
ABSTRACT
How cancer disseminates and metastasizes remains an outstanding open question. Emerging evidence indicates that membrane trafficking is frequently harnessed by tumors of epithelial origin to acquire a mesenchymal program of invasiveness. However, the critical molecular hubs used by cancer cells this context have only began to be elucidated. Here, we discussed the results of a recent phenotypic screening that led to the identification of the small GTPase RAB2A, not previously involved in cancer dissemination, as pivotal for the acquisition of pericellular proteolysis, cell dissemination and distant metastatic spreading of human breast cancer. At the cellular levels, RAB2A controls both canonical polarized Golgi-to-Plasma membrane trafficking of the junctional protein E-cadherin, and post-endocytic trafficking of the membrane-bound metalloprotease, MT1-MMP. This finding reveals an unexpected plasticity in the control of diverse trafficking routes exerted by RAB2A through canonical (Golgi stacking) and non-canonical (late endosome recycling) functional interactions, contributing to break established membrane trafficking dogma on the rigorous molecular distinction between polarized Golgi and post endocytic routes. Finally, they suggest that epithelial cancers may specifically select for those molecules that enable them to control multiple trafficking routes, in turn essential for the regulation of activities necessary for acquisition of mesenchymal traits.
Introduction
Metastasis remains the undefeated, leading reason of mortality in cancer patient. The metastatic phenotype is complex, featuring a wide range of flexible cellular processes that enable tumors to adapt to and overcome microenvironmental barriers to migration and invasion.Citation1 Cell migration, a hallmark of metastasis, is a typical case in point. A number of recent studies have revealed how tumor cells use different motility modes to disseminate.Citation1 Each of these modes is driven and controlled by distinct molecular pathways.Citation2 In one of this strategy, tumor of epithelial origin acquire prototypical mesenchymal features that enable cells to breach the basement membrane, navigate within the surrounding, collagen-rich stromal tissue and loose tight cell-cell adhesion.Citation3 This mode of motility and invasiveness typical begins with the formation of elongated and adherent, actin-rich protrusions driven by polarized RAC activity, not dissimilar to the one guiding the motility of cells onto 2 dimensional substrate.Citation4 Within 3-dimensional interstitial tissues, however, tumor cells must also learn to navigate within and deal with dense, desmoplastic stromal matrices that represent formidable physical, barriers to free dissemination. Tumors overcome this barrier either by tearing and pulling on ECM fibers to widen pre-existing gaps and channels through active, contractile forces and by chemical remodelling the extracellular space through the acquisition of pericellular-associated or locally secreted proteolytic enzyme. In some cases, these processes are executed by in a non-cell autonomous fashion by tumor associated host cells, such as fibroblasts, that have been seen to cooperate with cancer cells to drive their invasiveness by remodelling of extracellular matrix.Citation5
Deregulation of matrix-degrading proteases, including matrix metalloproteinases (MMPs), is indeed one of the mechanisms used by cancer to remodel the ECM.Citation6 Although secreted MMPs have been implicated in cancer, membrane-anchored MMPs, and most notably membrane type 1 (MT1)-MMP have been shown to exert a pivotal role.Citation7-9 Consistently MT1-MMP expression is frequently elevated in various invasive carcinomas, and genetic evidence in murine models further support its critical role in the process.Citation10-12 In addition to deregulation of total protein and mRNA levels of MT1-MMP, subversion of the activity of this protease is also frequently brought about by alterations of its intracellular trafficking routes.
