
ABSTRACT
The small GTPases of the Ras family play a pivotal role in the regulation of cell proliferation and motility, both in normal and transformed cells. In particular, the 3 genes encoding for the N-, H- and K-Ras are frequently mutated in human cancer and their inappropriate regulation, expression and subcellular localization can drive tumor onset and progression. Activation of the Ras-MAPK pathway directly signals on the cell cycle machinery by regulating the expression and/or localization of 2 key cell cycle player, Cyclin D1 and p27Kip1. We recently reported that in normal fibroblasts, following mitogenic stimuli, p27Kip1 translocates to the cytoplasm where it regulates H-Ras localization and activity. This regulatory mechanism ensures that cells pass beyond the restriction point of the cell cycle only when the proper level of stimulation is reached. Here, we comment on this new evidence that possibly represents one of the ways that cells have developed during evolution to ensure that the cell decision to divide is taken only when time and context are appropriate.
The response of the cell to extracellular stimuli and the integration of different stimuli are at the base of the development of each living organism.Citation1 One of the clearest examples of this integration is the cell decision to divide (or not) in presence of mitogenic signals. It is well accepted that a Restriction (R) point exists in the regulation of cell cycle progression and that a threshold of activation of specific signaling pathway must be reached and overcome before the R point can be bypassed.
Following mitogenic stimuli, one of the most critical signaling pathway activated is the Ras-mitogen activated protein kinases (MAPK) pathway that transduces the information from the plasma membrane to the nucleus and regulates the cell cycle progression, by regulating the expression of cyclin D1 and the localization of the cyclin-dependent kinase (CDK) inhibitor p27Kip1.Citation2 Accordingly, deregulation and/or mutation of RAS, RAF, MAPK (e.g. MEK, ERK), CCND1 (the gene encoding for cyclin D1) and, less frequently, CDKN1B (the gene encoding for p27Kip1) are frequently implicated in the onset and progression of many human cancers. We recently showed that one of the downstream regulated mediator of this pathway, p27Kip1, is also in charge of controlling the activity and subcellular localization of one upstream regulator, H-Ras, eventually ensuring a second level of cell cycle control.Citation3
Here, we discuss the biological and translational implications of this new scientific evidence, also in consideration of the fact that not all Ras proteins are “created equal.”
Ras proteins: Signaling hubs finely regulated at post-translational level
The central role of Ras in driving hyper-proliferation and tumor onset and progression has been widely demonstrated over the past 40 y.Citation4 Yet, each mammalian Ras gene seems to play unique roles in specific tissues. In mice, only the knock-out (KO) of K-Ras results in embryonic death, while H-Ras and N-Ras KO mice display more restricted phenotypes.Citation4 In particular, N-Ras KO results in decreased T-cell proliferation and altered CD8+ cells maturation, accompanied by a specific decrease of MAPK activation.Citation4,5 Knockout of H-Ras, although partially compensated by the increased expression of K-Ras and N-Ras, results in decreased carcinogen-induced tumor formation.Citation4,6
Accordingly, the 3 RAS genes are differently mutated in the diverse types of human disease. Activating mutation of H-Ras is the cause of the Costello syndrome in humans, characterized by mental retardation, high birth weight and predisposition to both benign and malignant tumors.Citation4,7 K-Ras mutations are prominent in pancreatic, colorectal and gastric cancer, H-Ras mutations are more frequent in head and neck squamous carcinomas and bladder cancer while N-Ras ones in acute myeloid leukemia and malignant melanomas, suggesting that different tissues differently rely on the function of each Ras protein.Citation8
A central role in the regulation of Ras protein functions is played by their membrane targeting. H-Ras, N-Ras and K-Ras4B (alternative splicing of exone 4 of K-Ras gene) are highly homolog proteins (82–90% amino acid identity in the first 165 amino acids) with a hyper variable C-terminus (24 aa), which comprises the membrane targeting sequence.Citation9 These different hyper variable regions result in several unique features, such as the control of sub-cellular localization and functional regulation. Membrane targeting of H-Ras and N-Ras requires the farnesylation of the CCAX motif and the palmitoylation of 2 (C181 and C184) or 1 cystein (C181) residue, for H-Ras and N-Ras, respectively.Citation10 K-Ras membrane targeting relies on the farnesylation of the CAAX motif and on the presence of the polybasic region comprised between the K175-K180 amino acidic stretch.Citation10 These different C-terminal sequence modifications result in the recycling of H-Ras and N-Ras, but not K-Ras4B, into the endosome compartment. H-Ras and N-Ras, but not K-Ras4B, require this recycling step to be fully activated.Citation9,11
