
ABSTRACT
Pancreatic ductal adenocarcinoma (PDAC) is a highly aggressive and therapy resistant malignancy. Mutant K-Ras, found in >90% of refractory PDAC, acts as a molecular switch activating Rho GTPase signaling that in turn promotes a plethora of pro-survival molecules and oncogenic microRNAs. We investigated the impact of Rho GTPase effector protein p21 activated kinase 4 (PAK4) inhibition on pro-survival p-Bad and oncogenic miRNA signaling. We demonstrate that the dual NAMPT and PAK4 modulators (KPT-9274 and KPT-9307) inhibit PDAC cell proliferation through downregulation of Bad phosphorylation and upregulation of tumor suppressive miRNAs (miR-145, let-7c, let-7d, miR-34c, miR320 and miR-100). These results suggest that targeting PAK4 could become a promising approach to restore pro-apoptotic function of Bad and simultaneously activate tumor suppressive miRNAs in therapy resistant PDAC.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is a highly aggressive malignancy with poor overall survival. The American Cancer Society estimates that, in the United States, about 53,000 people (slightly more men than women) will be newly diagnosed with pancreatic cancer and about 43,000 will die of it in 2017.Citation1 The 5-year overall survival (OS) of patients with pancreatic cancer is only 8% with early stage (stage IA) patients experiencing a somewhat higher 5-year survival rate of 14%.Citation1 However, patients with advanced pancreatic cancer (stage IV) have 5-year survival rate around 1%. Since PDAC is typically diagnosed at a late stage, it is very aggressive and spreads rapidly at this point and is a major reason why PDAC has become one of the leading causes of cancer related deaths. In addition, the aggressive growth and high mortality of PDAC could, in part, be due to the ability of these cancer cells to become drug resistant from numerous alterations in a variety of signal transduction pathways.
K-Ras mutations are found in more than 90% of PDAC tumors.Citation2 Mutant K-Ras acts as a molecular switch that activates critical cellular signaling such as survival, proliferation and cell division. One of the important signaling pathways downstream of K-Ras activated in drug resistant PDAC is Rho GTPase signaling.Citation3 Rho GTPases play a central role in the regulation of gene transcription, cell cycle entry, apoptosis and cell survival.Citation4,Citation5 Members of the p21-activated kinase (PAK) family are effectors of this signaling and include group I (PAK1–3) and II (PAK4–6) kinases. Studies have shown that PAK4 is aberrantly expressed in PDAC and not in normal human pancreatic ductal epithelial cells.Citation6 The PAK4 kinase influences a variety of cellular functions including apoptosis. It has been reported that PAK4 inhibits caspase activation and protects cells from apoptosis.Citation7 Furthermore, increased expression of PAK4 up-regulates the phosphorylation of the pro-apoptotic protein Bad.Citation7 Bad is a pro-apoptotic Bcl-2 family member and phosphorylation of the Bad protein inhibits its pro-apoptotic function. The alterations in the ratio of p-Bad/Bad signaling determine the survival of cancer cells through apoptotic machinery. The hyper-phosphorylation of Bad also critically influences cancer chemoresistance.Citation8-Citation10 Therefore, targeting PAK4/Bad signaling could be a promising strategy for overcoming PDAC drug resistance. Not much is known about the mechanisms by which group II PAKs augment drug resistance and cell survival. The role of PAK4 in the regulation of Bad signaling is not currently understood.
MicroRNAs (miRNAs), which are aberrantly expressed in PDAC, have been implicated in the regulation of several cell survival and apoptotic pathways that include Ras, PAK and Bad.Citation11,Citation12 Improper expression of miRNAs have been clearly linked to therapy resistance in PDAC.Citation13 Several Rho GTPase effector proteins such as ROCK1 have been shown to promote oncogenic miRNAs and block tumor suppressive miRNAs.Citation14,Citation15 Emerging experimental evidence have shown that tumor suppressive miRNAs regulate Ras-activated signaling in PDAC, leading to the induction of apoptosis and inhibition of drug resistance.Citation16-Citation18 Therefore, understanding the role of miRNAs in PAK4 regulation is critical for the development of therapies against drug resistance in PDAC. However, the molecular mechanisms underlying miRNA-regulated apoptosis and drug-resistance through PAK4 signaling remain unclear.
