *p < 0.05 vs control, n = 3–6 **p < 0.05 n = 4.
The GDIα KD podocytes also showed RhoA and Cdc42 hyperactivity that was statistically significant when compared with control podocytes (). However the degree of RhoA/Cdc42 hyperactivation was much less striking compared with Rac1 and re-expression of the WT GDIα-GFP decreased RhoA/Cdc42 activity (). Podocytes with the mutant forms of GDIα generally had higher RhoA and Cdc42 activities compared to control podocytes; however the degree of hyperactivation was somewhat variable and therefore, only the RhoA activity in ΔD185 GDIα podocytes remained statistically different compared with control. Thus, the loss of GDIα in podocytes had the strongest impact on Rac1 activity among the 3 prototypical Rho-GTPases. The three disease-causing GDIα mutants failed to suppress the high Rac1 activity found in GDIα KD podocytes, demonstrating that the abnormal GDIα proteins are unable to bind to Rac1 and inactivate it.
Mutant GDIα proteins do not protect the Rho-GTPases from proteosomal degradation
It was reported previously that Rho-GTPases bound to GDIα are protected from proteosomal degradation.Citation14,15 Thus, we examined the total levels of Rho-GTPases in the various podocyte lines. The GDIα KD podocytes had decreased levels of Rac1, RhoA, and Cdc42 compared with control podocytes (). This decrease persisted in podocytes with mutant GDIαs, indicating that the mutant forms of GDIα fail to protect the Rho-GTPases from proteosomal degradation, even in the presence of the G173V protein that is able to maintain binding to Rho-GTPases. In contrast, rescue with the WT GDIα –GFP restored the expression levels of Rho-GTPases (). The Rho-GTPase levels tended to remain lower in WT GDIα-GFP podocytes compared with control but the differences were not statistically significant. The inability of the mutant forms of GDIα to prevent proteosomal degradation of Rho-GTPases further supports the argument that these mutations result in a loss-of-function.
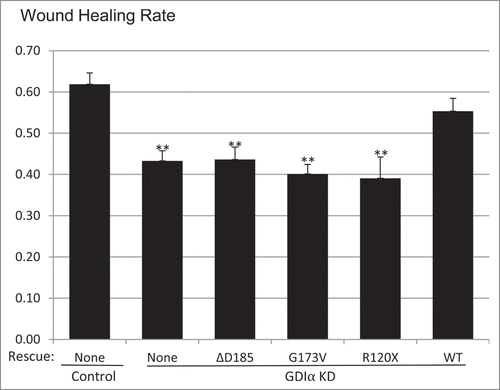
Mutant GDIα podocytes show decreased motility
It has been suggested that altered motility of podocytes, either increased or decreased, contributes to podocyte dysfunction and proteinuria.Citation16-18 Thus we next studied the motility of the mutant podocyte cell lines, using a wound healing assay. The GDIα KD podocytes showed decreased wound healing rates as compared with control (). This impaired motility was not restored in cells with the mutant forms of GDIα. The GDIα KD and the mutant podocytes' wound healing rates showed statistical significance when compared to the control, but fell short of statistical significance when compared to the WT GDIα-GFP podocytes. However, there was a clear trend of decreasing wound healing rate in the GDIα KD and the mutant GDIα podocytes.
Figure 5. Mutant GDIα podocytes show decreased motility. Wound healing assays (described in Methods) show that the GDIα KD and the mutant GDIα podocytes had decreased motility. Re-expression of the WT GDIα-GFP rescued the GDIα KD cells from this phenotype. Wound healing rate was measured as the fraction of the scratched area which has recuperated. **p < 0.01 vs. control, n = 7–44
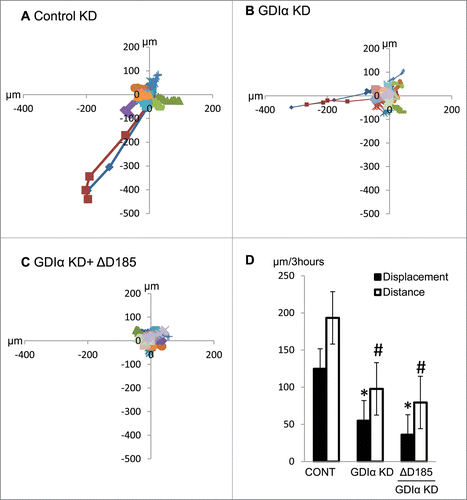
GDIα knockdown impairs coordinated movement of podocytes
A decrease in wound healing may reflect either impaired motility in general or impaired directional movement. To determine the nature of motility dysfunction in mutant podocytes, podocytes were stimulated with EGF over 3 hours and then the cells' movements were tracked by live cell video imaging. Both the distance of the trajectory and the displacement from the starting point were tracked. Both the GDIα KD and the ΔD185 GDIα podocytes traveled a shorter distance, resulting in less displacement from the starting point and confirming their reduced motility ().