Recent studies have further suggested that H-Ras and N-Ras, but not K-Ras4B, signaling activities are negatively regulated by ubiquitination.Citation12 Mono- and/or bi-ubiquitination of H/N-Ras are location-specific and mediated by the E3-ligase Rabex-5, eventually promoting Ras endosomal localization and suppression of ERK activation.Citation13 This modification has important functional consequences since inhibition of H/N-Ras ubiquitination results in their hyperactivity in vitroCitation12,13 and in overgrowth phenotypes in vivo.Citation14 Accordingly, targeting Ras post-translational modification and/or subcellular localization are currently explored as possible anticancer strategies.Citation15
Ras and p27 share the stage
Modulation of Ras activity is of primary importance in the regulation of cellular response to mitogenic signals. Ras proteins represent real hubs in the cellular protein network, connecting different extracellular stimuli to several signal transduction pathways.Citation9,10,15
We recently observed that p27Kip1 regulates, in concert with stathmin, the response to mitogenic extracellular stimuli, both in in vitro and in vivo models.Citation3,16 Our experiments highlighted that Ras were involved in this regulatory mechanism and we demonstrated that the interaction between p27Kip1 and stathmin affects H-Ras subcellular localization and mono-bi ubiquitination. Accordingly, p27Kip1 null cells and organs from p27Kip1 KO animals display increased activation of the Ras-MAPK pathway, faster entrance into the cell cycle and augmented proliferation rates.Citation3 At mechanistic level, we observed that following mitogenic signals p27Kip1 is displaced in the cytoplasm where it binds and restrains stathmin activity, eventually altering MT stability and H-Ras endosomal localization.
In normal mammalian cells, at least 2 subsequent growth factor stimuli are necessary for the G1-S phase transition, via the overcoming of a threshold that requires prolonged activation of ERK and induction of Egr-1.Citation17 Our data suggest that cytoplasmic p27Kip1 could be involved in the control of this threshold. From our results, it is conceivable that the first mitogenic stimulus is necessary to shuttle p27Kip1 from the nucleus to the cytoplasm. In the absence of a second impulse p27Kip1 blocks Ras-MAPK activity via stathmin, thereby preventing full cyclin-CDK complex activation. If a second stimulus is present and/or persists, p27Kip1 will be eventually degraded, giving the green light to the full activation of the MAPK pathway and of the cyclin-CDK complexes.
In accord with this hypothesis, while almost all of the hyper-proliferative phenotypes observed in p27Kip1 null mice and cells were rescued by co-ablation of stathmin,Citation3,16 neither the deletion of CDK4Citation18 nor cyclin D1Citation19,20 in p27 null mice did the same. For instance, thymus hyperplasia is still present in cyclin D1/p27 double knock-out (DKO) mice probably because proliferation in the thymus mostly relies on cyclin D2 and D3 and CDK6, rather than on cyclin D1 and CDK4.Citation20
On the other side, the decreased MAPK activation, T-cell proliferation and altered CD8+ cell maturation observed in N-Ras knockout miceCitation4,5 is a phenotype highly reminiscent of the one observed in p27KO mice.Citation16,21 Similarly, the phenotype of predisposition to benign and malignant tumors observed in H-Ras transgenic (H-RasG12V) miceCitation4,6,Citation7,22 are reminiscent of the ones observed in p27KO mice.Citation16,23-25 Overall, the insights coming from genetically modified animals support the possibility that the regulation of H/N-Ras by p27Kip1-stathmin interaction is of physiological relevance. Based on all the above considerations, we expect that p27Kip1-stathmin interaction will play a minor role in the control of K-Ras driven proliferation. Understanding the role of these regulations during cell transformation and tumor progression, in the context of mutated H-Ras versus K-Ras, will be of primary relevance in the field of cancer research.
p27Kip1, a master regulator of small GTPases activity?
