
ABSTRACT
Successful cancer metastasis relies on the ability of cancer cells to survive independently of attachment to the extracellular matrix (ECM) and to overcome ECM-detachment-induced death programs. This can be accomplished through activating mutations in cellular oncogenes that subsequently lead to the inhibition of anoikis and to alterations in productive metabolism. One example of such an oncogene is Ras which is found to be mutated and hyperactivated in a variety of distinct cancers. Despite numerous studies on Ras, the precise molecular mechanisms that facilitate survival during ECM-detachment remain poorly understood. Recently, we discovered that ECM-detached cells harboring oncogenic Ras mutations require signaling through the PI(3)K/SGK1 signaling axis to promote survival. Furthermore, we found that oncogenic Ras can concurrently diminish PHLPP1 phosphatase levels, which results in a decrease in p38 MAPK-mediated activation of anoikis. Thus, our data suggest that cancer cells with activating Ras mutations can survive during ECM-detachment using downstream effector molecules that modulate distinct pathways. Overall, these data suggest that new therapeutic interventions that aim to mitigate SGK1 signaling and activate the p38 MAPK activity may aid in specifically targeting and eliminating metastatic cancer cells.
KEYWORDS:
Anoikis and metabolic alterations during ECM-detachment: 2 barriers to the survival of cancer cells
A significant number of disparate cell types require integrin-mediated attachment to the extracellular matrix (ECM) to ward off the induction of detachment-induced cell death programs.Citation1,Citation2 In direct contrast, cancer cells acquire the capability to forego this requirement and thus can robustly survive during detachment from the ECM. This capacity for survival during ECM-detachment is acutely critical during the metastatic cascade, where cancer cells will be exposed to numerous periods of ECM-detachment and abnormal ECM environments during the dissemination of cancer cells throughout the body. Thus, it can be argued that ECM-detached cancer cells must evade detachment-induced cell death mechanisms at every step of the metastatic cascade; from localized invasion, to survival in the circulation, to colonizing a secondary site. Perhaps the most well-described cell death pathway that cancer cells evade during ECM-detachment is anoikis, which is defined as caspase-dependent apoptosis that is induced by loss of integrin-mediated attachment to ECM.Citation3 Much of our current understanding of anoikis has been accrued from a 3-dimensional cell culture model of mammary acini where a hollow lumen is generated (in part) by anoikis of centrally located cells.Citation4-Citation6 Using this model, it has become abundantly clear that inhibition of anoikis is not sufficient to promote survival of ECM-detached cells in the luminal space. More specifically, ECM-detached mammary epithelial cells develop metabolic alterations that lead to diminished viability during ECM-detachment and resolving these metabolic deficiencies leads to the survival of ECM-detached cells in the luminal spaceCitation7,Citation8
A novel mechanism downstream of oncogenic ras that facilitates survival of ECM-detached cells
Strategies used by cancer cells to overcome metabolic deficiencies and evade anoikis have been revealed to be multi-faceted and diverse.Citation2,Citation9-Citation11 However, downstream signaling emanating from activated oncogenes is now appreciated to function to antagonize the induction of anoikis and to promote energy production. This phenomenon was first observed with ErbB2,Citation8 which is overexpressed in ∼30% of breast cancers and can activate a variety of distinct downstream signaling pathways. We sought to expand on these studies by examining whether distinct oncogenic mutations would converge on specific downstream effector pathways to promote survival during ECM-detachment.Citation12,Citation13 Given that hyperactivating mutations in the small GTPase Ras (via mutations in codon 12, 13, or 61), occur in approximately 30% of all cancers (most commonly in pancreatic (90%), colon (50%), and lung (30%), we sought to investigate the molecular mechanisms underlying the ability of oncogenic Ras to facilitate both anoikis evasion and productive metabolism during ECM-detachment.Citation14,Citation15 In addition, these oncogenic mutations in Ras can plays a critical role promoting tumor progression, metastasis, and cell survival.Citation16,Citation17
As expected, we discovered that the overexpression of oncogenic Ras is sufficient to drive ATP generation during ECM-detachment while simultaneously blocking anoikis. However, when investigating the molecular mechanisms responsible for promoting ATP generation, we found that while phosphatidylinositol 3-kinase (PI(3)K) signaling was critically important, ATP generation did not require downstream signaling through AKT1.Citation12 Instead, we determined that the activity of serum and glucocorticoid-regulated kinase-1 (SGK1), a serine/threonine kinase known to function downstream of PI(3)K, is critical for ATP generation during ECM-detachment. SGK1 shares approximately 50% similarity with AKT1 in the catalytic domain. It is activated by mTORC2-dependent phosphorylation in the hydrophobic motif (S422) and phosphorylation of the activation loop (T256) by PDK1.Citation18,Citation19 While SGK1 has been previously linked to glucose metabolism,Citation20,Citation21 the pathology of certain metabolic syndromes,Citation22 and regulation of small GTPases,Citation23 multiple recent studies suggest an emerging role for SGK1 in tumorigenesis.Citation24 More specifically, several reports have observed elevated expression/activation of SGK1 in malignant tumors,Citation25-Citation30 and thus our findings that SGK1 functions downstream of PI(3)K to promote ATP generation during ECM-detachment provide a possible mechanism by which SGK1 can contribute to cancer pathogenesis.
