
ABSTRACT
The ubiquitously expressed small GTPase RhoA is essential for embryonic development and mutated in different cancers. Functionally, it is well described as a regulator of the actin cytoskeleton, but its role in gene regulation is less understood. Using primary mouse keratinocytes with a deletion of the RhoA gene, we have now been exploring how the loss of RhoA affects gene expression. Performing transcription factor reporter assays, we found a significantly decreased activity of a RAR luciferase reporter in RhoA-null keratinocytes. Inhibition of the RhoA effector ROCK in control cells reproduced this phenotype. ATRA and retinal, but not retinol increased RAR reporter activity of keratinocytes with impaired RhoA/ROCK signaling, suggesting that retinol metabolism is regulated by RhoA/ROCK signaling. Furthermore a significant percentage of known ATRA target genes displayed altered expression in RhoA-null keratinocytes. These data reveal an unexpected link between the cytoskeletal regulator RhoA and retinoid signaling and uncover a novel pathway by which RhoA regulates gene expression.
KEYWORDS:
Introduction
The small GTPase RhoA is an important regulator of the actin cytoskeleton.Citation1,2 In its activated, GTP-bound state RhoA interacts with different effector molecules, which mediate the biological effects of RhoA activation.Citation3 Thus, RhoA GTP promotes actin polymerization via formins of the mDia family, inhibits severing of F-actin via phosphorylation of cofilin, and increases cell contraction via phosphorylation of MLC. The latter 2 effects are mediated by the serine-threonine kinase ROCK. RhoA is crucial for the early embryonic development in mice, and mutations in the effector domain of RhoA have recently been identified as a driver mutation in head-and-neck cancer and gastric cancer.Citation4,5
In contrast to the intensely studied role of RhoA in cytoskeletal organization, much less is known on the effects of RhoA on gene expression. In fact, the only well described RhoA-dependent regulation of transcription factor activity is a consequence of RhoA-induced actin polymerization, which releases the transcriptional co-activator MRTF from monomeric G-actin.Citation6 Free MRTF then translocates to the nucleus, where it binds to the SRF transcription factor and activates gene transcription. However, also other regulators of actin polymerization, such as Rac1 and Cdc42 were shown to affect MRTF-SRF activity and in some cells RhoA appears to play only a minor role in this pathway.Citation7,8 RhoA was also described to regulate GATA-4 and c-Jun transcription factors via p38 dependent phosphorylation, but the molecular details of this pathway are unclear.Citation9,10
To look for novel mechanisms of RhoA-dependent regulation of gene expression, we took advantage of primary keratinocytes with a deletion of the RhoA gene.Citation11 Freshly isolated, these cells show reduced phosphorylation of MLC and cofilin in epidermal lysates, suggesting decreased ROCK activation.Citation11 Cultured RhoA-null keratinocytes display a number of defects including enlarged cell area, decreased contraction, increased adhesion, impaired directional migration, and a partial defect in cytokinesis. Conceivably, most of these defects are related to the role of RhoA as a master regulator of actin cytoskeletal organization.
We tested now the activity of selected transcription factors relevant for different skin diseases in RhoA-null keratinocytes using luciferase reporter constructs.
Surprisingly, we did not observe RhoA-dependent changes in MRTF-SRF activation, but revealed a crucial role for RhoA in the metabolism of retinol and, thus, in the regulation of RAR dependent gene expression. This function is mediated by the RhoA effector ROCK and contributes to many changes in gene expression observed in RhoA-null keratinocytes. These data demonstrate for the first time that RhoA/ROCK activation is a major regulator of retinoid signaling, which might contribute to the importance of RhoA during embryonic development and in tumors.
Results
RhoA regulates RAR activity in keratinocytes
In order to identify transcription factors regulated by RhoA in keratinocytes, we transfected reporters expressing the luciferase gene under the control of a promoter containing binding sites for specific transcription factors in primary keratinocytes with a deletion of the RhoA gene and in control cells (). In addition to reporters for SRF and SRF-MRTF, which were earlier described to be regulated by RhoA-dependent changes of the actin cytoskeleton,Citation6 we determined the activation of luciferase reporters for the transcription factors NFAT, STAT1, STAT3, SMAD and RAR, which are important in inflammatory skin diseases and skin tumor.
Figure 1. Decreased RAR reporter activity in RhoA-null keratinocytesCultured control (white bars) and RhoA-null keratinocytes (black bars) were transfected with luciferase reporters for the indicated transcription factors as described in “Materials and Methods.” Shown is the firefly luciferase activity of the reporters normalized to the renilla luciferase activity indicating transfection efficiency (n: 3-15/3-15; ***: p < 0,001).
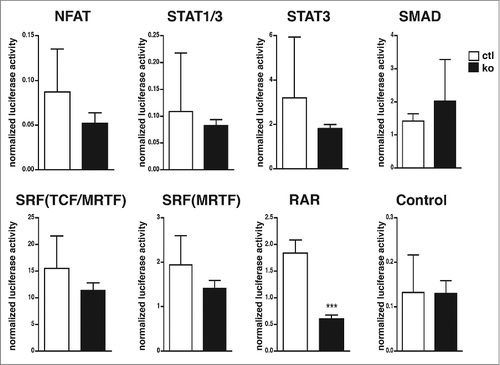
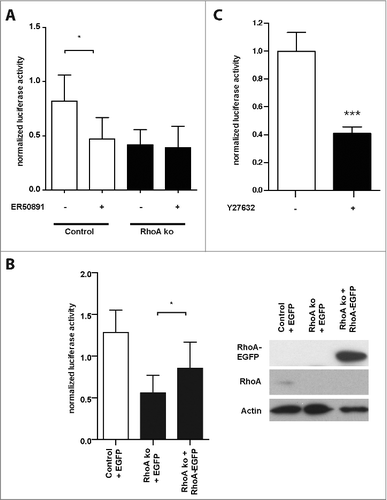
Interestingly, we could not observe a significant alteration in the activation of the reporters for SRF, SRF-MRTF, NFAT, STAT1, STAT3, and SMAD in the absence of RhoA (). All reporters responded to activation of the corresponding signaling pathways when transfected in HEK293 cells (Fig. S1). However, the luciferase reporter for RAR displayed a 3-fold activity decrease in keratinocytes lacking RhoA (). To prove the specificity of the RAR reporter, we repeated the experiment in the presence and absence of the RAR inhibitor ER50891.Citation12 ER50891 inhibited RAR reporter activity in control cells to the level of RhoA-ko keratinocytes, while it showed no significant effect on RhoA-null cells (). Expression of a RhoA-EGFP fusion protein in primary RhoA-null keratinocytes partially rescued the phenotype ().