Invadopodia are a cell culture proxy of pericellular proteolysis and cancer invasion.Citation13 These structures are adhesive protrusions, rich in filamentous actin, capable to degrade ECM in a highly controlled and spatially confined fashion.Citation14 Invadopodia are primarily found on ventral membrane protrusions formed by cancer cells during invasion and migration.Citation15,16 The assembly of invadopodia can be stimulated by growth factors, and many signaling-and actin-related proteins are actively transported to invadopodia. Among these factors, MT1-MMP has emerged as the critical and indispensable enzyme necessary to confer to invadopodia their ECM degradation activity. MT1-MMP is a single pass transmembrane protein. It is synthesized as an inert pro-enzyme, which is processed in the Golgi by protein convertases and transported to the PM.Citation17 At this location MT1-MMP dimerizes and is rapidly inactivated by binding to tissue inhibitors of metalloproteases (TIMP I and II), which are also abundantly expressed in cancer.Citation18 The short cytoplasmic tail of membrane-bound MT1-MMP directly links this protein to internalization machineries.Citation19,20 Accordingly, MT1-MMP is constantly undergoing clathrin dependent and independent endocytosis, is transported to early and late endosome from where it can actively and directly recycle back to the PM.Citation21 This endocytic/exocytic cycles are essential to ensure both a constant flux of active molecules that exert their transient proteolytic activity before being inactivated, as well as the delivery of MT1-MMP to the actin rich core of invadopodia. Not surprisingly, local exocytic burst of intracellular MT1-MMP have been documented by real time microscopy analysis to occur at invadopodia in contact with fibrillar collagen type I,Citation22 while direct interaction of the cytoplasmic tail of MT1-MMP to filamentous actin may serve as docking or tethering mechanisms.Citation23 In addition to recycling from early and late endosomes,Citation24 targeting of MT1-MMP to invadopodia can also be achieved through direct polarized Golgi-to-PM traffickingCitation25 and exosomes release,Citation26 enlarging the range of potential trafficking regulatory routes that can be exploited to control ECM degradation and that can go awry in a tumor setting.Citation8 The sum of these evidence indicate that membrane trafficking determinants are crucial to control MT1-MMP pericellular proteolysis, and further suggest that the factors so far identified likely represent only the tip of the iceberg of much larger endocytic networks essential to tune the activity of this protease. It is also important to point out that while the trafficking routes through which MT1-MMP is precisely delivered have, by and large, been identified, we have only began to decipher and characterize the key molecular determinants and mechanisms that normal and more relevantly tumors cells utilizes to control the delivery of this cargo. Even less clear is whether the various membrane trafficking routes taken by MT1-MMP are coordinated.
Here, we will describe a recent experimental strategy we used to address some of these issues and further discuss how, unexpectedly, the small GTPase, RAB2A, though to be exclusively involved in endoplasmic reticulum (ER)-to-Golgi transport, may be hijacked to control post-endosomal recycling trafficking, motility and ultimately the PM delivery of MT1-MMP. We will further highlight how RAB2A is harnessed by aggressive, highly metastatic human breast cancer to promote a mesenchymal invasive program, and, ultimately, the acquisition of metastatic properties.
Identification of RAB2A as a critical regulator of invasive programs in BC (Breast Cancer) lines and patients
We used invadopodia as “easy-to-follow” read outs for imaging, RNAi-based, phenotypic screenings of the whole set of RAB family GTPases, which are key hubs of membrane trafficking networks.Citation27 More specifically, we used a stepwise siRNA screening strategy that led to identification of RAB2A, not previously involved in tumor progression, as critical for matrix degradation and BC cell invasion. ISH analysis of a cohort of invasive breast cancers in Tissue MicroArray further revealed that RAB2A elevated expression is a powerful and independent predictor of BC recurrence and metastatic dissemination in BC patients,Citation28 supporting the notion that this protein is key for the acquisition of an invasive BC phenotype. Notably, ectopic RAB2A expression significantly increased matrix degradation suggesting that RAB2A is not only required for, but also sufficient to promote ECM digestion.
Figure 1. Working model of the impact of RAB2A trafficking routes on E-cadherin junctional homeostasis and MT1-MMP late endosomal recycling. RAB2A acts at the ER-Golgi intermediate stages and control Golgi stacking, in turn influencing the processing and trafficking from Golgi-to-plasma membrane of E-Cadherin. In its active form (not shown), RAB2A localizes to late endosomes, which are positive for the membarne-bound metalloprotease, MT1-MMP. At this site, RAB2A, together with VPS39, a member of homotypic fusion and vacuole protein sorting (HOPS) complex, controls MT1-MMP recycling to invadopodia, ensuring that the activity of this proteolytic enzyme is spatially confined. We propose that RAB2A may be a central hub for the control of distinct trafficking routes, in turn essential for the regulation of different cargos, such as MT1-MMP and E-cadherin, which are critical for the acquisition of mesenchymal invasive properties in breast cancer. Not suprisingly, RAB2A is elevated in human breast cancerand is a powerful and independent predictor of distant metastatic spreading. (Figure modified with permission from Kajiho H. et al EMBO R. 2016Citation28).