In subsequent examination of the importance of SGK1 kinase signaling on the survival of ECM-detached cells, we found that SGK1 activation promotes cell viability during ECM-detachment owing to its capacity to promote glucose uptake and ATP generation.Citation12 In support of this conclusion, expression of a constitutively active SGK1 (S422D) promotes luminal filling in a 3-dimensional cell culture model of mammary morphogenesis. Given these data, we next investigated the impact of SGK1-mediated ATP generation in enabling the survival of ECM-detached cancer cells that harbor mutations in oncogenic Ras. Indeed, inhibition of SGK1 signaling (via both pharmacological and shRNA based approaches) in HCT116 cells (a colon cancer cell line that contains an oncogenic mutation in K-Ras) substantially compromises metabolic activity and survival during ECM-detachment. Furthermore, we found that loss of SGK1 signaling significantly hinders anchorage independent growth, a classic hallmark of tumorigenic capacity. In aggregate, these data suggest that cancer cells that contain oncogenic Ras mutations can utilize PI(3)K/SGK1 signaling to rectify ECM-detachment-induced metabolic deficiencies in a fashion that promotes cell survival. In agreement with this inference, we observed a positive correlation between protein levels of oncogenic K-Ras levels and SGK1 kinase activity in colon cancer patients.Citation12 Thus, our findings suggest that SGK1 could be a potential therapeutic target to specifically compromise anchorage-independent growth in cancer cells containing oncogenic Ras mutations. In line with this possibility, other studies have already demonstrated that SGK1 activity is sufficient to overcome cell death mediated by PI(3)K and AKT1 inhibitors.Citation32,Citation25 These findings suggest that tumor cells that acquire resistance to these inhibitors may do so through constitutive SGK1 activation. Furthermore, SGK1 inhibitors have begun to show promise in blocking cancer cell proliferationCitation33 and in potentiating the effects of radiotherapy.Citation34,Citation35
In addition to Ras-mediated promotion of ATP generation during ECM-detachment, hyperactivating mutations in Ras profoundly inhibit anoikis in a fashion that is independent of the SGK1-mediated effects on glucose metabolism. Given this, we were interested in discerning the effectors that operate downstream of Ras to potentiate anoikis evasion.Citation12 Interestingly, we found that ECM-detached cells expressing oncogenic Ras maintain phosphorylation of AKT1 (at S473) in the presence of robust inhibition of PI(3)K. These data suggest that Ras activation may antagonize phosphatase activity toward AKT1 and thus facilitate evasion of anoikis. AKT1 is known to be dephosphorylated at S473 by the phosphatase PHLPP1 (PH Domain Leucine Rich Repeat Protein Phosphatase 1),Citation36,Citation37 which has been shown to function as a tumor suppressor.Citation38-Citation41 In support of the possibility that Ras-mediated inhibition of AKT1 dephosphorylation blocks anoikis, we found that expression of oncogenic Ras results in a significant loss of PHLPP1 protein. Furthermore, shRNA-mediated reduction of PHLPP1 inhibits anoikis and can promote the survival of ECM-detached cells in the luminal space of mammary acini. Additionally, re-introduction of PHLPP1 into cells harboring oncogenic Ras mutations sensitizes ECM-detached cells to anoikis and hinders anchorage independent growth.