Figure 2. Decreased RAR reporter activity in RhoA-null keratinocytes is dependent on RhoA, ROCK, RARα. (A) Cultured control (white bars) and RhoA-null keratinocytes (black bars) were transfected with luciferase reporters for RAR and treated for 24 h with 10 μM ER50891, as indicated. Shown is the firefly luciferase activity of the reporters normalized to the renilla luciferase activity indicating transfection efficiency (n: 3; *: p < 0,05). (B) Left: Cultured control (white bars) and RhoA-null keratinocytes (black bars) were co-transfected with luciferase reporters for RAR and an EGFP-RhoA fusion protein or EGFP constructs, as indicated. Shown is the firefly luciferase activity of the reporters normalized to the renilla luciferase activity indicating transfection efficiency (n: 3; *: p < 0,05). Right: Representative blots showing efficient expression of EGFP-RhoA fusion protein and endogenous RhoA in control and RhoA-null keratinocytes. (C) Cultured control keratinocytes were transfected with luciferase reporters for RAR and incubated for 24 h with 25 μM of the ROCK inhibitor Y27632 (black bar) or left untreated (white bar). Shown is the firefly luciferase activity of the reporters normalized to the renilla luciferase activity indicating transfection efficiency (n: 15; ***: p < 0,001).
These data demonstrate that loss of RhoA decreases RAR activation in keratinocytes.
RhoA controls RAR activation via ROCK
To understand the mechanism of the decreased RAR activation, we tested whether inhibition of the RhoA effector ROCK will reduce RAR activation, since it was shown previously that ROCK activation is lowered in freshly isolated RhoA-null keratinocytes compared to controls.Citation11 Indeed, the ROCK inhibitor Y27632 effectively diminished RAR activity in control keratinocytes, similarly to the deletion of the RhoA gene, suggesting that RhoA controls RAR activity via ROCK ().
RhoA/ROCK signaling controls RAR ligand availability
RAR activity can be controlled at the level of the RAR transcription factor or at the level of the retinoic acid ligands, which activate RAR. To distinguish between these possibilities, we tried to rescue the impairment of RAR activation caused by the deletion of the RhoA gene or the ROCK inhibitor by increasing the amount of RAR ligands. To this end, we added retinol, retinal, or ATRA to the medium. ATRA can directly bind to RAR, while retinol and retinal require oxidation to be metabolized to ATRA.
Retinal and ATRA strongly activated RAR reporter activity in Y27632 treated control cells () and in RhoA-ko keratinocytes (), indicating that RhoA/ROCK signaling controls the availability of RAR ligands and neither amount nor modification of RAR. Interestingly, we observed an increased toxicity of retinal to RhoA-ko keratinocytes compared to control cells, suggesting an impaired retinal metabolism in the mutant cells ().
Figure 3. Retinal and ATRA rescue reduced RAR-signaling activity in ROCK inhibitor treated keratinocytes. (A-C) Cultured control keratinocytes were transfected with luciferase reporters for RAR-mediated signaling and treated for 24 h with 25 μM Y27632, 10 μM retinol (A), 2 μM retinal (B), 1 μM ATRA (C), or left untreated, as indicated. Shown is the firefly luciferase activity of the reporters normalized to the renilla luciferase activity indicating transfection efficiency. All the samples were statistically compared to untreated cells. (n: 4-14; **: p < 0,01; ***: p < 0,001). (D-F) Cultured RhoA-null keratinocytes were transfected with luciferase reporters for RAR-mediated signaling and treated for 24 h with 25 μM Y27632, 10 μM retinol (D), 2 μM retinal (E), 1μM ATRA (F), or left untreated, as indicated. Shown is the firefly luciferase activity of the reporters normalized to the renilla luciferase activity indicating transfection efficiency. All the samples were statistically compared to untreated cells (n: 4-14; **: p < 0,01; ***: p < 0,001). (G) Treatment for 24 h with 2 μM retinal induced increased cell death in RhoA-null keratinocytes. Shown are representative pictures of keratinocytes in culture treated as indicated (size bar = 50mm).
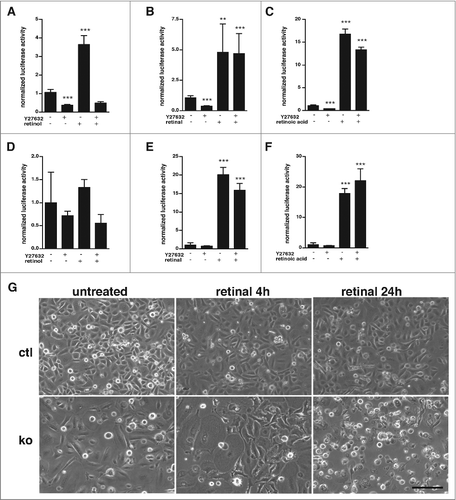
In contrast, Y27632 cells treated and RhoA-ko cells were not responding to retinol, which in control cells caused a 3.5-fold stimulation of RAR reporter activity (). These data suggest that RhoA/ROCK signaling may control the oxidation of retinol to retinal.
Oxidation of retinol to retinal can be catalyzed by many different enzymes belonging the class of alcohol dehydrogenases and short-chain dehydrogenases/reductases (SDRs). Overexpression experiments in human skin rafts suggested an important function for the SDR gene RDH10 in the regulation of retinoic acid responsive genes.Citation13 We therefore tested whether deletion of the RDH10 gene in primary keratinocytes would result in defective RARE reporter activation similar to ablation of RhoA. However, despite efficient CRISPR/Cas9 mediated inactivation of the RDH10 gene we did not observe altered RARE reporter activation, indicating involvement of other genes than RDH10 (Fig. S2A, B).