Accumulating evidence supports the notion that p27Kip1 regulates the activity of several members of the small GTPases family, independently on its ability to bind and inhibit the cyclins/CDK complexes. Different groups have reported that p27Kip1 inhibits the activity of Rac,Citation26 RhoCitation27,28 and H-Ras,Citation3,29 either by direct binding or by modulating the microtubule stability. In all cases, these p27Kip1 functions have been linked to its cytoplasmic localization and, often, to the regulation of cell motility. The C-terminal domain of p27Kip1 seems to be necessary for the regulation of these small GTPases and, accordingly, this part of the protein is frequently altered by mutations in human cancer (for a review see ref. Citation30).
Overall, these data suggest that to fully integrate different external stimuli cells may have developed a mechanism of super-control in which few critical players are in charge of taking the “big” decisions, such as to migrate or to duplicate, once that all intracellular and environmental conditions are satisfied. It is possible that the regulation of membrane-associated small GTPases by p27Kip1 is one of the first identified signaling hub of such type, strictly required to couple the control of proliferation with motility. The fact that p27Kip1 can regulate microtubule stability, either via direct tubulin bindingCitation31 or via interaction with stathmin,Citation32 coupled with the fact that microtubules serve as tracks for cargos, such as organelles and vesicles, strongly support that the regulated transport of small GTPases along microtubules might be responsible for the spatial regulation of their activity. Accordingly, the inhibition of microtubule-dependent transport by taxol, dynamin or dynein inhibitors impaired the full activation of the MAPK cascade.Citation3
Conclusions and future directions
In the last 40 years, many critical aspects of the regulation of small GTPases of the Ras family have been clarified, teaching us how they play a central role in both onset and progression of human cancer. We now need to better clarify why each specific gene of this family is altered in restricted subtypes of cancer and not in others and whether their temporal and spatial regulation could represent a promising strategy of intervention in our battle against cancer. Our recent work, linking p27Kip1 cytoplasmic displacement with the response to mitogenic stimuli and H-Ras regulation has contributed to unveil a previously unexpected level of reciprocal regulation (). Exploiting these new findings to address and possibly meet unsolved clinical questions is now our more critical task.
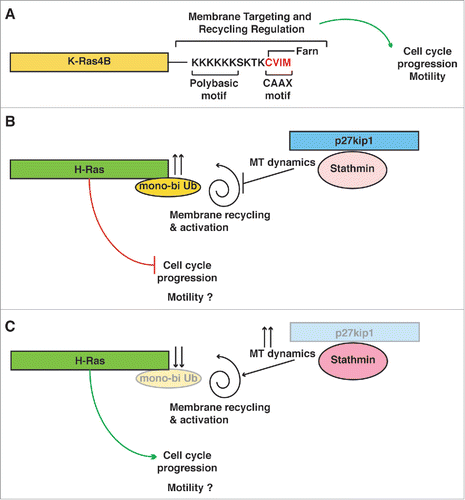
Figure 1. p27 restrains H-Ras activation via stathmin. (A) Schematic representation of K-Ras. K-Ras4B (alternative splicing of exone 4 of K-Ras gene) is highly homolog to H-Ras (83% amino acid identity, in the first 165 amino acids), but includes a hyper variable C-terminus, comprising the membrane targeting sequence. The different hyper variable region results in several unique features, such as that K-Ras-4B activation and membrane targeting does not require recycling in the endosomal compartment and is not affected by mono-bi ubiquitination. (B) Schematic representation of H-Ras functioning in cells expressing normal levels of p27. Following extracellular stimuli, such as growth factor, p27 shuttles to the cytoplasm. There, p27 can interact with stathmin, leading to modulation of microtubules dynamics and of endosomal vesicles trafficking. H-Ras gets mono-bi ubiquitinated, losing part of its activity. In this setting, the cell cycle does not progress and cells do not proliferate. It is implied that, if the external stimulus persists, then p27 will be ubiquitinated and degraded, releasing this block on H-Ras and cell cycle. (C) Schematic representation of H-Ras functioning in cells with low or absent expression p27. Stathmin activity is increased and so is MT dynamics, releasing the brake from full H-Ras activation and leading to faster cell cycle progression and proliferation.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to acknowledge all authors of the original report.Citation3
Funding
This work was funded by Associazione Italiana Ricerca sul Cancro (AIRC) Grants IG 16865 (to G.B.) and IG 15902 (to B.B.).