Figure 6. GDIα KD and ΔD185 GDIα podocytes have impaired coordinated movement and overall motility. Cells were stimulated with EGF (100nM) for 3 hours and then tracked by live cell video imaging. (A) (B) and (C) shows the trajectory of control, GDIα KD, and ΔD185 GDIα cells respectively. Axes signify distance from starting point (in μm); different colors represent individual units. (D) shows the total cell displacement and distance traveled over 3 hours in μm. *p < 0.05 vs control n = 12–27 # p < 0.05 vs control n = 3.
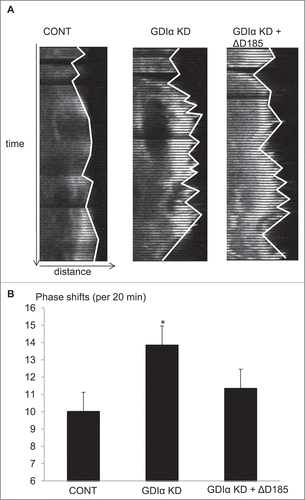
In 2011, Akilesh et al. used kymographic analysis to show protrusion and retraction of the leading edge of the cell (phase switching) in podocytes.Citation18 Similarly, kymographic analysis was used to examine the behavior of the leading edge of the GDIα KD and ΔD185 GDIα podocytes. Podocytes transfected with mCherry-actin were stimulated with EGF and images were captured every 20 seconds over 20 minutes using fluorescent microscopy. Kymographs were generated from the leading edge of the cells as described in the methods and the number of phase shifts was counted manually. The GDIα KD podocytes had significantly more phase shifts than control cells (). The ΔD185 GDIα cells also had more phase shifts than the control cells, although the difference was not statistically significant (). The results suggest that dis-coordinated actin remodeling at the leading edge resulted in ineffective movement of the GDIα KD and mutant podocytes.
Figure 7. Kymographic analysis shows GDIα KD and ΔD185 GDIα cells have ineffective directional movement. (A) Podocytes were transfected with mCherry-actin and stimulated with EGF (100nM). The pictures show the leading edge of cells over 20 min. (B) Images of the podocytes were captured every 20 seconds over 20 minutes and the total number of phase shifts was counted. The GDIα KD cells had more frequent phase shifts than control cells, reflective of ineffective directional movement. The ΔD185 GDIα cells also had more phase shifts than control; however, the difference was not statistically significant. *p < 0.05 vs control n = 3, 3–4 cells examined per experiment and 1–4 ruffling areas were analyzed per cell.
Mutant GDIα podocytes have decreased actin polymerization, smaller cell size and more cellular protrusions
A well-organized actin cytoskeleton within foot processes is the hallmark of healthy podocytes. To determine the impact of GDIα mutations on the actin cytoskeleton, the degree of actin polymerization was assessed by quantifying the ratio of filamentous (F) to globular (G) actin. The F/G-actin ratio was significantly lower in GDIα KD podocytes as compared to both control and WT GDIα-GFP podocytes; the ratio was not restored by expression of the 3 GDIα mutants (). To our surprise, when we transfected mouse podocytes with constitutively-active Rac1, the F/G actin ratio increased compared to control mouse podocytes transfected with GFP alone (control: 1.67 ± 0.071 n = 34, CA-Rac1: 2.65 ± 0.138 n = 27)(see below for discussion).
Figure 8. Mutant GDIα podocytes have decreased actin polymerization, smaller cell size, and more cellular protrusions. (A) Actin polymerization was quantified using the ratio of filamentous (F) to globular (G) actin and values were normalized to control. GDIα KD and mutant GDIα podocytes had decreased F/G ratios (n = 60–126 cells from 7 experiments). Re-expression of WT GDIα-GFP rescued the F/G ratio (n = 56–82 cells from 4 experiments). (B) Graphic representation of podocytes' average surface area. GDIα KD and GDIα mutant podocytes had decreased surface area (n = 40–74 from 4 experiments). Re-expression of WT GDIα restored cell size (n = 55–81 cells from 4 experiments). (C) Graphical representation of number of cellular projections. The GDIα KD podocytes had more cellular protrusions than the control podocytes, although the difference was not statistically significant. However, the 3 mutant GDIαs had increased number of cellular protrusions that did reach statistical significance. Re-expression of WT GDIα decreased the number of projections back to control levels. *p < 0.05 vs control **p < 0.001 vs control. Between 49 and 74 cells quantified.
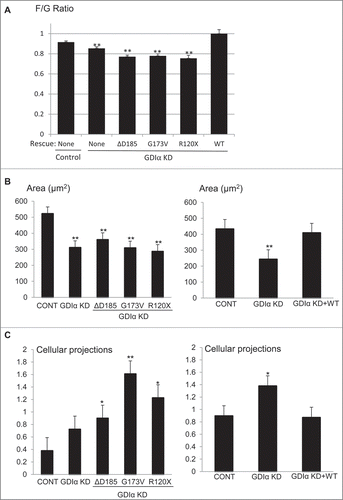
Actin polymerization is known to be necessary for the spreading of the cellCitation19; thus, we hypothesized that a decreased F/G ratio would give rise to smaller cell size. Indeed, when the cross-sectional area of the cells was quantified, GDIα KD cells had smaller areas as compared with control cells. Similar to the F/G actin ratio, the cell size was restored by the re-expression of the WT GDIα but not by the 3 GDIα mutants ().