Altered expression of all-trans retinoic acid regulated genes in RhoA-null keratinocytes
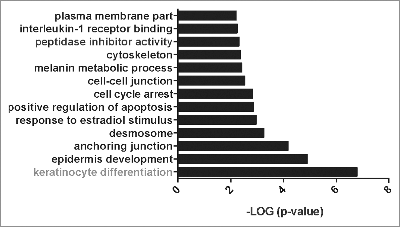
To assess to which extent the decreased RAR activation contributes to changes in gene expression observed in RhoA-null keratinocytes, we first analyzed gene expression in primary RhoA-null and control keratinocytes by microarray analysis. Excluding lowly expressed genes, we identified 311 differentially expressed genes (Table S1). Functional annotation of the 183 genes upregulated in RhoA-null keratinocytes using the DAVID programCitation14 revealed a particular increase of genes related to keratinocyte differentiation () such as loricrin, involucrin, or filaggrin, suggesting an altered terminal differentiation in the absence of RhoA. Since retinoic acid is known to reduce terminal differentiation of keratinocytes, increased expression of these genes is expected if retinoid signaling is reduced. Interestingly, we could not observe any differential expression of genes involved in retinoid signaling or metabolism (data not shown).
Figure 4. Gene ontology of upregulated genes in RhoA-ko keratinocytes in vitro Significantly enriched gene ontology categories of upregulated genes in RhoA-ko keratinocytes in vitro according to EASE Score, a modified Fisher Exact P-Value (p < 0.01).
Previous studies revealed a number of genes decreased in keratinocytes treated with ATRA.Citation15-17 Out of 47 genes analyzed, 18 (39%) were increased in RhoA-null keratinocytes, which suggests reduced RAR activation (S2 Table). Furthermore, of 18 genes shown to be upregulated by ATRA, 3 (17%) were decreased in RhoA-deficient keratinocytes. These data suggest that diminished RAR activation contributes partially to the altered gene expression observed in RhoA-deficient cells compared to controls.
Next, we analyzed 6 suggested ATRA target genes regulated by RhoA (Flg, Cdsn, Dsc1, Krt6b, Lor, Dsg1b) and 3 additional genes upregulated in RhoA-null cells according to the microarray gene expression (Stfa3, Gjb2, Tnc; Table S1). First, we validated by qRT-PCR the increased expression of all these genes in RhoA-null keratinocytes () and that they are all regulated in primary mouse keratinocytes by ATRA (). Then we assessed their dependency on ROCK signaling by inactivating the genes for both ROCK1 and ROCK2 in primary cultured mouse keratinocytes by CRISPR/Cas9 genome editing. Six of the genes (Cdsn, Dsc1, Krt6b, Lor, Gjb2, Stfa) were significantly increased in ROCK1/2-ko keratinocytes compared to control cells (). Flg and Dsg1b were not changed, while Tnc was decreased.
Figure 5. ROCK inhibition increases expression of retinoic acid dependent genes in keratinocytes. (A) qRT-PCR of RNA samples from cultured control and RhoA-null keratinocytes. Shown are fold inductions from RhoA-ko compared to control keratinocytes (n > 3; *: p < 0.05; **: p < 0,01; ***: p < 0,001). (B) Cultured RhoA-null keratinocytes were treated for 24 h with 1 μM ATRA. RNA was isolated and qRT-PCR was carried out for the indicated genes. Shown are fold inductions from treated keratinocytes compared to untreated keratinocytes. All the samples were statistically compared to untreated cells (n > 3; *: p < 0.05; **: p < 0,01; ***: p < 0,001). (C) qRT-PCR of RNA samples from cultured control and ROCK1/2-ko keratinocytes. Shown are fold inductions from ko compared to control keratinocytes (n > 3; *: p < 0.05; **: p < 0,01; ***: p < 0,001). (D) Representative blot showing efficient loss of ROCK1 and ROCK2 protein in ROCK1 and/or ROCK2-ko keratinocytes.
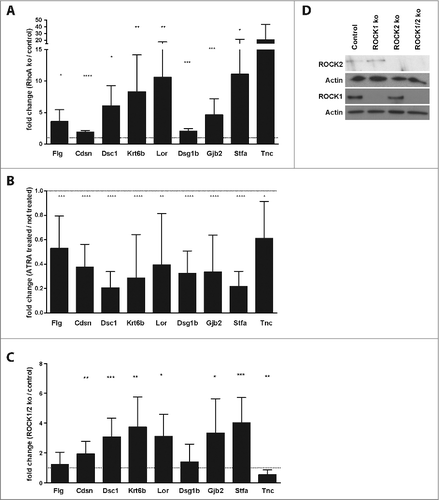
Taken together these data demonstrate that RhoA-ROCK dependent regulation of RAR activity is an important pathway by which RhoA regulates gene expression in keratinocytes.
Loss of RhoA increases the size of the stem cell population in skin
Even though retinoic acid inhibits terminal differentiation of keratinocytes, a number of studies demonstrate that retinoic acid promotes differentiation of stem cells.Citation18,19 In head-and neck cancer cell lines and embryonic stem cells retinoic acid reduces the expression of stem cell markers and suppresses Wnt/β-catenin signaling.Citation20,21 We therefore hypothesized that the decreased retinoid signaling in RhoA-null keratinocytes in vivo might lead to an expansion of keratinocyte stem cells, which are dependent on Wnt/β-catenin.Citation22 To address this question, we assessed the percentage of the α6 integrin+ CD34+ cell population in freshly isolated primary keratinocytes from mice with a keratinocyte-specific knockout of RhoA and in controls by FACS. Indeed, this cell population, which contains skin stem cells, increased in the absence of RhoA from 4% to more than 7% (), suggesting that loss of RhoA in keratinocytes might increase the stem cell population in skin.