Recently, we have reported that the dual PAK4 and NAMPT allosteric modulators (KPT9274 and KPT-9307 which inhibit the activity of PAK4 and NAMPT) have remarkable anti-tumor activity in PDAC and other cancers in vitro and in vivo.Citation19-Citation21 NAMPT plays important roles in the regulation of metabolism, stress response, aging and cancers. NAMPT inhibition could overcome gemcitabine resistance by decreasing the NAD level and suppressing glycolytic activity.Citation22 We investigated the effect these novel PAK4/NAMPT inhibitors have on the regulation of PAK4/Bad and miRNA signaling. In this brief report, we demonstrate that these compounds inhibit p-Bad leading to re-sensitization of PDAC cells to gemcitabine. Treatment with these inhibitors caused upregulation of Bad and caspase 3, downregulation of Bcl-2, and enhancement in tumor suppressive miRNAs. These results provide strong indications that targeting PAK4/p-Bad and miRNA signaling could be a potent therapeutic strategy to overcome drug resistance in PDAC.
Results
PAK4 and p-Bad proteins are increased in drug resistant PDAC cell lines when compared with normal pancreatic ductal epithelial cells
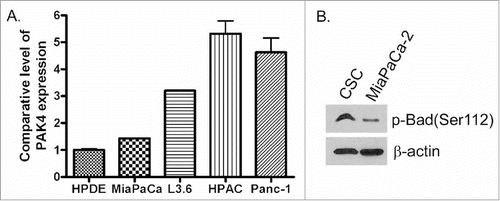
By conducting real-time PCR, we found that PDAC cells expressed a much higher level of PAK4, compared with normal human ductal epithelial (HPDE) cells (), which is consistent with others' report.Citation23 We have previously reported that PDAC cells sorted for stem like markers (CD33+CD44+EpCAM+ termed cancer stem cells or CSCs) demonstrate marked resistance to standard chemotherapeutics such as gemcitabine and oxaliplatin.Citation21 These flow-sorted CSCs harbor mesenchymal markers and have a propensity to form spheroids in long-term culture conditions. Highlighting the critical role of PAK4 in the PDAC stem cell biology, we also reported enhancement of PAK4 mRNA in gemcitabine resistant MiaPaCa-2 cells (MiaPaCa-2-GR) as well as in MiaPaCa-2 CSCs.Citation21 Knocking down PAK4 in these CSCs using RNAi suppressed their propensity to form spheroids. More importantly, we found that the expression of p-Bad in CSCs was significantly upregulated when compared with the parental PDAC cells (). Collectively, our data suggests a role for PAK4 in PDAC and CSC subsistence as well as a role for p-Bad in PDAC drug resistance.
Figure 1. PDAC cells expressed much higher level of PAK4 and p-Bad, especially drug resistant PDAC, compared with normal pancreatic ductal epithelial cells. (A) The expression level of PAK4 in HPDE, MiaPaCa-2, L3.6pl, HPAC and Panc-1 cells were measured by real-time RT-PCR. (B) The expression of p-Bad protein in MiaPaCa-2 and CSC cells was accessed by Western Blot analysis.