As we assessed the morphology of the cells, we noted that GDIα KD and mutant GDIα podocytes had more cellular protrusions. Thus, we next quantified the number of cellular protrusions as described in the Methods. The GDIα KD podocytes had more cellular protrusions as compared with control cells, although the difference did not reach statistical significance (). Re-expression of the WT GDIα decreased the number of cellular protrusions to the level of control podocytes. In striking contrast, each of the mutant GDIα podocyte lines exhibited an even greater number of cellular protrusions than observed in control cells.
GDIα is upregulated as podocytes mature in vivo
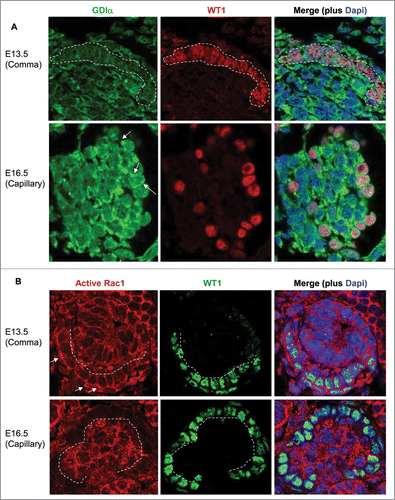
To determine if expression of GDIα correlates with podocyte maturation, we analyzed GDIα mRNA expression in podocytes during different stages of glomerular development using the microarray data from the Genito Urinary Development Molecular Anatomy Project (GUDMAP). Compared to podocytes from embryonic day (E) 13.5 mouse kidneys, GDIα mRNA expression increased by 3.5 fold and 4 fold in E15.5 and adult kidneys, respectively (). We next studied the expression of GDIα by performing immunohistochemistry on embryonic mouse kidneys. In WT1-positive podocyte precursor cells found in immature (comma-shaped) glomeruli, GDIα protein expression was minimal (). In contrast, GDIα protein was clearly observed in the cytosol of podocytes in a more developed (capillary stage) glomerulus (). A similar analysis of embryonic kidneys was performed using an antibody that recognizes the active form of Rac1, but not the inactive form. Active Rac1 protein was clearly detected at the plasma membrane in WT1-positive podocyte precursors in a comma-shaped glomerulus (). However, in the capillary stage glomerulus, active Rac1 staining was limited to the center of the glomerulus (where developing capillaries reside) and the podocytes were virtually devoid of positive signal. The results suggest that GDIα protein is upregulated as podocytes mature, whereas Rac1 activity, although high at an early developmental stage, decreases as podocytes mature. It is likely that the mutant GDIα fails to suppress the Rac1 activity at a later stage of podocyte development, thereby leading to abnormal maturation of podocytes.
Figure 9. GDIα mRNA expression in podocytes is increased during kidney development. FlexArray analysis of microarray data in podocytes isolated from developing mouse kidneys at embryonic day (E) 13.5, 15.5 and adult kidneys showed that GDIα mRNA increases during development (n = 3 for each stage). *p < 0.01.
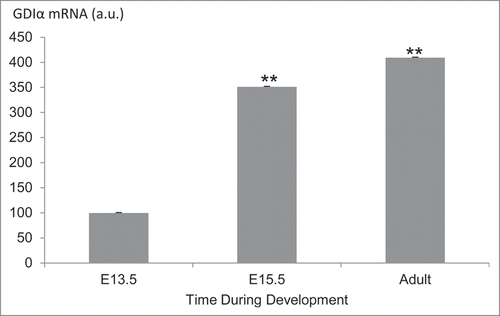
Figure 10. GDIα protein expression increases during kidney development, whereas Rac1 activity decreases. (A) Mouse embryonic kidney sections underwent immunofluorescent analysis with anti-GDIα antibody, WT1 antibody and Dapi. Dotted lines surround podocyte precursors in E13.5 pictures. Arrows show the positive staining for GDIα protein. GDIα protein expression in podocytes was higher at E16.5 than E13.5 (60x magnification). (B) Mouse embryonic kidneys were stained with an anti-WT1 antibody and an antibody that recognizes the active Rac1. There is less active Rac1 at E16.5 than at E13.5. Dotted lines mark the border between podocytes and developing capillaries. Arrows indicate areas of high Rac1 activity at E13.5.