Figure 6. α6highCD34high stem cells increased in the absence of RhoA. (A) Representative dot plot and quantification of FACS analysis for α6highCD34high cells in control and RhoA-ko freshly isolated keratinocytes from epidermis. (n: 4; *: p < 0.05). (B) Clonogenic assay of α6highCD34high sorted epidermal cells from control mice treated with 1 μM ATRA, 25 μM Y27632 or 10 μM ER50891 as indicated (n: 3 in triplicate; *: p < 0.05; ***: p < 0,001).
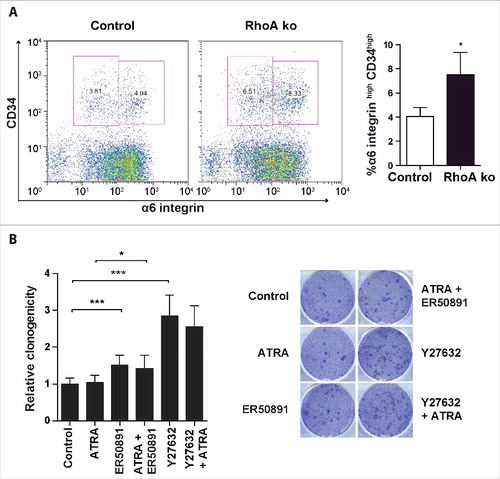
To investigate the respective role of ROCK and retinoic acid signaling on keratinocyte stem cell growth in vitro, we performed a clonogenic growth assay with primary α6 integrin+ CD34+ keratinocytes from control mice. While stimulation of retinoic acid signaling by ATRA did not affect the clonogenicity of primary mouse keratinocytes, inhibition of RARα by ER50891 lead to a small, but significant increase, indicating that basic retinoic acid signaling in culture keratinocytes inhibits clonogenicity (). Inhibition of ROCK by Y27632 strongly increased clonogenicity, which was slightly, but non-significantly decreased by RARα inhibition. These data suggest that decreased retinoic acid signaling might contribute to the increased clonogenicity induced by ROCK inhibition. However, most of this increase seems independent of retinoic acid signaling.
Discussion
Retinoid signaling has important functions during development and in adults and has also been implicated in cancer.Citation23,24 We show now that RhoA/ROCK signaling is a potent regulator of retinoid signaling by controlling metabolism of retinol ().
Figure 7. RhoA/ROCK regulates RA signaling in primary keratinocytes RhoA/ROCK signaling regulates the oxidation of retinol to retinal.
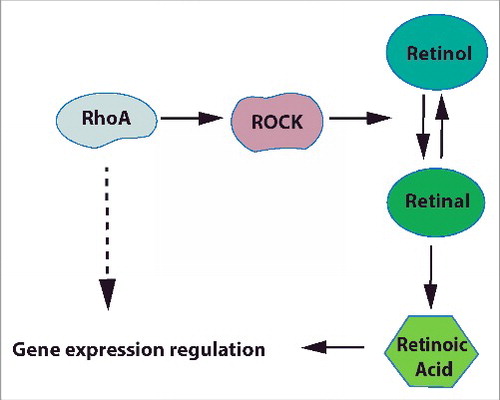
Since retinoic acid and retinal, but not retinol rescued the loss of RhoA/ROCK inhibitor induced defect in RAR activity in keratinocytes, the major target of RhoA/ROCK signaling appears to be the oxidation of retinol to retinal, which already previously has been described as the rate limiting step of retinoic acid metabolism.Citation25 Also the increased toxicity of retinal to RhoA-ko keratinocytes in cell culture points to an impaired equilibrium between toxic retinal and less toxic retinol. As a consequence of the defective retinol metabolism, retinoic acid levels should be reduced in RhoA-ko keratinocytes. We tried to test this, but in accordance with earlier publications, retinoid levels in RhoA-ko and control keratinocytes as determined by HPLC were below detection limit (data not shown).Citation26
Retinoid metabolism is very complex and involves, in addition to the oxidation of retinol to retinal and retinoic acid, the transport of retinol into the cell, storage, and degradation.Citation27 Moreover, a large number of enzymes were reported to be able to oxidize retinol to retinal or to catalyze the reverse reaction. Since no specific inhibitors are available for the different enzymes, knockdown or knockout screens have to be applied to identify potential target molecules for the RhoA/ROCK dependent regulation of retinoid metabolism. Since ROCK is a kinase, it might influence the activity or turnover of retinol oxidising enzymes by phosphorylation, without affecting the mRNA expression level. Mass spectroscopy analysis revealed threonine-phosphorylation of, for example, the retinol oxidase RDH10 (phosphosite.org), but it has not been tested whether this influences the respective enzyme activities.
Mice lacking the 2 isoforms of ROCK, ROCK1 and ROCK2, die before embryonic day 9.5 showing defects in vascular remodelling in the yolk sac, which could be modeled by Y27632 treatment of wild-type embryos.Citation28 Interestingly, also retinoid acid signaling affects vascular remodelling during embryogenesis.Citation23,29 It seems possible that defective RA signaling is contributing to the early embryonic death of mice with a constitutive knockout of RhoA (Brakebusch et al., unpublished results).
In cell culture, ROCK inhibitor treatment is known to increase the number of keratinocytes with stem cell properties and preventing their senescence.Citation30,31 The underlying mechanism for this effect is not clear, but could involve the decreased retinoid acid signaling observed in Y27632 treated keratinocytes. Retinoic acid promotes stem cell differentiation and, thus, a reduction of stem cell numbers in different systems.Citation18,19 Importantly, we show now expansion of a keratinocyte stem cell population in RhoA-deficient mice, which would be in line with a decreased retinoid signaling in vivo. Since an increased number of stem cells and decreased senescence are both suggested to enhance the susceptibility to tumor formation,Citation32,33 our data would predict that mice with a keratinocyte-restricted deletion of the RhoA gene are more prone to chemically induced skin tumor formation. Indeed, retinoids reduce growth of head-and-neck squamous cell carcinoma in nude mice.Citation34
Potentially inactivating mutations of RhoA have been found in head-and neck cancer in humans.Citation4 Since loss of RhoA function might result in decreased retinoid signaling, this patient group might benefit from treatment with retinoic acid, which could reduce the cancer stem cell population.