The dual PAK4 and NAMPT modulators inhibited p-Bad and induced pro-apoptotic signaling in PDAC cells
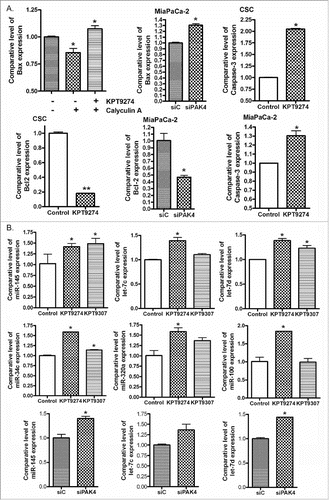
To better understand the connection between Bad and PAK4 in PDAC CSCs, we investigated the impact of PAK4 inhibition on Bad signaling. RNAi knockdown of PAK4 or treatment with KPT-9274 (a dual modulator of NAMPT and PAK4 recently initiated in a phase 1 clinical trial of patients with advanced solid malignancies; NCT02702492) or KPT-9307 resulted in the re-expression of Bad in MiaPaCa-2 and CSC cells (). In addition, we discovered that KPT-9274 and KPT-9307 effectively decreased the levels of phosphorylated PAK4 and phosphorylated Bad in PDAC cells (), suggesting the regulation of Bad signaling by PAK4 inhibition. Most interestingly, KPT-9274 also abrogated Calyculin A (a potent protein phosphatase inhibitor) induced hyper-phosphorylation of Bad protein in PDAC cells (). Examining total RNA from these cells, we clearly observed re-expression of Bad and Bax that otherwise were suppressed by Calyculin A treatment ( and ). Further analysis revealed that KPT-9274 treatment or RNAi knockdown of PAK4 resulted in activation of caspases-3, upregulation of Bax, and reduction of Bcl-2 (). Collectively, these results demonstrate that targeting PAK4 can effectively induce apoptosis through the reduction of p-Bad and induction of Bad and Bax mRNA.
Figure 2. PAK4 modulators induced Bad expression and inhibited phosphorylation of Bad, activating Bad signaling. Knockdown of PAK4 by siRNA, KPT-9274 or KPT-9307 treatment induced the expression of Bad mRNA tested by real-time PCR (A). The PAK4 inhibitors significantly inhibited the phosphorylation of PAK4 and Bad protein tested by Western Blot analysis (B). PAK4 inhibitors abrogated the Bad phosphorylation stimulated by Calyculin A (C). PAK4 inhibitors also abrogated the downregulation of Bad mRNA stimulated by Calyculin A (D). (*: p<0.05).
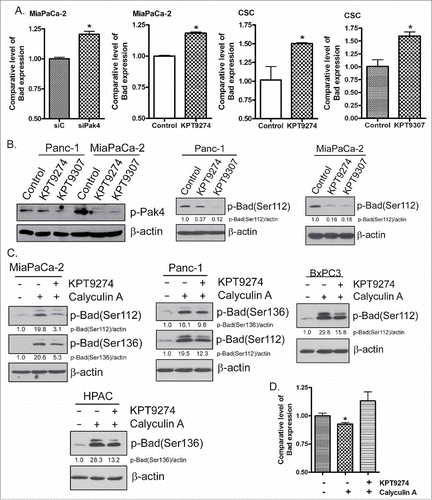
Figure 3. KPT-9274 regulated the expression of Bax, caspase 3, Bcl-2, and tumor suppressive miRNAs involved in apoptotic signaling. (A) MiaPaCa-2 and CSC cells were treated with 500 nM KPT-9274 or transfected with PAK4 siRNA. The expression of Bax, caspase 3 and Bcl-2 mRNAs was accessed by real-time RT-PCR. (B) MiaPaCa-2 cells were treated with 500 nM KPT-9274 or KPT-9307 for 48 hours or transfected with PAK4 siRNA. The expression level of miR-145, let-7c, let-7d, miR-34c, miR-320 and miR-100 was measured by real-time RT-PCR. (*: p<0.05).
KPT-9274 and KPT-9307 induced tumor suppressive miRNAs related to apoptosis and survival signaling in PDAC cells
While the role of group I PAKs on miRNAs is well understood, there is little known about the PAK4-miRNA regulatory mechanism in PDAC. We found, for the first time, that KPT-9274 or KPT-9307 treatment or RNAi knockdown of PAK4 could increase the expression of tumor suppressive miRNAs (miR-145, miR-34c, let-7c, let-7d, miR-320a and miR-100) which control apoptosis and cell survival pathways (). Nevertheless, the impact of these inhibitors on the entire set of miRNAs is not known and needs to be further evaluated.