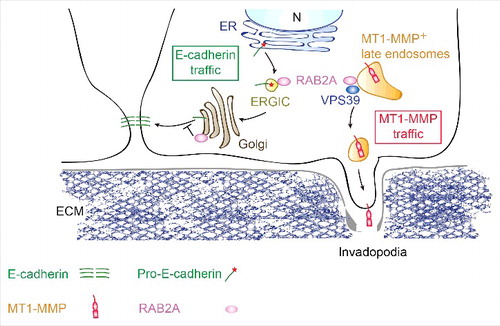
RAB2A and VPS39 in MT1-MMP trafficking in late endosomes
Next, we focused our attention on uncovering the molecular mechanism of invasive BC program regulated by RAB2A. RAB2A mainly localizes to ER-Golgi intermediate complex (ERGIC), and regulates vesicular transport from ERGIC to both ER and Golgi via COP I vesicles.Citation29,30 Tisdale et al.Citation31,32 established GAPDH as an effector of RAB2A, and further showed that this complex is required for the transport between ER and Golgi. However, silencing of RAB2A did not decrease the secretion of either soluble MMPs or cell surface MT1-MMP in BC cell lines. Additionally, silencing of GAPDH did not inhibit ECM degradation activity. These results indicate that the mechanism of action of RAB2A in invasive program is, unexpectedly, independent from its “conventional role” in ER-to-Golgi or ERGIC.
Endo/exocytic circuitries of MT1-MMP are critical to restrict in a spatio-temporal defined fashion the activity of this proteolytic enzyme.Citation22,24,33,34 The molecular mechanisms driving polarized trafficking have therefore been subject of intense investigation. Recently, for example, it has been reported that WASH, ARF6, and its effectors JIP3/JIP4 regulate tumor cell invasion by modulating MT1-MMP exocytosis from late endosomes to PM. This axis was shown to control microtubule-driven transport of vesicles of invasive BC,Citation22,35 reinforcing the notion that post-late endosomal trafficking is frequently hijacked by disseminating tumors. Based on this set of evidence, we explore the possibility that RAB2A may also impinge on this transport route. We found that: i) active RAB2A causes a significant enlargement of late endosomal vesicles where both RAB2A and MT1-MMP localize; ii) we further showed that transient exocytic outburst of MT1-MMP are significantly reduced by RAB2A silencing. Thus RAB2A appear to control pericellular ECM degradation through regulation of post-endocytic MT1-MMP trafficking. This latter results are consistent with recent observations indicating that the ortholog of RAB2A in Drosophila, RAB2, is enriched in late endosome when in its active, GTP-bound form, and was found to interact with VPS39, a component of the homotypic fusion and vacuole protein sorting (HOPS) complex involved in fusion events between late endosomes and lysosomes.Citation36 We extended this latter finding and confirmed that also mammalian RAB2A and VPS39 physically interact. Furthermore, VPS39 silencing attenuated ECM degradation and invasive activity in BC cell lines, and prevented the increased ECM digestion caused by ectopic expression of RAB2A. Thus, RAB2A and its effector VPS39 may be part of a novel trafficking axis regulating late endosomal recycling of MT1-MMP, providing a plausible molecular explanation as to why RAB2A is hijacked by cancer cells to promote their invasive program. RAB2A, however, is primarily involved in regulating ERGIC/Golgi polarized transport rather than post-endocytic trafficking, begging the question as to how this GTPase acts at distinct trafficking steps. Two possibilities can be envisioned: first, only GTP-bound RAB2A is found in late endosome, indicating that activation of this GTPases might also be critical to restrict a fraction of this protein to this compartment. Alternatively, ERGIC/Golgi and late endosomes may be functional, if not physically interconnected through trafficking intermediate or transient contact sites, in keeping with the emerging plasticity and flexibility of the vesicular membrane transport. Additional experiments are clearly needed to clarify these issues.