RhoA-null keratinocytes showed differential expression also of genes, which are not regulated by retinoic acid signaling. A possible pathway contributing to this could be YAP/TAZ signaling that transduces mechanical and cytoskeletal cues and is partially dependent on Rho signaling.Citation35 Furthermore, we found among the upregulated genes in RhoA-null cells RORα, which is a transcription factor inducing keratinocyte differentiationCitation36 that could contribute to the increased expression of keratinocyte differentiation markers. RhoA-null keratinocytes display increased protein amounts and activation of RhoB,Citation11 which might affect gene expression of RhoA-null cells. However, our data indicate that the increased the RhoB activation is not able to fully replace RhoA and to maintain normal gene expression in RhoA-null keratinocytes.
In conclusion, we identified a novel pathway through which RhoA/ROCK signaling controls retinol metabolism and thus retinoic acid dependent gene expression, which is involved in keratinocyte differentiation and clonogenicity.
Materials and methods
Mice
Mice with a keratinocyte-restricted deletion of the RhoA (RhoA fl/fl K5 cre) gene were generated using the Cre-loxP system as describe previously and kept as 129Sv/C57BL6 outbred.Citation11 The protocol for the generation and breeding of the mice was approved by the National Animal Research Committee of Denmark. Animals were kept according to national and European animal welfare laws in an AAALAC accredited animal house under SPF conditions. Any approval was needed to perform this study, according to the Danish legislation (Bekendtgørelse af dyreværnsloven. 1343, 04/12/2007, Bekendtgørelse af lov om dyreforsøg. 253, 08/03/2013, Bekendtgørelse om dyreforsøg 31. januar 2013). Cervical dislocation was used as the method of sacrifice and no experimental procedures were carried out prior to sacrifice.
Cell culture
Isolation of primary keratinocytes from adult mice and subsequent in vitro culture was carried out as described previously.Citation37 For in vitro experiments, subconfluent, growing keratinocyte cultures were used. When indicated, keratinocytes were treated for specified times with 25 μM Y27632 (Calbiochem), 1 µM all-trans retinoic acid, 10 μM retinol, 2 μM retinal (all Sigma), or 10 μM ER50891 (Tocris Bioscience).
HEK293T cells were grown in DMEM medium (Invitrogen) supplemented with 10% fetal bovine serum (Hyclone) and penicillin/streptomycin (Invitrogen).
CRISPR/Cas9 knockout keratinocytes
Single guide RNAs (sgRNA) targeting ROCK1, ROCK2, and RDH10 coding regions were cloned into the lentiviral vector lentiCRISPRv2 (GeCKO; Addgene) according to manufacture´s instructions. The oligonucleotide sequences corresponding to the sgRNA were: ROCK1: F: 5′-GCCGATTTGGGATCCCGCAGC-3′, R: 5′-GCTGCGGGATCCCAAATCGGC-3′; ROCK2: F: 5′ -GCGGGCATTTTCCCCGTCGGC- 3′, R: 5′-GCCGACGGGGAAAATGCCCGC-3′. RDH10_5p: F: 5′-CACCGTTACCTACACTTGTGACGTG-3′, R: 5′-AAACCACGTCACAAGTGTAGGTAAC-3′; RDH10_3p: F: 5′-CACCGTGCGTGGCAGTTGACCATCA-3′, R: 5′AAACTGATGGTCAACTGCCACGCAC 3′.
For the lentivirus production, HEK293T cells were used by transfection of lentiCRISPRv2 with the corresponding sgRNA or the empty vector, together with pCMV-VSV-G and psPAX2 using polyethylenimine as previously described.Citation38 The following day, HEK293T medium was exchange by keratinocyte medium. After 48 hours, supernatant containing the viral particles was collected, mixed with fresh medium (1:1) containing 8 μg/ml polybrene (Sigma), and added to the target cells. After 48h, keratinocytes were selected with 3 μg/ml puromycin for 5 d for ARN collection, or for 4 d for luciferase assays.
Biochemical analysis
Cell lysis and Western blotting was performed according to standard protocols. The following antibodies were used: mouse anti RhoA (sc-179; Santa Cruz), rabbit anti ROCK1 (sc-5560; Santa Cruz), rabbit anti ROCK2 (sc-5561; Santa Cruz), and mouse anti Actin (ab6276, abcam). Horseradish-peroxidase-coupled goat anti-rabbit and goat anti-mouse antibodies were used as secondary antibodies (all Jackson ImmunoResearch).
Microarray gene expression analysis and qRT PCR
For microarray analysis, RNA was isolated from cultured primary keratinocytes (3 independent biological samples from control and RhoA-null keratinocytes) using the RNeasy kit (Qiagen). Hybridization to GeneChip Mouse Genome 430 2.0 Array (Affymetrix) was carried out at the Copenhagen University Hospital Microarray Center using standard protocols.
The microarray data discussed in this publication have been deposited in NCBI's Gene Expression OmnibusCitation39 and are accessible through GEO Series accession number GSE64714 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE64714).