Interestingly, in the aforementioned experiments, we did not detect alterations in the phosphorylation of AKT1 (at S473) when PHLPP1 was reintroduced into cancer cells with Ras mutations. This appears to be due to Ras-mediated downregulation of the scaffold protein FKBP5, which is required to target PHLPP1 to S473 on AKT1.Citation42 Thus, our findings suggest that PHLPP1-mediated induction of sensitivity to anoikis involves a pathway distinct from AKT1. Indeed, we found that PHLPP1-mediated induction of anoikis is a direct result of downstream activation of the p38 MAPK pathway. This stimulation of p38 MAPK by PHLPP1 is conceivably due to PHLPP1-mediated dephosphorylation of MST1, which increases MST1 kinase activity.Citation43 MST1 activity has been previously shown to function upstream of kinases that can activate p38 MAPK. However, the precise mechanism by which PHLPP1 activity leads to p38 MAPK and anoikis has yet to be fully delineated. That being said, we did observe a negative correlation between oncogenic K-Ras levels and p38 MAPK activation in colon cancer patients,Citation12 suggesting that inhibition of p38 MAPK due to Ras-mediated downregulation of PHLPP1 may be operative in patient populations.
Conclusions and perspectives
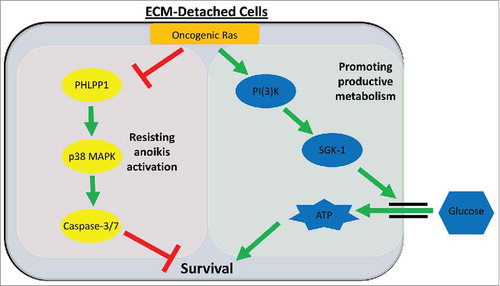
Given that cancer metastasis accounts for approximately 90% of cancer related deaths,Citation44 there is an ardent need to develop novel therapeutic strategies aimed at specifically eliminating metastatic cancer cells. Additionally, one of the most commonly activated proteins across a diversity of distinct cancers is Ras, yet targeted therapeutics against Ras mutant cells are limited or poorly effective.Citation17 In our recent study (summarized in ),Citation12 we have found that SGK1 is critically important downstream of Ras/PI(3)K activation to promote ATP generation and cancer cell survival during ECM-detachment. At the same time, oncogenic Ras suppresses PHLPP1 to overcome p38 MAPK-mediated anoikis induction. These data unveil new targets for therapeutic intervention in cancer cells with activating Ras mutations. More specifically, our data suggest that simultaneous inhibition of SGK1 activity and activation of p38 MAPK signaling, could be an efficacious therapeutic strategy for patients whose cancers are driven by oncogenic Ras mutations. Furthermore, since the SGK1 and p38 MAPK-mediated effects of Ras are uniquely important in ECM-detached cells, such a regimen may be particularly helpful in antagonizing tumor progression and metastasis. Additional pre-clinical studies aimed at assessing the efficacy of this potential strategy will be important to the ultimate translation of these findings into meaningful clinical outcomes.