References
- Scott JD, Pawson T. Cell signaling in space and time: where proteins come together and when they're apart. Science 2009; 326:1220-4; PMID:19965465; http://dx.doi.org/10.1126/science.1175668
- Meloche S, Pouysségur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene 2007; 26:3227-39; PMID:17496918; http://dx.doi.org/10.1038/sj.onc.1210414
- Fabris L, Berton S, Pellizzari I, Segatto I, D'Andrea S, Armenia J, Bomben R, Schiappacassi M, Gattei V, Philips MR, et al. p27kip1 controls H-Ras/MAPK activation and cell cycle entry via modulation of MT stability. Proc Natl Acad Sci U S A 2015; 112:13916-21; PMID:26512117; http://dx.doi.org/10.1073/pnas.1508514112
- Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer 2003; 3:459-65; PMID:12778136; http://dx.doi.org/10.1038/nrc1097
- Pérez de Castro I, Diaz R, Malumbres M, Hernández M-I, Jagirdar J, Jiménez M, Ahn D, Pellicer A. Mice deficient for N-ras: impaired antiviral immune response and T-cell function. Cancer Res 2003; 63:1615-22; PMID:12670913
- Ise K, Nakamura K, Nakao K, Shimizu S, Harada H, Ichise T, Miyoshi J, Gondo Y, Ishikawa T, Aiba A, et al. Targeted deletion of the H-ras gene decreases tumor formation in mouse skin carcinogenesis. Oncogene 2000; 19:2951-6; PMID:10871846; http://dx.doi.org/10.1038/sj.onc.1203600
- Aoki Y, Niihori T, Kawame H, Kurosawa K, Ohashi H, Tanaka Y, Filocamo M, Kato K, Suzuki Y, Kure S, et al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet 2005; 37:1038-40; PMID:16170316; http://dx.doi.org/10.1038/ng1641
- Der CJ. Are All RAS Proteins Created Equal in Cancer? Natl. Cancer Inst. [cited 2016 Feb 16]; Available from: http://www.cancer.gov/research/key-initiatives/ras/ras-central/blog/ras-proteins-created-equal
- Hancock JF. Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol 2003; 4:373-85; PMID:12728271; http://dx.doi.org/10.1038/nrm1105
- Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Regulating the regulator: post-translational modification of RAS. Nat Rev Mol Cell Biol 2012; 13:39-51; http://dx.doi.org/10.1038/nrm3255
- Roy S, Wyse B, Hancock JF. H-Ras signaling and K-Ras signaling are differentially dependent on endocytosis. Mol Cell Biol 2002; 22:5128-40; PMID:12077341; http://dx.doi.org/10.1128/MCB.22.14.5128-5140.2002
- Jura N, Scotto-Lavino E, Sobczyk A, Bar-Sagi D. Differential modification of Ras proteins by ubiquitination. Mol Cell 2006; 21:679-87; PMID:16507365; http://dx.doi.org/10.1016/j.molcel.2006.02.011
- Xu L, Lubkov V, Taylor LJ, Bar-Sagi D. Feedback regulation of Ras signaling by Rabex-5-mediated ubiquitination. Curr Biol 2010; 20:1372-7; PMID:20655225; http://dx.doi.org/10.1016/j.cub.2010.06.051
- Yan H, Jahanshahi M, Horvath EA, Liu H-Y, Pfleger CM. Rabex-5 ubiquitin ligase activity restricts Ras signaling to establish pathway homeostasis in Drosophila. Curr Biol 2010; 20:1378-82; PMID:20655224; http://dx.doi.org/10.1016/j.cub.2010.06.058
- Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission Possible? Nat Rev Drug Discov 2014; 13:828-51; PMID:25323927; http://dx.doi.org/10.1038/nrd4389
- Berton S, Pellizzari I, Fabris L, D'Andrea S, Segatto I, Canzonieri V, Marconi D, Schiappacassi M, Benevol S, Gattei V, et al. Genetic characterization of p27(kip1) and stathmin in controlling cell proliferation in vivo. Cell Cycle 2014; 13:3100-11; PMID:25486569; http://dx.doi.org/10.4161/15384101.2014.949512