RAB2A in E-cadherin trafficking from Golgi to PM
When cancer cells penetrate within ECM by increasing invasive activity and motility, they often loosen cell-cell adhesion at adherence junction (AJ). One of the most important AJs proteins in normal epithelial cells is E-cadherin, whose expression typically decreases during epithelial-mesenchymal transition (EMT).Citation37 We found that silencing of RAB2A in various BC cell lines increases cell surface E-cadherin and promotes cell compaction. Conversely, elevated expression of RAB2A in normal and tumorigenic human mammary epithelial cell lines decreases surface E-cadherin, and promotes the extension of elongated, mesenchymal cellular protrusions, recapitulating, at least at the phenotypic level, an epithelial-to-mesenchymal phenotypic transition. However, this EMT-like transition is accompanied by neither an E-cadherin/N-cadherin switch nor by the upregulation of typical mesenchymal transcription factors, indicating that RAB2A expression promotes mesenchymal-like features without affecting canonical EMT transcriptional programs.
Classical cadherins, including E-cadherin, are single membrane-spanning proteins. They are synthesized in the ER as immature, protein with the addition of a pro-region appendage.Citation38 The pro-region needs to be cleaved in the Golgi by pro-protein convertase family proteins, such as furin, to enable the transport of the now mature form of E-cadherin to the PM.Citation39 By exploiting the retention using selective hooks (RUSH) system,Citation40 we monitored in real time the trafficking of E-cadherin from donor to its destined compartment. We found that RAB2A elevation delays significantly the transport of E-cadherin from Golgi to PM.Citation28 As a consequence of this trafficking blockade, Golgi-mediated cleavage of pro-E-cadherin is also compromised, and cell surface levels of E-cadherin are diminished, likely accounting for the reduced cell compaction observed in RAB2A expressing BC. RAB2A regulates E-cadherin trafficking independently from VPS39-mediated post-endocytic transport, but, likely, by controlling the size of Golgi stacks, which appears enlarged and extended following RAB2A expression. The precise molecular mechanisms that RAB2A uses in this context remain, however, ill-defined and it is unclear which of the downstream effectors are essential for this function. This notwithstanding, this latter set of finding together with the emerging post-endocytic role of RAB2A suggest that this protein may be a central hub for the control of distinct trafficking routes, in turn essential for the regulation of different cargos, such as MT1-MMP and E-cadherin, which are critical for the acquisition of mesenchymal invasive properties.
Conclusion
Our data point to unexpected multifunctional roles of RAB2A in the control of distinct membrane trafficking processes that are exploited to promotes e mesenchymal programs of BC invasion.
Recently Luo et al.Citation41 showed that RAB2A elevation induces also BC stemness, by promoting canonical mesenchymal transcriptional programs through aberrant regulation of endosomally-located ERK1/2 activity. We, on the other hand, report here that elevation of RAB2A decreases surface E-cadherin, but without any detectable changes in the transcriptional program associated to EMT. These apparently diverse results may be interpreted in light of the notion that interconversion among epithelial and mesenchymal states is not only achieved through alteration of transcriptional programs, but also by exploiting transient biochemical and cellular processes, including trafficking networks, including the one controlled by RAB2A.Citation42 Within this context, elevation of RAB2A may tip the balance toward more mesenchymal features by acting both on a ERK axis impacting on gene regulation, but also by rewiring membrane trafficking routes.