Treatment of PDAC cells with KPT-9274 induced apoptotic cell death and growth inhibition
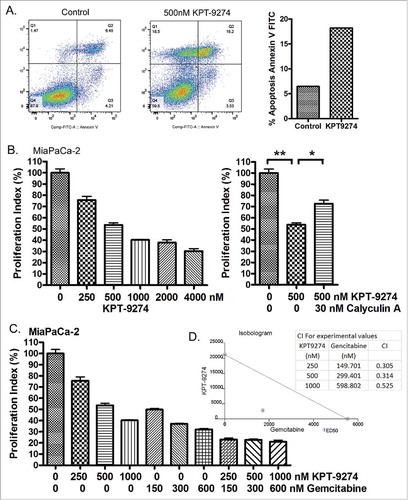
Using an Annexin V FITC assay, the effect of KPT-9274 on apoptotic cell death was measured. We found that KPT-9274 treatment significantly induced apoptosis of PDAC cells (). A standard MTT assay showed that KPT-9274 treatment inhibited proliferation of PDAC cells in a dose-dependent manner (). We found that Calyculin A treatment could protect PDAC cells from growth inhibition induced by KPT-9274 () perhaps due to increased p-Bad stimulated by Calyculin A. More importantly, the combination treatment of KPT-9274 with gemcitabine demonstrated a synergistic inhibitory effect on PDAC cell proliferation with all CI (combination index) values < 1 ( and ). These results clearly demonstrate that inhibition of PAK4 can induce apoptosis and suppress PDAC cell proliferation through the reduction of p-Bad and induction of tumor suppressive miRNAs. Therefore, PAK4 inhibition by KPT-9274 may be a beneficial therapy for drug resistant PDAC patients.
Figure 4. Treatment of PDAC cells with KPT-9274 induced apoptotic cell death and growth inhibition, and synthesized the anti-cancer effect of Gemcitabine. (A) MiaPaCa-2 cells were treated with 500 nM KPT-9274 for 72 hours and the apoptotic cell death was detected by Annexin V FITC. (B) MiaPaCa-2 cells were treated with 250 to 4000 nM KPT-9274 for 72 h or along with 30 nM Calyculin A for 18 hours. The cell proliferation index was accessed by MTT assay. (*: p<0.05; **: p<0.01). (C) MiaPaCa-2 cells were treated with 250 to 1000 nM KPT-9274 and/or 150 to 600 nM gemcitabine for 72 hours. The cell proliferation index was accessed by MTT assay. (D) The combination index (CI) and isobologram were created by CalcuSyn software based on the MTT results.
Discussion
It is known that most chemotherapies induce apoptosis of cancer cells, leading to tumor growth inhibition. However, most PDACs are resistant to standard treatments because PDAC cells have evolved a plethora of molecular mechanisms to escape apoptosis induction and ensure survival. In PDAC, aberrant expression of Bcl-2 family members causes apoptotic resistance and metastasis.Citation24 The Bcl-2 protein family includes an anti-apoptotic subgroup (i.e. Bcl-2 and Bcl-XL) and 2 pro-apoptotic subgroups, the Bax-like subgroup (i.e., Bax and Bak) and the BH3-only subgroup (i.e., Bad and Bim).Citation25 The BH3-only protein, Bad serves as a sensor of cellular stress to initiate apoptosis. However, de-regulation of Bad through phosphorylation renders it ineffective as an apoptosis provocateur.Citation26 Bad function is modulated by phosphorylation at 2 sites, serine 112 (Ser112) and serine 136 (Ser136).Citation27 Phosphorylation at these sites has been correlated with binding and sequestering of Bad by the 14–3–3 protein, prevention of Bcl-2 and Bcl-XL binding and thus promoting cell survival.Citation27,Citation28 Moreover, the mutant Ras pathway, altered in >90% of PDAC, is known to induce and maintain Bad phosphorylation, suggesting a role for Ras-regulated Bad phosphorylation in drug resistance.Citation29 In addition, Akt, a molecule downstream of Ras signaling, could directly phosphorylate Bad at Ser-136 in vitro and in vivo resulting in suppression of apoptosis and promotion of cell survival in various cancers.Citation9 Our results showed that dual PAK4 and NAMPT modulators (KPT-9274 and KPT-9307) induce Bad expression, inhibit Bad phosphorylation and abrogate the Bad phosphorylation induced by Calyculin A. This data suggests that PAK4 inhibition could sensitize PDAC cells to apoptosis induced by conventional chemotherapeutics. Indeed, our study also shows the synergistic inhibitory effects of KPT-9274 and Gemcitabine treatment of PDAC cells.