For qRT-PCR analysis, RNA was reverse transcribed using the RevertAid H Minus First Strand cDNA Synthesis kit (Thermo Scientific). qRT-PCR was performed on the Applied Biosystems 7300 Real Time PCR system using SYBR green incorporation following standard protocols. The primers used were: Gjb2: F: 5′-ATCCTCGGGGGTGTCAACAA-3′, R: 5′-AGACAAAATCGGCTTGCTCATC-3′; Tnc: F: 5′-ACGGCTACCACAGAAGCTG-3′, R: 5′-ATGGCTGTTGTTGCTATGGCA-3′; Stfa3: F: 5′-GAGCAAACCAATGAGAAATA-3′, R: 5′-CAAGGAGAAGAATCAGGTCA-3′; Flg: F: 5′-GGACAACTACAGGCAGTCTTGAAGA-3′, R: 5′-CATTTGCATGAAGACTTCAGCG-3′; Cdsn: F: 5′- GTTTCTGCCTGGTTGTGCTT-3′, R: 5′-GAGGGAACTGGGAGTCTGTG-3′; Dsc1: F: 5′-TGGTATTATTTTGTGATGCCTGT-3′, R: 5′-GGCTGGGTGATTTGAGACAC-3′; Krt6b: F: 5′-GCACTGAAATCCCAACTACCA-3′, R: 5′-GAGAAGCGAGAGGACACAGC-3′; Lor: F: 5′-CTCATCTTCCCTGGTGCTTC-3′, R: 5′-AGGTCTTTCCACAACCCACA-3′; Dsg1: F: 5′-CTGCCCTGCTGCTCACTT-3′, R: 5′-CCATTCTCTTTTCTGCCTTCTG-3′; Cypa: F: 5′-TTCTGCTGTCTTTGGAACTTTGTC-3′, R: 5′-ATGGTCAACCCCACCGTGT-3′. The Ct value was calculated based on duplicates and normalized to the housekeeping gene Cypa.
Luciferase assay
Primary keratinocytes were transfected at 30-40% confluence with pGL3-RARE-luciferase construct (Addgene 13548; “RAR”), 3D.A-Luc (kindly obtained by G. Posern, Martinsried, Germany; “SRF (MRTF)”), pGL3-NFAT luciferase (Addgene 17870; “NFAT”), SBE4-Luc (Addgene 16495; “SMAD”), 3xLy6e-pZLuc-TK (Addgene 8689; “STAT1/3”), P4RTK GL4.10 (Addgene 16057; “control” for testing the luciferase assay), SIE-th-lucSTAT3 (kindly obtained by G. Schmidt, Freiburg, Germany; “STAT3”), pSRE-luc construct (M. Frödin, Copenhagen, Denmark; “SRF (TCF/MRTF)”) and pRL-TK-Renilla (Promega) using TransIT-Keratinocyte Transfection Reagent (Mirus) following the instructions of the manufacturer. A total of 1 μg of DNA was used for transfection in 24 well plates or 5 μg was used in 6 well plates (2 Luc: 1 Ren). Two days later cells were harvested and Renilla and firefly luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega) following the instructions of the manufacturer using a Lumat CB9597 (Berthold Technologies).
Functionality of the luciferase reporters was tested in HEK293T cells. A total of 50 ng of DNA was used for transfection in 12 well plates (2 Luc: 1 Ren) using polyethylenimine as previously described.Citation38 Cells transfected with SBE4-Luc construct were pre-treated with 0,5% serum medium for 6 hours, following 24h treatment with 10% serum medium supplemented with 25 ng/ml of TGFβ. Cells transfected with 3xLy6e-pZLuc-TK or SIE-th-lucSTAT3 constructs were pre-treated with 0,5% serum medium for 6 hours, following 24h treatment with 10% serum medium supplemented with 10 ng/ml of human Interleukin 6 (hIL6). Cells transfected with 3D.A-Luc or pSRE-luc constructs were pre-treated with 0,5% serum media for 18 hours, following 6h treatment with 15% serum media. Cells transfected with pGL3-NFAT luciferase construct were pre-treated with 0,5% serum medium for 18 hours, following 6h treatment with 10 ng/ml of phorbol 12-myristate 13- acetate (PMA) and 0,5 μM Ionomycin (Iono).
For a rescue experiment, primary keratinocytes in 6 well plates were co-transfected with pGL3-RARE-luciferase construct (2 μg), pRL-TK-Renilla (1 μg) and vectors expressing an EGFP-RhoA fusion protein or EGFPCitation11 (2 μg) using the TransIT-Keratinocyte Transfection Reagent (Mirus). After two days, cells were selected for 2 extra days with geneticin (25 ug/ml), following by luciferase activity measurement as indicated above.
Fluorescence activated cell sorter (FACS) analysis and clonogenic assay
Surface marker expression of single cells suspensions from epidermisCitation37 was analyzed by FACS. Freshly isolated keratinocytes were labeled with fluorescein isothiocyanate–conjugated CD49f antibody (integrin 6 chain; 555735, BD Biosciences) and anti mouse CD34 eFluor 660 (50-0341-80, eBioscience). Data were collected by FACScalibur flow cytometry system (BD Bioscience) using the BD CellQuest Pro software and analyzed by Flowjo.
For the clonogenic assay, α6highCD34high freshly isolated primary keratinocytes from control mice were sorted by FACS Aria and plated in clonal density, as previously described.Citation40 Briefly, 5000 of the flow sorted epidermal cells were plated in 6 well plates previously covered by a monolayer of Mitomycin C-inactivated 3T3 J2 feeder cells. When indicated, epidermal cells were treated since the seeding time during 24h. After 14 days, colonies were washed in PBS, fixed with 70% ethanol for 20 min, and stained with 0,5% crystal violet solution for 15 min. Crystal violet excess was removed by 6 washed with water. Colonies were counted.
Statistics
Data are presented as means with error bars representing standard deviation. Statistical significance was determined by the 2-tailed Student's t-test. For all statistical analyses, P values are denoted as: ***P < 0.001, **P < 0.01 and *P < 0.05.