Discussion
The key finding of the current study is that all 3 NS-causing GDIα mutations result in consistent and prominent Rac1 hyperactivity in podocytes, while hyperactivation of other Rho-GTPases (i.e. RhoA and Cdc42) was much less striking and inconsistent (). This is in accordance with the current prevailing notion that hyperactive Rac1 plays a role in podocyte dysfunction and NS. Our results demonstrate that podocytes respond to a loss of GDIα with a decrease in motility, a decrease in coordinated movements, less actin polymerization, smaller cellular size, and more cellular protrusions. We also demonstrate that GDIα is upregulated, while active Rac1 is downregulated as podocytes mature during development. These observations begin to provide important clues as to how hyperactivation of Rac1 may lead to podocyte injury and NS.
In 2008, Shibata et al. showed that deleting GDIα in mice led to Rac1 hyperactivation in the kidney along with proteinuria (protein leakage in the urine), that was alleviated by the Rac1 inhibitor.Citation20 Inducing the expression of constitutively active Rac1 specifically in podocytes in mice caused podocyte damage and proteinuria.Citation21 Loss-of-function mutations in GTPase-activating protein (GAP) Rho-GAP 24 (Arhgap24) allow increased Rac1 activity; such mutations have been found in patients with hereditary focal segmental glomerulosclerosis (FSGS), a kidney disease that is characterized by podocyte dysfunction.Citation18 Furthermore, human immunodeficiency virus type 1 (HIV1) protein Nef has been reported to activate Vav2, a Rac1 activator. This Rac1 hyperactivity leads to morphological changes in podocytes, which may play a role in HIV1-associated nephropathy (HIVAN).Citation22 While Rac1 hyperactivity in podocytes is harmful, Rac1 inactivity can actually be beneficial during acute kidney injury. Podocyte-specific Rac1 knockout mice live to adulthood and show no renal dysfunction or abnormal podocyte morphology.Citation23,24 In fact, Rac1 deletion actually renders podocytes resistant to the disruption of the morphology of foot processes induced by protamine sulfate.Citation23 The results of the current study are consistent with these reports and further highlight the pathological role of Rac1 activation in podocyte injury and NS.
Compared with Rac1, hyperactivation of RhoA and Cdc42 was much less impressive in GDIα KD and GDIα mutant podocytes. In fact, as compared to control podocytes, RhoA hyperactivation was statistically significant only in GDIα KD and ΔD185 GDIα cells and Cdc42 hyperactivity only in GDIα KD cells. Prominent hyperactivation of Rac1 and modest or no hyperactivation of RhoA result in imbalanced activity of RhoA and Rac1 during actin polymerization,Citation25 which manifested as decreased actin polymerization in the GDIα KD and mutant GDIα podocytes. The impaired actin polymerization likely contributes to podocyte injury and NS. Considering the GDIα KD podocytes and GDIα mutant podocytes have increased Rac1 activity, it was surprising to find that transiently expressing constitutively active Rac1 in mouse podocytes increased filamentous actin. A possible explanation could be that the endogenously hyperactivated Rho-GTPases found in the GDIα KD and mutant GDIα podocytes interfere with one another, giving rise to a different phenotype than the exogenously overexpressed constitutively active Rac1. When we studied actin polymerization in fibroblasts derived from a patient who carries the ΔD185 GDIα mutation or from control subjects, we noted that ΔD185 GDIα fibroblasts had more prominent stress fibers and quantitatively higher amounts of F-actin, as compared with control fibroblasts (not shown). Thus, the imbalance of Rho-GTPase activities and the impact on actin polymerization caused by GDIα loss may differ from cell to cell and podocytes may be particularly sensitive to the GDIα loss. This is consistent with the observation in humans Citation11,12 and in mice Citation26 that heavy proteinuria with kidney failure is the prominent phenotype observed with the systemic loss of GDIα.
Since Rac1 activation is generally associated with increased cell motility,Citation27,28 we were surprised to find that GDIα KD and mutant GDIα podocytes all showed impaired motility. We initially hypothesized that GDIα KD and mutant GDIα cells might move more randomly than directionally, resulting in overall slower wound healing. However, when live cells were video-tracked, we found that GDIα KD and ΔD185 GDIα podocytes moved significantly less than control cells in any direction. When the actin dynamics at the moving front was observed by kymograph, GDIα KD podocytes, and to a lesser extent ΔD185 podocytes, showed more phase shifts, suggesting that actin turnover is not translating into effective cell movement. It is noteworthy that both increased and decreased podocyte motility have been associated with the pathogenesis of NS.Citation16-18 Thus, impaired cell migration during podocyte development likely contributes to the pathogenesis of NS in patients with GDIα mutations. In addition, it may impair the ability of podocytes to adapt to environmental stressors such as increased capillary blood pressure.
The mutant podocytes also displayed other morphological abnormalities. We previously reported that the expression of constitutively active Rac1 in mouse podocytes increases the number of cellular processes.Citation29 Thus, it was not surprising that the GDIα KD and the mutant GDIα podocytes, which had increased Rac1 activity, also displayed an increased number of cellular projections. The increased number of cellular protrusions may result in an aberrant podocyte morphology that alters the morphology of the slit diaphragms, thereby allowing protein leakage into the urine. The GDIα KD and mutant GDIα podocytes also had decreased surface area; this may make it more difficult for cells to bridge the intercellular distance and form a proper junction with neighboring cells. All of these cellular changes may contribute to a leaky glomerular filtration barrier and proteinuria.