It is of note that the action of RAB2A is specifically restricted to the control of post-endocytic transport of MT1-MMP that is internalized from PM. Mechanistically, how RAB2 controls this step remains to be fully investigated. Our preliminary evidence, however, indicates that loss of RAB2A as well as VPS39 dramatically impairs the motility of MT1-MMP-positive late endosomes, which aberrantly accumulate around the nucleus, ultimately preventing them to reach peripheral site for the final delivery of this protease to the PM. These findings suggest that RAB2A is critical for the motility and spatial regulations of late endosomes, a process primarily controlled by microtubule-mediated transport. Thus, similarly to the roles played by yet another small GTPase, ARF6,Citation35 it is tantalizing to speculate that also active RAB2A in conjunction with VPS39 might be necessary to link late endosomal vesicles to motor machineries ultimately mediating the proper positioning and motility of vesicles. The identification of the component of this machinery will likely illuminate on more general roles of this GTPases in post-endocytic membrane transport and provide insights into how tumor cells exploit the endosomal network to plastically adapt to the micro-environment and acquire aggressive traits.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
The following funding has supported the work highlighted in this commentary:
GS was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC #10168 and #18621), MIUR (the Italian Ministry of University and Scientific Research), the Italian Ministry of Health, Worldwide Cancer Research (AICR- 14–0335); the European Research Council (Advanced-ERC-#268836).
HK was supported by a Marie Curie/AIRC TRAIN fellowship.
YK was supported by Grant-in-Aid for Japan Society for the Promotion of Science (JSPS) Fellows.
References
- Friedl P, Wolf K. Plasticity of cell migration: a multiscale tuning model. J Cell Biol 2010; 188:11-9; PMID:19951899; https://doi.org/10.1083/jcb.200909003
- Friedl P, Sahai E, Weiss S, Yamada KM. New dimensions in cell migration. Nat Rev Mol Cell Biol 2012; 13:743-7; PMID:23072889; https://doi.org/10.1038/nrm3459
- Friedl P, Locker J, Sahai E, Segall JE. Classifying collective cancer cell invasion. Nat Cell Biol 2012; 14:777-83; PMID:22854810; https://doi.org/10.1038/ncb2548
- Madsen CD, Sahai E. Cancer dissemination–lessons from leukocytes. Dev Cell 2010; 19:13-26; PMID:20643347; https://doi.org/10.1016/j.devcel.2010.06.013
- Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, Harrington K, Williamson P, Moeendarbary E, Charras G, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol 2013; 15:637-46; PMID:23708000; https://doi.org/10.1038/ncb2756
- Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell 2010; 141:52-67; PMID:20371345; https://doi.org/10.1016/j.cell.2010.03.015
- Hotary K, Li XY, Allen E, Stevens SL, Weiss SJ. A cancer cell metalloprotease triad regulates the basement membrane transmigration program. Genes Dev 2006; 20:2673-86; PMID:16983145; https://doi.org/10.1101/gad.1451806
- Hotary KB, Allen ED, Brooks PC, Datta NS, Long MW, Weiss SJ. Membrane type I matrix metalloproteinase usurps tumor growth control imposed by the 3-dimensional extracellular matrix. Cell 2003; 114:33-45; PMID:12859896; https://doi.org/10.1016/S0092-8674(03)00513-0
- Ferguson SM, Brasnjo G, Hayashi M, Wolfel M, Collesi C, Giovedi S, Raimondi A, Gong LW, Ariel P, Paradise S, et al. A selective activity-dependent requirement for dynamin 1 in synaptic vesicle endocytosis. Science 2007; 316:570-4; PMID:17463283; https://doi.org/10.1126/science.1140621