Abbreviations
| ATRA | = | all-trans retinoic acid |
| Cdsn | = | Corneodesmosin |
| DAVID | = | Database for Annotation, Visualization and Integrated Discovery |
| Dsg1b | = | desmoglein 1 β |
| Dsc1 | = | Desmocollin 1 |
| FACS | = | Fluorescence activated cell sorter |
| Flg | = | Filaggrin |
| Gjb2 | = | Gap Junction Protein Beta 2 |
| hIL6 | = | human Interleukin 6 |
| HPLC | = | High Performance Liquid Chromatography |
| Iono | = | Ionomycin |
| Krt6b | = | Keratin 6B |
| Lor | = | Loricrin |
| MLC | = | Myosin Light Chain |
| MRTF | = | Myocardin-related transcription factors |
| NFAT | = | Nuclear factor of activated T-cells |
| PMA | = | phorbol 12-myristate 13- acetate |
| RAR | = | Retinoic acid receptor |
| RDH10 | = | Retinol Dehydrogenase 10 |
| ROCK | = | Rho-associated protein kinase |
| SDRs | = | Short-chain dehydrogenases/reductases |
| SRF | = | Serum response factor |
| STAT | = | Signal Transducers and Activators of Transcription |
| Stfa3 | = | Stefin A3 |
| TGFβ | = | Transforming growth factor β |
| Tnc | = | Tenascin C |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental_materials.zip
Download Zip (1.2 MB)Acknowledgments
We would like to thank Volkan Turan for excellent technical help, and the Drs. Morten Frödin, Gudula Schmidt and Guido Posern for kindly providing reporter constructs. We also want to acknowledge Drs. Michael Underhill, Jim Darnell, Jerry Crabtree, Bert Vogelstein and Robert Benezra for their constructs obtained from Addgene. We thank Dr. John Couchman for helpful comments on the manuscript.
Funding
This work was supported by the Danish Research Council, the Novo Nordisk Foundation, the Lundbeck Foundation, and by the Marie-Curie Fellowship program of the European Community.
Related Research Data
References
- Jaffe AB, Hall A. Rho GTPases: Biochemistry and biology. Annu Rev Cell Dev Bi 2005; 21:247-69; https://doi.org/10.1146/annurev.cellbio.21.020604.150721
- Pedersen E, Brakebusch C. Rho GTPase function in development: how in vivo models change our view. Exp Cell Res 2012; 318:1779-87; PMID:22659168; https://doi.org/10.1016/j.yexcr.2012.05.004
- Bustelo XR, Sauzeau V, Berenjeno IM. GTP-binding proteins of the Rho/Rac family: regulation, effectors and functions in vivo. BioEssays 2007; 29:356-70; PMID:17373658; https://doi.org/10.1002/bies.20558
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014; 505:495-501; PMID:24390350; https://doi.org/10.1038/nature12912
- Wang K, Yuen ST, Xu J, Lee SP, Yan HH, Shi ST, Siu HC, Deng S, Chu KM, Law S, et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat Genet 2014; 46:573-82; PMID:24816253; https://doi.org/10.1038/ng.2983
- Posern G, Treisman R. Actin' together: serum response factor, its cofactors and the link to signal transduction. Trends Cell Biol 2006; 16:588-96; PMID:17035020; https://doi.org/10.1016/j.tcb.2006.09.008
- Busche S, Descot A, Julien S, Genth H, Posern G. Epithelial cell-cell contacts regulate SRF-mediated transcription via Rac-actin-MAL signalling. J Cell Sci 2008; 121:1025-35; PMID:18334560; https://doi.org/10.1242/jcs.014456
- Sebe A, Erdei Z, Varga K, Bodor C, Mucsi I, Rosivall L. Cdc42 regulates myocardin-related transcription factor nuclear shuttling and alpha-smooth muscle actin promoter activity during renal tubular epithelial-mesenchymal transition. Nephron Exp Nephrol 2010; 114:E117-25; PMID:20016221; https://doi.org/10.1159/000265550
- Marinissen MT, Chiariello M, Gutkind JS. Regulation of gene expression by the small GTPase Rho through the ERK6 (p38 gamma) MAP kinase pathway. Genes Dev 2001; 15:535-53; PMID:11238375; https://doi.org/10.1101/gad.855801
- Charron F, Tsimiklis G, Arcand M, Robitaille L, Liang Q, Molkentin JD, Meloche S, Nemer M. Tissue-specific GATA factors are transcriptional effectors of the small GTPase RhoA. Genes Dev 2001; 15:2702-19; PMID:11641276; https://doi.org/10.1101/gad.915701
- Jackson B, Peyrollier K, Pedersen E, Basse A, Karlsson R, Wang Z, Lefever T, Ochsenbein AM, Schmidt G, Aktories K, et al. RhoA is dispensable for skin development, but crucial for contraction and directed migration of keratinocytes. Mol Biol Cell 2011; 22:593-605; PMID:21209320; https://doi.org/10.1091/mbc.E09-10-0859
- Kikuchi K, Tagami K, Hibi S, Yoshimura H, Tokuhara N, Tai K, Hida T, Yamauchi T, Nagai M. Syntheses and evaluation of quinoline derivatives as novel retinoic acid receptor alpha antagonists. Bioorg Med Chem Lett 2001; 11:1215-8; PMID:11354380; https://doi.org/10.1016/S0960-894X(01)00177-9
- Lee SA, Belyaeva OV, Wu L, Kedishvili NY. Retinol dehydrogenase 10 but not retinol/sterol dehydrogenase(s) regulates the expression of retinoic acid-responsive genes in human transgenic skin raft culture. J Biol Chem 2011; 286:13550-60; PMID:21345790; https://doi.org/10.1074/jbc.M110.181065
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44-57; PMID:19131956; https://doi.org/10.1038/nprot.2008.211
- Bernard FX, Pedretti N, Rosdy M, Deguercy A. Comparison of gene expression profiles in human keratinocyte mono-layer cultures, reconstituted epidermis and normal human skin; transcriptional effects of retinoid treatments in reconstituted human epidermis. Exp Dermatol 2002; 11:59-74; PMID:11952828; https://doi.org/10.1034/j.1600-0625.2002.110107.x
- Lee DD, Stojadinovic O, Krzyzanowska A, Vouthounis C, Blumenberg M, Tomic-Canic M. Retinoid-Responsive Transcriptional Changes in Epidermal Keratinocytes. J Cell Physiol 2009; 220:427-39; PMID:19388012; https://doi.org/10.1002/jcp.21784
- Torma H. Regulation of keratin expression by retinoids. Dermato-endocrinology 2011; 3:136-40; PMID:22110773; https://doi.org/10.4161/derm.15026
- Duester G. Retinoid signaling in control of progenitor cell differentiation during mouse development. Semin Cell Dev Biol 2013; 24:694-700; PMID:23973941; https://doi.org/10.1016/j.semcdb.2013.08.001
- Gudas LJ. Retinoids induce stem cell differentiation via epigenetic changes. Semin Cell Dev Biol 2013; 24:701-5; PMID:23973942; https://doi.org/10.1016/j.semcdb.2013.08.002
- Osei-Sarfo K, Gudas LJ. Retinoic acid suppresses the canonical Wnt signaling pathway in embryonic stem cells and activates the noncanonical Wnt signaling pathway. Stem Cells 2014; 32:2061-71; PMID:24648413; https://doi.org/10.1002/stem.1706
- Lim YC, Kang HJ, Kim YS, Choi EC. All-trans-retinoic acid inhibits growth of head and neck cancer stem cells by suppression of Wnt/beta-catenin pathway. Eur J Cancer 2012; 48:3310-8; PMID:22640830; https://doi.org/10.1016/j.ejca.2012.04.013
- Collins CA, Watt FM. Dynamic regulation of retinoic acid-binding proteins in developing, adult and neoplastic skin reveals roles for beta-catenin and Notch signalling. Dev Biol 2008; 324:55-67; PMID:18805411; https://doi.org/10.1016/j.ydbio.2008.08.034
- Rhinn M, Dolle P. Retinoic acid signalling during development. Development 2012; 139:843-58; PMID:22318625; https://doi.org/10.1242/dev.065938
- Tang XH, Gudas LJ. Retinoids, retinoic acid receptors, and cancer. Annu Rev Pathol 2011; 6:345-64; PMID:21073338; https://doi.org/10.1146/annurev-pathol-011110-130303
- Kumar S, Sandell LL, Trainor PA, Koentgen F, Duester G. Alcohol and aldehyde dehydrogenases: Retinoid metabolic effects in mouse knockout models. Bba-Mol Cell Biol L 2012; 1821:198-205.