Among the 3 GDIα mutations that cause NS, G173V appears to have the least severe phenotype; patients with the G173V mutation developed NS later in childhood (at one year of age or older) compared to patients with the ΔD185 and R120X mutations (within the first 3 months of life). When we characterized the interaction of GDIα and Rho-GTPases biochemically, we found that the ΔD185 and the R120X mutations abrogated the interaction, while the G173V mutation did not. Despite the G173V GDIα binding to the Rho-GTPases, Rac1 remained hyperactivated in the G173V GDIα podocytes, indicating that G173V GDIα cannot properly inhibit Rac1. The ability of G173V GDIα to partially interact with Rho-GTPases likely explains the less severe clinical phenotype observed in these patients.
A previous report by Gee et al. studied the effects of the G173V and R120X GDIα mutations in podocytes and, similar to the current study, suggested that the 2 mutations cause loss-of-function.Citation12 However, there are several differences between that report and the current results. Firstly, they observed that the GDIα mutations increased podocyte motility, rather than impairing it. These differences may be explained by the WT GDIα present in the human podocytes used by Gee et al. since they overexpressed the mutants without knocking down the endogenous GDIα; while we first knocked down the endogenous WT GDIα and replaced it with the mutant forms of GDIα. Secondly, in their study, G173V failed to bind Rac1, RhoA or Cdc42; whereas we found that there is at least partial binding to these proteins. This can be explained by the different expression levels of the proteins. They transfected podocytes with the mutant GDIα and co-immunoprecipitated with endogenous Rho-GTPases. However, podocytes are known for low transfection efficiency; thus, it is possible that a low expression level of transfected GDIα and the endogenous Rho-GTPase levels were not sufficient to detect a partial interaction. In contrast, we transfected HEK293T cells (which have a much higher transfection efficiency than podocytes) with the GDIα and exogenous Rho-GTPases and then co-immunoprecipitated them. Higher expression levels of both GDIα and Rho-GTPases may have uncovered a partial interaction of G173V and Rho-GTPases.
In summary, we found that the 3 NS-causing GDIα mutations cause hyperactivation of Rac1 and impaired actin polymerization and motility in podocytes. Further research on Rac1 activity regulation in podocytes will likely shed light on pathological mechanisms of NS in general since Rac1 hyperactivity appears to cause a wide range of proteinuric diseases and should be considered as a potential therapeutic target for NS.
Materials and methods
Cell culture, plasmids, and transfection
The original immortalized mouse podocytes were obtained from Dr. Shankland (University of Seattle).Citation30 The mouse podocytes were maintained at 33°C in RPMI (Wisent Inc., 350–000-CL) supplemented with 10% FBS (Wisent Inc., 920–040) and interferon γ (10 U/mL) (Life Technology, PHC4031). Cells were cultured at 37°C for differentiation for 10–14 d.Citation31
Plasmids encoding the GFP-tagged WT and ΔD185 GDIα were constructed as previously reported.Citation11 The R120X and G173V mutants were similarly constructed by PCR and cloned at the C-terminus of the GFP.
GDIα KD podocytes were established using shRNA that targets the 3′ non-coding region of GDIα mRNA with puromycin selection, as previously described.Citation11 The GDIα KD podocytes were further transfected with one of the GDIα mutants or the WT GDIα and selected with puromycin (Wisent Inc., 400–160-UG) and G418 (750 μg/mL) (Wisent Inc., 400-130-IG).
HEK293T cells were transiently transfected using Lipofectamine 2000 (Invitrogen, 1645048) and maintained in Dulbecco's Modified Eagle Medium (DMEM) (Wisent Inc., 319–005-CL).
Mouse podocytes were transfected using lentivirus packaged into HEK293T cells, as previously described.Citation11 Virus-containing supernatants were added to mouse podocytes in permissive conditions for 16 hours. Puromycin was added 48 hours later and puromycin-resistent cells were pooled for further experiments.
Rho-GTPase pull down assay and immunoblotting
Active RhoA was pulled down using the Rho-binding domain of rhotekin (RBD) fused to glutathione S-transferase (GST) beads (GE Healthcare Bio-Sciences, 17-5279-01) to make GST-RBD; active Rac1 and Cdc42 were pulled down using the Cdc42-Rac1 Interactive Binding domain fused to GST beads (GST-CRIB), as previously described.Citation32 These proteins only bind the active forms of Rho-GTPases, but not the inactive forms.
Mouse podocytes were lysed and mixed with the beads at 4°C. These beads were then subjected to SDS-PAGE followed by immunoblotting. Total cell lysates were also immunoblotted for RhoGDIα (Santa Cruz Biotechnology, sc-360) and tubulin (Santa Cruz Biotechnology, sc-53646). Signals were visualized with enhanced chemiluminescent (ECL) (Thermo Scientific, 34077) using horseradish peroxidase as substrate.