- Zwang Y, Sas-Chen A, Drier Y, Shay T, Avraham R, Lauriola M, Shema E, Lidor-Nili E, Jacob-Hirsch J, Amariglio N, et al. Two phases of mitogenic signaling unveil roles for p53 and EGR1 in elimination of inconsistent growth signals. Mol Cell 2011; 42:524-35; PMID:21596316; http://dx.doi.org/10.1016/j.molcel.2011.04.017
- James MK, Ray A, Leznova D, Blain SW. Differential modification of p27Kip1 controls its cyclin D-cdk4 inhibitory activity. Mol Cell Biol 2008; 28:498-510; PMID:17908796; http://dx.doi.org/10.1128/MCB.02171-06
- Tong W, Pollard JW. Genetic evidence for the interactions of cyclin D1 and p27(Kip1) in mice. Mol Cell Biol 2001; 21:1319-28; PMID:11158317; http://dx.doi.org/10.1128/MCB.21.4.1319-1328.2001
- Geng Y, Yu Q, Sicinska E, Das M, Bronson RT, Sicinski P. Deletion of the p27Kip1 gene restores normal development in cyclin D1-deficient mice. Proc Natl Acad Sci U S A 2001; 98:194-9; PMID:11134518; http://dx.doi.org/10.1073/pnas.98.1.194
- Jatzek A, Tejera MM, Singh A, Sullivan JA, Plisch EH, Suresh M. p27(Kip1) negatively regulates the magnitude and persistence of CD4 T cell memory. J Immunol 2012; 189:5119-28; PMID:23071285; http://dx.doi.org/10.4049/jimmunol.1201482
- Viosca J, Schuhmacher AJ, Guerra C, Barco A. Germline expression of H-RasG12V causes neurological deficits associated to Costello syndrome. Genes Brain Behav 2009; 8:60-71; PMID:18823404; http://dx.doi.org/10.1111/j.1601-183X.2008.00443.x
- Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM, et al. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell 1996; 85:733-44; PMID:8646781; http://dx.doi.org/10.1016/S0092-8674(00)81239-8
- Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature 1998; 396:177-80; PMID:9823898; http://dx.doi.org/10.1038/24179
- Philipp J, Vo K, Gurley KE, Seidel K, Kemp CJ. Tumor suppression by p27Kip1 and p21Cip1 during chemically induced skin carcinogenesis. Oncogene 1999; 18:4689-98; PMID:10467416; http://dx.doi.org/10.1038/sj.onc.1202840
- McAllister SS, Becker-Hapak M, Pintucci G, Pagano M, Dowdy SF. Novel p27(kip1) C-terminal scatter domain mediates Rac-dependent cell migration independent of cell cycle arrest functions. Mol Cell Biol 2003; 23:216-28; PMID:12482975; http://dx.doi.org/10.1128/MCB.23.1.216-228.2003
- Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev 2004; 18:862-76; PMID:15078817; http://dx.doi.org/10.1101/gad.1185504
- Berton S, Belletti B, Wolf K, Canzonieri V, Lovat F, Vecchione A, Colombatti A, Friedl P, Baldassarre G. The tumor suppressor functions of p27(kip1) include control of the mesenchymal/amoeboid transition. Mol Cell Biol 2009; 29:5031-45; PMID:19596789; http://dx.doi.org/10.1128/MCB.00144-09
- Moeller SJ, Head ED, Sheaff RJ. p27Kip1 inhibition of GRB2-SOS formation can regulate Ras activation. Mol Cell Biol 2003; 23:3735-52; PMID:12748278; http://dx.doi.org/10.1128/MCB.23.11.3735-3752.2003
- Belletti B, Baldassarre G. New light on p27(kip1) in breast cancer. Cell Cycle 2012; 11:3701-2; PMID:22895010; http://dx.doi.org/10.4161/cc.21573
- Godin JD, Thomas N, Laguesse S, Malinouskaya L, Close P, Malaise O, Purnelle A, Raineteau O, Campbell K, Fero M, et al. p27(Kip1) is a microtubule-associated protein that promotes microtubule polymerization during neuron migration. Dev Cell 2012; 23:729-44; PMID:23022035; http://dx.doi.org/10.1016/j.devcel.2012.08.006
- Baldassarre G, Belletti B, Nicoloso MS, Schiappacassi M, Vecchione A, Spessotto P, Morrione A, Canzonieri V, Colombatti A. p27Kip1-stathmin interaction influences sarcoma cell migration and invasion. Cancer Cell 2005; 7:51-63; PMID:15652749; http://dx.doi.org/10.1016/j.ccr.2004.11.025