- Kang S, Duell EA, Fisher GJ, Datta SC, Wang ZQ, Reddy AP, Tavakkol A, Yi JY, Griffiths CE, Elder JT, et al. Application of retinol to human skin in vivo induces epidermal hyperplasia and cellular retinoid binding proteins characteristic of retinoic acid but without measurable retinoic acid levels or irritation. J Invest Dermatol 1995; 105:549-56; PMID:7561157; https://doi.org/10.1111/1523-1747.ep12323445
- Kedishvili NY. Enzymology of retinoic acid biosynthesis and degradation. J Lipid Res 2013; 54:1744-60; PMID:23630397; https://doi.org/10.1194/jlr.R037028
- Kamijo H, Matsumura Y, Thumkeo D, Koike S, Masu M, Shimizu Y, Ishizaki T, Narumiya S. Impaired vascular remodeling in the yolk sac of embryos deficient in ROCK-I and ROCK-II. Genes Cells 2011; 16:1012-21; PMID:21895889; https://doi.org/10.1111/j.1365-2443.2011.01546.x
- Bohnsack BL, Lai L, Dolle P, Hirschi KK. Signaling hierarchy downstream of retinoic acid that independently regulates vascular remodeling and endothelial cell proliferation. Gene Dev 2004; 18:1345-58; PMID:15175265; https://doi.org/10.1101/gad.1184904
- Terunuma A, Limgala RP, Park CJ, Choudhary I, Vogel JC. Efficient procurement of epithelial stem cells from human tissue specimens using a Rho-associated protein kinase inhibitor Y-27632. Tissue Eng Part A 2010; 16:1363-8; PMID:19912046; https://doi.org/10.1089/ten.tea.2009.0339
- Chapman S, McDermott DH, Shen K, Jang MK, McBride AA. The effect of Rho kinase inhibition on long-term keratinocyte proliferation is rapid and conditional. Stem Cell Res Ther 2014; 5:60; PMID:24774536; https://doi.org/10.1186/scrt449
- Singh SR. Stem cell niche in tissue homeostasis, aging and cancer. Curr Med Chem 2012; 19:5965-74; PMID:22963571; https://doi.org/10.2174/0929867311209065965
- Pincelli C, Marconi A. Keratinocyte stem cells: friends and foes. J Cell Physiol 2010; 225:310-5; PMID:20568222; https://doi.org/10.1002/jcp.22275
- Shalinsky DR, Bischoff ED, Gregory ML, Gottardis MM, Hayes JS, Lamph WW, Heyman RA, Shirley MA, Cooke TA, Davies PJ, et al. Retinoid-induced suppression of squamous cell differentiation in human oral squamous cell carcinoma xenografts (line 1483) in athymic nude mice. Cancer Res 1995; 55:3183-91; PMID:7541715
- Low BC, Pan CQ, Shivashankar GV, Bershadsky A, Sudol M, Sheetz M. YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS Lett 2014; 588:2663-70; PMID:24747426; https://doi.org/10.1016/j.febslet.2014.04.012
- Dai J, Brooks Y, Lefort K, Getsios S, Dotto GP. The retinoid-related orphan receptor RORalpha promotes keratinocyte differentiation via FOXN1. PLoS One 2013; 8:e70392; PMID:23922987; https://doi.org/10.1371/journal.pone.0070392
- Pedersen E, Basse A, Lefever T, Peyrollier K, Brakebusch C. Rho GTPase knockout induction in primary keratinocytes from adult mice. Methods Mol Biol 2012; 827:157-66; PMID:22144274; https://doi.org/10.1007/978-1-61779-442-1_11
- Ehrhardt Christina, Schmolke Mirco, Matzke Andreas, Knoblauch Alexander, Will Carola, Wixler Viktor, Ludwig S. Polyethylenimine, a cost-effective transfection reagent. Signal Transduction 2006; 6:179-84; https://doi.org/10.1002/sita.200500073
- Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 2002; 30:207-10; PMID:11752295; https://doi.org/10.1093/nar/30.1.207
- Jensen KB, Driskell RR, Watt FM. Assaying proliferation and differentiation capacity of stem cells using disaggregated adult mouse epidermis. Nat Protoc 2010; 5:898-911; PMID:20431535; https://doi.org/10.1038/nprot.2010.39