Wound healing assay
To quantify cell mobility, we used a wound healing assay that has been previously described.Citation33 In this assay, cells were serum starved in RPMI containing 0.5% FBS (non-proliferative conditions) overnight prior to experiment. Using a sterile 10 μl pipette tip, straight scratches were made on a confluent layer of cells. The cell layer was washed with phosphate buffered saline (PBS) (Wisent Inc., 311-010-CL) to remove the detached cells. Photographs were taken at 0h and 24h and used to measure the wound surface area and calculate the percentage of the wound closure. The average of 4 wound areas per dish was used to calculate the average wound healing for 4 to 6 sets of experiments.
Live cell tracking
Following overnight serum starvation cells, were imaged every 10 minutes for 3 hours using a Bright Field Axiovert microscope. Their positions through the time were traced and the total distance along the trajectories and displacement (distance between the initial and final points) were measured using Zen Imaging Software (ZEISS Efficient Navigation). The average cell displacement and distance were calculated for each cell line.
Kymographs
Mouse podocytes were transfected with mCherry-tagged actin using Lipofectamine 2000 (Invitrogen, 1645048). After the basal images were acquired, EGF (100 nM) (Sigma, E-4127) was added and live cell images were acquired every 15 seconds for 20 minutes using the Zeiss LSM 780 confocal microscope. The Multiple Kymograph plug-in for ImageJ was used to generate kymographs. The plug-in and the instructions needed to create and analyze the kymographs can be found on http://www.embl.de/eamnet/html/body_kymograph.html.
Immunostaining
In preparation for staining, mouse podocytes were plated on glass cover slips coated with collagen type I (1 μg/cm2) (Sigma, C3867-1VL), serum starved overnight and fixed in 4% paraformaldehyde (PFA) (Thermo Scientific, 28908) in PBS before being permeabilized with Triton 0.1% (Sigma, T-9284). Cells were treated with Alexa Fluor-555 phalloidin (1: 1000) (Cell Signaling Technology, 8953) to stain for F-actin and Alexa Fluor 594 DNase I (0.3 μM) (Molecular Probes, D-12372) for G-actin for 25 minutes. After being washed, cells were stained with 40,6-diamidino-2-phenylindole (DAPI) (Invitrogen, D1306) and mounted on slides that were left to dry overnight at 4°C. Pictures of the cells were taken at 40x using Zeiss LSM 780 confocal microscope using 2 lasers (564 and 631 nm to avoid overlaps between Phalloidin staining and DNase I). Once acquired, the pictures were analyzed using ImageJ. The intensity of the phalloidin and DNAse staining per cell line was measured by individually selecting cells. Once we acquired the intensity of phalloidin (F- actin) and DNase I (G-actin) we divided F-actin values by G-actin values respectively to give the F/G ratio.
Statistical analysis
Data are presented as the mean ± SEM for multiple experiments. For comparison between 2 groups, statistical significance was assessed by a t test using IBM SPSS Software (Armonk, New York) or Microsoft Excel. For the comparison among more groups, statistical significance was assessed by One-way Anova. Post Hoc analysis (Tukey's range test, the IBM SPSS Software) was performed when significant difference was found among groups at p < 0.05.
Microarray analysis
Microarray data for E13.5, E15.5, and adult podocytes were obtained from the GenitoUrinary Molecular Anatomy Project (www.gudmap.org). The expression profiles studied were of E13.5 developing podocytes (GUDMAP series ID: 32, Series GEO ID: GSE 17148), E15.5 developing podocytes (GUDMAP series ID: 33, Series GEO ID: GSE 17145), and adult visceral epithelium (GUDMAP series ID: 31, Series GEO ID: GSE 17142). The expression data was analyzed by FlexArrayCitation34 software package using affimetrix power tool (APT) for normalization.
Immunohistochemistry of the kidney
Kidneys were isolated from embryonic mice at E13.5 and E16.5, fixed in 100% methanol for 30 minutes at −20°C, paraffin embedded and sliced to give rise to 4 μm thick sections. Citrate treatment was performed for antigen retrieval. Sections were stained with RhoGDIα antibody (1: 50) (Santa Cruz Biotechnology, sc-360) and Alexa Fluor 488 anti-rabbit IgG (1: 1000) (Cell Signaling Technology, 4412S). WT1 was visualized using the WT1 antibody (1: 200) (Santa Cruz Biotechnology, sc-192 and sc-7385) and either Alexa Fluor 555 anti-mouse IgG (1: 1000) (Cell Signaling Technology, 4409) or Alexa Fluor 488 anti-rabbit IgG (1: 1000) (Cell Signaling Technology, 4412). Nuclei were stained with Dapi (1: 500) (Invitrogen, D1306). Finally, sections were stained using anti-active Rac1-GTP antibody (1: 50) (NewEast Biosciences, 26903) and Alexa Fluor 555 anti-mouse IgG (1: 1000) (Cell Signaling Technology, 4409). A Zeiss Axiphot microscope was used to take the photographs at 60x magnification.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Drs. Elena Torban and Sima Babayeva for assistance in optimizing immunofluorescence staining of the kidney.