- Rozanov DV, Savinov AY, Williams R, Liu K, Golubkov VS, Krajewski S, Strongin AY. Molecular signature of MT1-MMP: transactivation of the downstream universal gene network in cancer. Cancer Res 2008; 68:4086-96; PMID:18519667; https://doi.org/10.1158/0008-5472.CAN-07-6458
- Sounni NE, Rozanov DV, Remacle AG, Golubkov VS, Noel A, Strongin AY. Timp-2 binding with cellular MT1-MMP stimulates invasion-promoting MEK/ERK signaling in cancer cells. Int J Cancer 2010; 126:1067-78; PMID:19551841
- Perentes JY, Kirkpatrick ND, Nagano S, Smith EY, Shaver CM, Sgroi D, Garkavtsev I, Munn LL, Jain RK, Boucher Y. Cancer cell-associated MT1-MMP promotes blood vessel invasion and distant metastasis in triple-negative mammary tumors. Cancer Res 2011; 71:4527-38; PMID:21571860; https://doi.org/10.1158/0008-5472.CAN-10-4376
- Linder S, Wiesner C, Himmel M. Degrading devices: invadosomes in proteolytic cell invasion. Ann Rev Cell Dev Biol 2011; 27:185-211; PMID:21801014; https://doi.org/10.1146/annurev-cellbio-092910-154216
- Sibony-Benyamini H, Gil-Henn H. Invadopodia: the leading force. Eur J Cell Biol 2012; 91:896-901; PMID:22633185; https://doi.org/10.1016/j.ejcb.2012.04.001
- Destaing O, Block MR, Planus E, Albiges-Rizo C. Invadosome regulation by adhesion signaling. Curr Opin Cell Biol 2011; 23:597-606; PMID:21550788; https://doi.org/10.1016/j.ceb.2011.04.002
- Buccione R, Orth JD, McNiven MA. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat Rev Mol Cell Biol 2004; 5:647-57; PMID:15366708; https://doi.org/10.1038/nrm1436
- Yana I, Weiss SJ. Regulation of membrane type-1 matrix metalloproteinase activation by proprotein convertases. Mol Biol Cell 2000; 11:2387-401; PMID:10888676; https://doi.org/10.1091/mbc.11.7.2387
- Osenkowski P, Toth M, Fridman R. Processing, shedding, and endocytosis of membrane type 1-matrix metalloproteinase (MT1-MMP). J Cell Physiol 2004; 200:2-10; PMID:15137052; https://doi.org/10.1002/jcp.20064
- Lehti K, Valtanen H, Wickstrom SA, Lohi J, Keski-Oja J. Regulation of membrane-type-1 matrix metalloproteinase activity by its cytoplasmic domain. J Biol Chem 2000; 275:15006-13; PMID:10748199; https://doi.org/10.1074/jbc.M910220199
- Uekita T, Itoh Y, Yana I, Ohno H, Seiki M. Cytoplasmic tail-dependent internalization of membrane-type 1 matrix metalloproteinase is important for its invasion-promoting activity. J Cell Biol 2001; 155:1345-56; PMID:11756481; https://doi.org/10.1083/jcb.200108112
- Remacle A, Murphy G, Roghi C. Membrane type I-matrix metalloproteinase (MT1-MMP) is internalised by 2 different pathways and is recycled to the cell surface. J Cell Sci 2003; 116:3905-16; PMID:12915589; https://doi.org/10.1242/jcs.00710
- Monteiro P, Rosse C, Castro-Castro A, Irondelle M, Lagoutte E, Paul-Gilloteaux P, Desnos C, Formstecher E, Darchen F, Perrais D, et al. Endosomal WASH and exocyst complexes control exocytosis of MT1-MMP at invadopodia. J Cell Biol 2013; 203:1063-79; PMID:24344185; https://doi.org/10.1083/jcb.201306162
- Murphy DA, Courtneidge SA. The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol 2011; 12:413-26; PMID:21697900; https://doi.org/10.1038/nrm3141
- Frittoli E, Palamidessi A, Marighetti P, Confalonieri S, Bianchi F, Malinverno C, Mazzarol G, Viale G, Martin-Padura I, Garré M, et al. A RAB5/RAB4 recycling circuitry induces a proteolytic invasive program and promotes tumor dissemination. J Cell Biol 2014; 206:307-28; PMID:25049275; https://doi.org/10.1083/jcb.201403127
- Bravo-Cordero JJ, Marrero-Diaz R, Megias D, Genis L, Garcia-Grande A, Garcia MA, Arroyo AG, Montoya MC. MT1-MMP proinvasive activity is regulated by a novel Rab8-dependent exocytic pathway. EMBO J 2007; 26:1499-510; PMID:17332756; https://doi.org/10.1038/sj.emboj.7601606