It has been reported that Bad could be phosphorylated by PKA or group I PAKs, leading to a loss of its pro-apoptotic function.Citation28,Citation30 Phosphorylation of Bad is regulated through either c-RAF (a substrate of different PAKsCitation31) or through PAK-mediated phosphorylation of c-RAF at Ser-338 causing c-RAF translocation to the mitochondria and subsequent binding to and phosphorylation of Bad.Citation32 Group I PAKs have also been shown to phosphorylate Bad directly,Citation28 suggesting that inhibitor for Group I PAKs could induce Bad-mediated apoptotic cell death. However, the group I PAK inhibitor, PF-3758309, was found to be clinically ineffective in cancers.Citation33 Therefore, the inhibitors for Group II PAKs (i.e., KPT-9274 and KPT9307) could have promising effects on PDAC. In addition, KPT-9273 and KPT-9307 are dual NAMPT and PAK4 modulators. By targeting PAK4 and NAMPT signaling, KPT-9274 has been shown to reduce G2-M transit, induce apoptosis and inhibit cell invasion and migration in cancers.Citation20
In PDAC, copy number analysis has revealed hyper-activation of Group II PAK (PAK4),Citation34 suggesting that the PAK4 gene is amplified and that PAK4 protein is overexpressed in PDAC. As an effector of Ras/Rho GTPases/CDC42 signaling, PAK4 regulates the function of several downstream proteins in a kinase dependent manner.Citation35 Moreover, PAK4 protects cells against apoptotic cell death through inhibition of caspase and Bad signaling.Citation7 In this study, we show that PAK4 is directly linked with Bad phosphorylation and drug resistance in PDAC cells. Inhibition of PAK4 by KPT-9274 abrogated p-Bad while knockdown of PAK4 by siRNA induced the pro-apoptotic potential of PDAC in vitro. These results demonstrate a distinct role for PAK4 in mediating drug resistance through Bad phosphorylation.
Several tumor suppressive miRNAs have been known to contribute to the regulation of apoptosis and cell survival in PDAC cells. The micro RNA, miR-145, has been found to regulate apoptosis in a hypoxia-dependent and TNF-α induced manner.Citation36,Citation37 Moreover, miR-145 could reverse drug resistance and sensitize cancer cells including PDAC to standard chemotherapies and radiotherapy.Citation38-Citation41 In distinct tumor models, miR-145 upregulation has been directly shown to inhibit tumor growth through the suppression of Rho-associated protein kinase 1 (ROCK1).Citation14,Citation15 These multiple lines of evidence indicate that the Rho family members, if not PAK4, can negatively influence miR-145 signaling and in turn promote cancer cell survival. This led us to hypothesize that targeting K-Ras-PAK4-microRNA axis could be a novel strategy to suppress predominantly oncogenic K-Ras-driven diseases such as PDAC.Citation11 In this study, we found that KPT-9274 increased the level of miR-145, which is consistent with apoptosis induction and increased PDAC drug sensitivity. The miRNA, let-7c, is critically involved in the regulation of cell survival and apoptosis. This miRNA can inhibit cancer cell proliferation and induce apoptotic cell death.Citation42,Citation43 More importantly, let-7c and let-7d can control chemo- and radio-resistance, and sensitize cancer cells to chemotherapies.Citation44-Citation46 Our results showed that KPT-9274 upregulated the level of let-7c and let-7d, which is correlated with induction of apoptosis, inhibition of cell proliferation and sensitization of PDAC cells. In addition, miR-34c, miR-320a and miR-100 have been shown to be tumor suppressive miRNAs that inhibit tumor growth, induce apoptosis and reverse drug resistance.Citation47-Citation50 The induction of these miRNAs by KPT-9274 were associated with the increased apoptosis, decreased proliferation and increased synergy with anti-cancer drugs in PDAC. Therefore, our results are consistent with other reports, suggesting the beneficial effects of PAK4 inhibition on the regulation of miRNA signaling.