Funding
The study was supported by a grant from the Canadian Institute for Health Research (MOP-126180, to TT and IRG).
References
- George B, Holzman LB. Signaling from the podocyte intercellular junction to the actin cytoskeleton. Semin Nephrol 2012; 32:307-18.
- He F-F, Chen S, Su H, Meng X-F, Zhang C. Actin-associated proteins in the pathogenesis of podocyte injury. Curr Genomics 2013; 14:477; PMID:24396279; http://dx.doi.org/10.2174/13892029113146660014
- Armelloni S, Corbelli A, Giardino L, Li M, Ikehata M, Mattinzoli D, Messa P, Pignatari C, Watanabe S, Rastaldi MP. Podocytes: recent biomolecular developments. Biomol Concepts 2014; 5:319-30; PMID:25372762; http://dx.doi.org/10.1515/bmc-2014-0020
- Saleem MA. New developments in steroid-resistant nephrotic syndrome. Pediat Nephrol 2013; 28:699-709; PMID:22782578; http://dx.doi.org/10.1007/s00467-012-2239-0
- Kari JA, Montini G, Bockenhauer D, Brennan E, Rees L, Trompeter RS, Tullus K, van't Hoff W, Waters A, Ashton E. Clinico-pathological correlations of congenital and infantile nephrotic syndrome over twenty years. Pediat Nephrol 2013:1-8.
- Hinkes BG, Mucha B, Vlangos CN, Gbadegesin R, Liu J, Hasselbacher K, Hangan D, Ozaltin F, Zenker M, Hildebrandt F. Nephrotic syndrome in the first year of life: two thirds of cases are caused by mutations in 4 genes (NPHS1, NPHS2, WT1, and LAMB2). Pediatrics 2007; 119:e907-e19; PMID:17371932; http://dx.doi.org/10.1542/peds.2006-2164
- Hinkes B, Wiggins RC, Gbadegesin R, Vlangos CN, Seelow D, Nürnberg G, Garg P, Verma R, Chaib H, Hoskins BE. Positional cloning uncovers mutations in PLCE1 responsible for a nephrotic syndrome variant that may be reversible. Nat Genet 2006; 38:1397-405; PMID:17086182; http://dx.doi.org/10.1038/ng1918
- Brown EJ, Schlöndorff JS, Becker DJ, Tsukaguchi H, Tonna SJ, Uscinski AL, Higgs HN, Henderson JM, Pollak MR. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet 2010; 42:72-6; PMID:20023659; http://dx.doi.org/10.1038/ng.505
- Kaplan JM, Kim SH, North KN, Rennke H. Mutations in ACTN4, encoding α-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet 2000; 24:251-6; PMID:10700177; http://dx.doi.org/10.1038/73456
- Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 2005; 308:1801-4; PMID:15879175; http://dx.doi.org/10.1126/science.1106215
- Gupta IR, Baldwin C, Auguste D, Ha KC, El Andalousi J, Fahiminiya S, Bitzan M, Bernard C, Akbari MR, Narod SA. ARHGDIA: a novel gene implicated in nephrotic syndrome. J Med Genet 2013; 50:330-8: jmedgenet-2012-101442
- Gee HY, Saisawat P, Ashraf S, Hurd TW, Vega-Warner V, Fang H, Beck BB, Gribouval O, Zhou W, Diaz KA. ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J Clin Invest 2013; 123:3243-53; PMID:23867502; http://dx.doi.org/10.1172/JCI69134
- DerMardirossian C, Bokoch GM. GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell Biol 2005; 15:356-63; PMID:15921909; http://dx.doi.org/10.1016/j.tcb.2005.05.001
- Boulter E, Garcia-Mata R, Guilluy C, Dubash A, Rossi G, Brennwald PJ, Burridge K. Regulation of Rho GTPase crosstalk, degradation and activity by RhoGDI1. Nat Cell Biol 2010; 12:477-83; PMID:20400958; http://dx.doi.org/10.1038/ncb2049
- Garcia-Mata R, Boulter E, Burridge K. The'invisible hand': regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol 2011; 12:493-504; PMID:21779026; http://dx.doi.org/10.1038/nrm3153
- Mele C, Iatropoulos P, Donadelli R, Calabria A, Maranta R, Cassis P, Buelli S, Tomasoni S, Piras R, Krendel M. MYO1E mutations and childhood familial focal segmental glomerulosclerosis. New Engl J Med 2011; 365:295-306; PMID:21756023; http://dx.doi.org/10.1056/NEJMoa1101273
- Mundel P, Reiser J. Proteinuria: an enzymatic disease of the podocyte&quest. Kidney Int 2009; 77:571-80; PMID:19924101; http://dx.doi.org/10.1038/ki.2009.424