- Hoshino D, Kirkbride KC, Costello K, Clark ES, Sinha S, Grega-Larson N, Tyska MJ, Weaver AM. Exosome secretion is enhanced by invadopodia and drives invasive behavior. Cell Rep 2013; 5:1159-68; PMID:24290760; https://doi.org/10.1016/j.celrep.2013.10.050
- Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol 2009; 10:513-25; PMID:19603039; https://doi.org/10.1038/nrm2728
- Kajiho H, Kajiho Y, Frittoli E, Confalonieri S, Bertalot G, Viale G, Di Fiore PP, Oldani A, Garre M, Beznoussenko GV, et al. RAB2A controls MT1-MMP endocytic and E-cadherin polarized Golgi trafficking to promote invasive breast cancer programs. EMBO Rep 2016; 17:1061-80; PMID:27255086; https://doi.org/10.15252/embr.201642032
- Tisdale EJ. A Rab2 mutant with impaired GTPase activity stimulates vesicle formation from pre-Golgi intermediates. Mol Biol Cell 1999; 10:1837-49; PMID:10359600; https://doi.org/10.1091/mbc.10.6.1837
- Tisdale EJ, Bourne JR, Khosravi-Far R, Der CJ, Balch WE. GTP-binding mutants of rab1 and rab2 are potent inhibitors of vesicular transport from the endoplasmic reticulum to the Golgi complex. J Cell Biol 1992; 119:749-61; PMID:1429835; https://doi.org/10.1083/jcb.119.4.749
- Tisdale EJ. Rab2 interacts directly with atypical protein kinase C (aPKC) iota/lambda and inhibits aPKCiota/lambda-dependent glyceraldehyde-3-phosphate dehydrogenase phosphorylation. J Biol Chem 2003; 278:52524-30; PMID:14570876; https://doi.org/10.1074/jbc.M309343200
- Tisdale EJ, Kelly C, Artalejo CR. Glyceraldehyde-3-phosphate dehydrogenase interacts with Rab2 and plays an essential role in endoplasmic reticulum to Golgi transport exclusive of its glycolytic activity. J Biol Chem 2004; 279:54046-52; PMID:15485821; https://doi.org/10.1074/jbc.M409472200
- Steffen A, Le Dez G, Poincloux R, Recchi C, Nassoy P, Rottner K, et al. MT1-MMP-dependent invasion is regulated by TI-VAMP/VAMP7. Curr Biol 2008; 18:926-31.
- Williams KC, Coppolino MG. Phosphorylation of membrane type 1-matrix metalloproteinase (MT1-MMP) and its vesicle-associated membrane protein 7 (VAMP7)-dependent trafficking facilitate cell invasion and migration. The Journal of biological chemistry 2011; 286:43405-16.
- Marchesin V, Castro-Castro A, Lodillinsky C, Castagnino A, Cyrta J, Bonsang-Kitzis H, et al. ARF6-JIP3/4 regulate endosomal tubules for MT1-MMP exocytosis in cancer invasion. J Cell Biol 2015; 211:339-58.
- Gillingham AK, Sinka R, Torres IL, Lilley KS, Munro S. Toward a comprehensive map of the effectors of rab GTPases. Dev Cell 2014; 31:358-73.
- Thompson EW, Torri J, Sabol M, Sommers CL, Byers S, Valverius EM, et al. Oncogene-induced basement membrane invasiveness in human mammary epithelial cells. Clin Exp Metastasis 1994; 12:181–94.
- Curtis MW, Johnson KR, Wheelock MJ. E-cadherin/catenin complexes are formed cotranslationally in the endoplasmic reticulum/Golgi compartments. Cell communication & adhesion 2008; 15:365–78.
- Posthaus H, Dubois CM, Laprise MH, Grondin F, Suter MM, Muller E. Proprotein cleavage of E-cadherin by furin in baculovirus over-expression system: potential role of other convertases in mammalian cells. FEBS Lett 1998; 438:306-10.
- Boncompain G, Divoux S, Gareil N, de Forges H, Lescure A, Latreche L, et al. Synchronization of secretory protein traffic in populations of cells. Nat Methods 2012; 9:493-8.
- Luo ML, Gong C, Chen CH, Hu H, Huang P, Zheng M, et al. The Rab2A GTPase promotes breast cancer stem cells and tumorigenesis via Erk signaling activation. Cell reports 2015; 11:111-24.
- Corallino S, Malabarba MG, Zobel M, Di Fiore PP, Scita G. Epithelial-to-Mesenchymal Plasticity Harnesses Endocytic Circuitries. Frontiers in oncology 2015; 5:45.