Figure 1. Oncogenic Ras promotes productive metabolism and blocks anoikis via divergent downstream effectors. During ECM-detachment, cells harboring oncogenic Ras mutations signal via a PI(3)K/SGK1-mediated pathway to increase glucose uptake, ATP generation, and survival (right, green box). Simultaneously, oncogenic Ras diminishes PHLPP1 levels to overcome p38 MAPK-mediated anoikis activation (left, red box).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- Buchheit CL, Rayavarapu RR, Schafer ZT. The regulation of cancer cell death and metabolism by extracellular matrix attachment. Semin Cell Dev Biol 2012; 23:402-11; PMID:22579674; http://dx.doi.org/10.1016/j.semcdb.2012.04.007
- Buchheit CL, Weigel KJ, Schafer ZT. Cancer cell survival during detachment from the ECM: multiple barriers to tumour progression. Nat Rev Cancer 2014; 14:632-41; PMID:25098270; http://dx.doi.org/10.1038/nrc3789
- Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol 1994; 124:619-26; PMID:8106557; http://dx.doi.org/10.1083/jcb.124.4.619
- Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer 2005; 5:675-88; PMID: 16148884; http://dx.doi.org/10.1038/nrc1695
- Debnath J, Mills KR, Collins NL, Reginato MJ, Muthuswamy SK, Brugge JS. The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell 2002; 111:29-40; PMID:12372298; http://dx.doi.org/10.1016/S0092-8674(02)01001-2
- Reginato MJ, Mills KR, Paulus JK, Lynch DK, Sgroi DC, Debnath J, Muthuswamy SK, Brugge JS. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat Cell Biol 2003; 5:733-40; PMID:12844146; http://dx.doi.org/10.1038/ncb1026
- Davison CA, Durbin SM, Thau MR, Zellmer VR, Chapman SE, Diener J, Wathen C, Leevy WM, Schafer ZT. Antioxidant enzymes mediate survival of breast cancer cells deprived of extracellular matrix. Cancer Res 2013; 73:3704-15; PMID:23771908; http://dx.doi.org/10.1158/0008-5472.CAN-12-2482
- Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Gao S, Puigserver P, Brugge JS. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009; 461:109-13; PMID:19693011; http://dx.doi.org/10.1038/nature08268
- Buchheit CL, Angarola BL, Steiner A, Weigel KJ, Schafer ZT. Anoikis evasion in inflammatory breast cancer cells is mediated by Bim-EL sequestration. Cell Death Differ 2015; 22:1275-86; PMID:25526094; http://dx.doi.org/10.1038/cdd.2014.209; PMID: 25526094; http://dx.doi.org/10.1038/cdd.2014.209
- Weigel KJ, Jakimenko A, Conti BA, Chapman SE, Kaliney WJ, Leevy WM, Champion MM, Schafer ZT. CAF-secreted IGFBPs regulate breast cancer cell anoikis. Mol Cancer Res 2014; 12:855-66; PMID:24803643; http://dx.doi.org/10.1158/1541-7786.MCR-14-0090
- Rayavarapu RR, Heiden B, Pagani N, Shaw MM, Shuff S, Zhang S, Schafer ZT. The role of multicellular aggregation in the survival of ErbB2-positive breast cancer cells during extracellular matrix detachment. J Biol Chem 2015; 290:8722-33; PMID:25681438; http://dx.doi.org/10.1074/jbc.M114.612754
- Mason JA, Davison-Versagli CA, Leliaert AK, Pape DJ, McCallister C, Zuo J, Durbin SM, Buchheit CL, Zhang S, Schafer ZT. Oncogenic Ras differentially regulates metabolism and anoikis in extracellular matrix-detached cells. Cell Death Differ 2016; 23:1271-82; PMID:26915296; http://dx.doi.org/10.1038/cdd.2016.15
- Mason JA, Schafer ZT. SGK-1 and PHLPP1: Ras-mediated effectors during ECM-detachment. Cell Cycle 2016; 15:2233-4; PMID:27267605; http://dx.doi.org/10.1080/15384101.2016.1194136
- Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res 2012; 72:2457-67; PMID:22589270; http://dx.doi.org/10.1158/0008-5472.can-11-2612
- Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer 2011; 11:761-74; PMID:21993244; http://dx.doi.org/10.1038/nrc3106
- Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer 2003; 3:11-22; PMID:12509763; http://dx.doi.org/10.1038/nrc969
- Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov 2014; 13:928-42; PMID:25435214; http://dx.doi.org/10.1038/nrd4281
- Bruhn MA, Pearson RB, Hannan RD, Sheppard KE. Second AKT: the rise of SGK in cancer signalling. Growth Factors 2010; 28:394-408; PMID:20919962; http://dx.doi.org/10.3109/08977194.2010.518616
- Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 2010; 11:9-22; PMID:20027184; http://dx.doi.org/10.1038/nrm2822
- Jeyaraj S, Boehmer C, Lang F, Palmada M. Role of SGK1 kinase in regulating glucose transport via glucose transporter GLUT4. Biochem Biophys Res Commun 2007; 356:629-35; PMID:17382906; http://dx.doi.org/10.1016/j.bbrc.2007.03.029
- Palmada M, Boehmer C, Akel A, Rajamanickam J, Jeyaraj S, Keller K, Lang F. SGK1 kinase upregulates GLUT1 activity and plasma membrane expression. Diabetes 2006; 55:421-7; PMID:16443776; http://dx.doi.org/10.2337/diabetes.55.02.06.db05-0720
- Lang F, Bohmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V. (Patho)physiological significance of the serum- and glucocorticoid-inducible kinase isoforms. Physiol Rev 2006; 86:1151-78; PMID:17015487; http://dx.doi.org/10.1152/physrev.00050.2005
- Amato R, Scumaci D, D'Antona L, Iuliano R, Menniti M, Di Sanzo M, Faniello MC, Colao E, Malatesta P, Zingone A, et al. Sgk1 enhances RANBP1 transcript levels and decreases taxol sensitivity in RKO colon carcinoma cells. Oncogene 2013; 32:4572-8; PMID:23108393; http://dx.doi.org/10.1038/onc.2012.470
- Talarico C, Dattilo V, D'Antona L, Menniti M, Bianco C, Ortuso F, Alcaro S, Schenone S, Perrotti N, Amato R. SGK1, the New Player in the Game of Resistance: Chemo-Radio Molecular Target and Strategy for Inhibition. Cell Physiol Biochem 2016; 39:1863-76; PMID:27771704; http://dx.doi.org/10.1159/000447885
- Castel P, Ellis H, Bago R, Toska E, Razavi P, Carmona FJ, Kannan S, Verma CS, Dickler M, Chandarlapaty S, et al. PDK1-SGK1 Signaling Sustains AKT-Independent mTORC1 Activation and Confers Resistance to PI3Kalpha Inhibition. Cancer Cell 2016; 30:229-42; PMID:27451907; http://dx.doi.org/10.1016/j.ccell.2016.06.004
- Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015; 163:506-19; PMID:26451490; http://dx.doi.org/10.1016/j.cell.2015.09.033
- Fagerli UM, Ullrich K, Stuhmer T, Holien T, Kochert K, Holt RU, Bruland O, Chatterjee M, Nogai H, Lenz G, et al. Serum/glucocorticoid-regulated kinase 1 (SGK1) is a prominent target gene of the transcriptional response to cytokines in multiple myeloma and supports the growth of myeloma cells. Oncogene 2011; 30:3198-206; PMID:21478911; http://dx.doi.org/10.1038/onc.2011.79
- Hall BA, Kim TY, Skor MN, Conzen SD. Serum and glucocorticoid-regulated kinase 1 (SGK1) activation in breast cancer: requirement for mTORC1 activity associates with ER-alpha expression. Breast Cancer Res Treat 2012; 135:469-79; PMID:22842983; http://dx.doi.org/10.1007/s10549-012-2161-y
- Szmulewitz RZ, Chung E, Al-Ahmadie H, Daniel S, Kocherginsky M, Razmaria A, Zagaja GP, Brendler CB, Stadler WM, Conzen SD. Serum/glucocorticoid-regulated kinase 1 expression in primary human prostate cancers. Prostate 2012; 72:157-64; PMID:21563193; http://dx.doi.org/10.1002/pros.21416
- Kach J, Conzen SD, Szmulewitz RZ. Targeting the glucocorticoid receptor in breast and prostate cancers. Sci Transl Med 2015; 7:305ps19; PMID:26378243; http://dx.doi.org/10.1126/scitranslmed.aac7531