- Akilesh S, Suleiman H, Yu H, Stander MC, Lavin P, Gbadegesin R, Antignac C, Pollak M, Kopp JB, Winn MP. Arhgap24 inactivates Rac1 in mouse podocytes, and a mutant form is associated with familial focal segmental glomerulosclerosis. J Clin Investigat 2011; 121:4127-37; PMID:21911940; http://dx.doi.org/10.1172/JCI46458
- Lessey EC, Guilluy C, Burridge K. From mechanical force to RhoA activation. Biochemistry 2012; 51:7420-32; PMID:22931484; http://dx.doi.org/10.1021/bi300758e
- Shibata S, Nagase M, Yoshida S, Kawarazaki W, Kurihara H, Tanaka H, Miyoshi J, Takai Y, Fujita T. Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney disease. Nat Med 2008; 14:1370-6; PMID:19029984; http://dx.doi.org/10.1038/nm.1879
- Yu H, Suleiman H, Kim AH, Miner JH, Dani A, Shaw AS, Akilesh S. Rac1 activation in podocytes induces rapid foot process effacement and proteinuria. Mol Cell Biol 2013; 33:4755-64; PMID:24061480; http://dx.doi.org/10.1128/MCB.00730-13
- Lu T-C, He JC, Wang Z-h, Feng X, Fukumi-Tominaga T, Chen N, Xu J, Iyengar R, Klotman PE. HIV-1 Nef disrupts the podocyte actin cytoskeleton by interacting with diaphanous interacting protein. J Biol Chem 2008; 283:8173-82; PMID:18234668; http://dx.doi.org/10.1074/jbc.M708920200
- Blattner SM, Hodgin JB, Nishio M, Wylie SA, Saha J, Soofi AA, Vining C, Randolph A, Herbach N, Wanke R. Divergent functions of the Rho GTPases Rac1 and Cdc42 in podocyte injury. Kidney Int 2013; 84:920-30; PMID:23677246.
- Scott RP, Hawley SP, Ruston J, Du J, Brakebusch C, Jones N, Pawson T. Podocyte-specific loss of Cdc42 leads to congenital nephropathy. J Am Soc Nephrol 2012; 23:1149-54; ASN. 2011121206; PMID:22518006; http://dx.doi.org/10.1681/ASN.2011121206
- DeMali KA, Wennerberg K, Burridge K. Integrin signaling to the actin cytoskeleton. Curr Opin Cell Biol 2003; 15:572-82; PMID:14519392; http://dx.doi.org/10.1016/S0955-0674(03)00109-1
- Togawa A, Miyoshi J, Ishizaki H, Tanaka M, Takakura A, Nishioka H, Yoshida H, Mizoguchi A, Matsuura N, Niho Y. Progressive impairment of kidneys and reproductive organs in mice lacking Rho GDIalpha. Oncogene 1999; 18:5373-80; PMID:10498891; http://dx.doi.org/10.1038/sj.onc.1202921
- Raftopoulou M, Hall A. Cell migration: Rho GTPases lead the way. Dev Biol 2004; 265:23-32; PMID:14697350; http://dx.doi.org/10.1016/j.ydbio.2003.06.003
- Asanuma K, Yanagida-Asanuma E, Faul C, Tomino Y, Kim K, Mundel P. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol 2006; 8:485-91; PMID:16622418; http://dx.doi.org/10.1038/ncb1400
- Attias O, Jiang R, Aoudjit L, Kawachi H, Takano T. Rac1 contributes to actin organization in glomerular podocytes. Nephron Exp Nephrol 2009; 114:e93-e106; PMID:19955829; http://dx.doi.org/10.1159/000262317
- Shankland S, Pippin J, Reiser J, Mundel P. Podocytes in culture: past, present, and future. Kidney Int 2007; 72:26-36; PMID:17457377; http://dx.doi.org/10.1038/sj.ki.5002291
- Wang L, Flannery PJ, Rosenberg PB, Fields TA, Spurney RF. Gq-dependent signaling upregulates COX2 in glomerular podocytes. J Am Soc Nephrol 2008; 19:2108-18; PMID:18667730; http://dx.doi.org/10.1681/ASN.2008010113
- Zhang H, Cybulsky AV, Aoudjit L, Zhu J, Li H, Lamarche-Vane N, Takano T. Role of Rho-GTPases in complement-mediated glomerular epithelial cell injury. Am J Physiol-Renal Physiol 2007; 293:F148-F56; PMID:17376765; http://dx.doi.org/10.1152/ajprenal.00294.2006
- Ma H, Togawa A, Soda K, Zhang J, Lee S, Ma M, Yu Z, Ardito T, Czyzyk J, Diggs L. Inhibition of podocyte FAK protects against proteinuria and foot process effacement. J Am Soc Nephrol 2010; 21:1145-56; PMID:20522532; http://dx.doi.org/10.1681/ASN.2009090991
- Blazejczyk M, Miron M, Nadon R. FlexArray: A statistical data analysis software for gene expression microarrays. Montreal, Canada: Genome Quebec, 2007.