- Skor MN, Wonder EL, Kocherginsky M, Goyal A, Hall BA, Cai Y, Conzen SD. Glucocorticoid receptor antagonism as a novel therapy for triple-negative breast cancer. Clin Cancer Res 2013; 19:6163-72; PMID:24016618; http://dx.doi.org/10.1158/1078-0432.CCR-12-3826
- Sommer EM, Dry H, Cross D, Guichard S, Davies BR, Alessi DR. Elevated SGK1 predicts resistance of breast cancer cells to Akt inhibitors. Biochem J 2013; 452:499-508; PMID:23581296; http://dx.doi.org/10.1042/bj20130342
- D'Antona L, Amato R, Talarico C, Ortuso F, Menniti M, Dattilo V, Iuliano R, Gigliotti F, Artese A, Costa G, et al. SI113, a specific inhibitor of the Sgk1 kinase activity that counteracts cancer cell proliferation. Cell Physiol Biochem 2015; 35:2006-18; PMID:25871776; http://dx.doi.org/10.1159/000374008
- Talarico C, D'Antona L, Scumaci D, Barone A, Gigliotti F, Fiumara CV, Dattilo V, Gallo E, Visca P, Ortuso F, et al. Preclinical model in HCC: the SGK1 kinase inhibitor SI113 blocks tumor progression in vitro and in vivo and synergizes with radiotherapy. Oncotarget 2015; 6:37511-25; PMID:26462020; http://dx.doi.org/10.18632/oncotarget.5527
- Talarico C, Dattilo V, D'Antona L, Barone A, Amodio N, Belviso S, Musumeci F, Abbruzzese C, Bianco C, Trapasso F, et al. SI113, a SGK1 inhibitor, potentiates the effects of radiotherapy, modulates the response to oxidative stress and induces cytotoxic autophagy in human glioblastoma multiforme cells. Oncotarget 2016; 7:15868-84; PMID:26908461; http://dx.doi.org/10.18632/oncotarget.7520
- Gao T, Furnari F, Newton AC. PHLPP: a phosphatase that directly dephosphorylates Akt, promotes apoptosis, and suppresses tumor growth. Mol Cell 2005; 18:13-24; PMID:15808505; http://dx.doi.org/10.1016/j.molcel.2005.03.008
- Newton AC, Trotman LC. Turning off AKT: PHLPP as a drug target. Annu Rev Pharmacol Toxicol 2014; 54:537-58; PMID:24392697; http://dx.doi.org/10.1146/annurev-pharmtox-011112-140338
- Cai J, Fang L, Huang Y, Li R, Yuan J, Yang Y, Zhu X, Chen B, Wu J, Li M. miR-205 targets PTEN and PHLPP2 to augment AKT signaling and drive malignant phenotypes in non-small cell lung cancer. Cancer Res 2013; 73:5402-15; PMID:23856247; http://dx.doi.org/10.1158/0008-5472.CAN-13-0297
- Chen M, Pratt CP, Zeeman ME, Schultz N, Taylor BS, O'Neill A, Castillo-Martin M, Nowak DG, Naguib A, Grace DM, et al. Identification of PHLPP1 as a tumor suppressor reveals the role of feedback activation in PTEN-mutant prostate cancer progression. Cancer Cell 2011; 20:173-86; PMID:21840483; http://dx.doi.org/10.1016/j.ccr.2011.07.013
- Wang P, Zhou Z, Hu A, Ponte de Albuquerque C, Zhou Y, Hong L, Sierecki E, Ajiro M, Kruhlak M, Harris C, et al. Both decreased and increased SRPK1 levels promote cancer by interfering with PHLPP-mediated dephosphorylation of Akt. Mol Cell 2014; 54:378-91; PMID:24703948; http://dx.doi.org/10.1016/j.molcel.2014.03.007
- Wen YA, Stevens PD, Gasser ML, Andrei R, Gao T. Downregulation of PHLPP expression contributes to hypoxia-induced resistance to chemotherapy in colon cancer cells. Mol Cell Biol 2013; 33:4594-605; PMID:24061475; http://dx.doi.org/10.1128/MCB.00695-13
- Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, Petersen G, Lou Z, Wang L. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 2009; 16:259-66; PMID:19732725; http://dx.doi.org/10.1016/j.ccr.2009.07.016
- Qiao M, Wang Y, Xu X, Lu J, Dong Y, Tao W, Stein J, Stein GS, Iglehart JD, Shi Q, et al. Mst1 is an interacting protein that mediates PHLPPs' induced apoptosis. Mol Cell 2010; 38:512-23; PMID:20513427; http://dx.doi.org/10.1016/j.molcel.2010.03.017
- Nguyen DX, Massague J. Genetic determinants of cancer metastasis. Nat Rev Genet 2007; 8:341-52; PMID:17440531; http://dx.doi.org/10.1038/nrg2101