In conclusion, our results indicate that KPT-9274 and KPT-9307 as novel therapeutic agents significantly induce apoptosis and inhibit PDAC cell proliferation through the downregulation of Bad phosphorylation and Bcl-2, and the upregulation of Bad, Bax, Caspase 3, and tumor suppressive miRNAs (miR-145, let-7c, let-7d, miR-34c, miR320 and miR-100) in PDAC cells. PAK4 inhibition by KPT-9274 also enhance the anti-cancer activity of gemcitabine in PDAC. These results suggest that KPT-9274 could be used in combination treatment with conventional chemotherapies as a promising therapy of drug resistant PDAC. However, more in vitro cellular experiments, in vivo animal studies and clinical trials are needed to better understand the value of using KPT-9274 as a targeted therapy for PDAC.
Materials and methods
Cell lines, reagents, and antibodies
MiaPaCa-2, HPAC, BxPC-3 and Panc-1 PDAC cells were purchased from American Type Culture Collection (ATCC, Manassas, VA) and maintained in DMEM (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg/mL streptomycin in a 5% CO2 atmosphere at 37°C. L3.6pl PDAC cells and human pancreatic duct epithelial (HPDE) cells were obtained from MD Anderson Cancer Center and cultured in DMEM/FBS or keratinocyte serum-free medium supplied with 5 ng/mL of epidermal growth factor and 50 μg/mL of bovine pituitary extract (Invitrogen). The cell lines have been tested and authenticated in core facility Applied Genomics Technology Center at Wayne State University. The method used for testing was short tandem repeat (STR) profiling using the PowerPlex® 16 System from Promega (Madison, WI). The dual PAK4 and NAMPT inhibitors including KPT-9274 and KPT-9307 (Karyopharm Therapeutics, Newton, MA) were dissolved in DMSO to make a 1 mM stock solution. Anti-phospho-Bad(Ser112) (Cell Signaling, Danvers, MA), anti-phospho-Bad(Ser136) (Cell Signaling), anti-phospho-Bad(Ser155) (Cell Signaling), and anti-β-actin (Sigma, St. Louis, MO) primary antibodies were used for Western Blot analysis.
Inhibition of PAK4 expression by siRNA in PDAC cells
MiaPaCa-2 cells were seeded in a 6 well plate (1.2 × 105 cells per well) and incubated at 37°C for 24 hours. The cells were then transfected with PAK4 siRNA (Santa Cruz) or control siRNA by DharmaFact Transfection Reagent (Dharmacon) for 72 hours. Then, the RNA was extracted and subject to mRNA RT-PCR.
RNA isolation and miRNA real-time RT-PCR
Total RNA was extracted and purified by using the miRNeasy Mini Kit and RNase-free DNase Set (QIAGEN, Valencia, CA) following the protocol provided by the manufacturer. The expression level of miR-145, miR-34c, let-7c, let-7d, miR-320 and miR-100 in KPT-9274 or KPT-9307 treated or un-treated PDAC cells was analyzed by using Universal cDNA Synthesis Kit (Exiqon, Woburn, MA), specific LNA™ PCR primer set (Exiqon), and SYBR Green RT-PCR Reagents (Applied biosystems). The PCR program was initiated by 10 min at 95°C before 40 thermal cycles, each of 15 s at 95°C and 1 min at 60°C. Data were analyzed according to the comparative Ct method and were normalized by RNU44 and RNU1a1 expression in each sample.
mRNA real-time RT-PCR
The expression level of Bad, PAK4, Caspase 3, Bcl-2 and Bax in KPT-9274 or KPT-9307 treated or un-treated and control siRNA or PAK4 siRNA transfected PDAC cells was analyzed by real-time RT-PCR using High Capacity cDNA Reverse Transcription Kit and SYBR Green Master Mixture from Applied Biosystems. The sequences of primers used were: Bad-F: CCGGAGGATGAGTGACGAGT; Bad-R: CCAAGTTCCGATCCCACCAG; PAK4-F: GTGCAAGAGAGCTGAGGGAG; PAK4-R: ATGCTGGTGGGACAGAAGTG; Caspase3-F: CTCTGGTTTTCGGTGGGTGT; Caspase3-R: TCCAGAGTCCATTGATTCGCT; Bcl2-F: TGAACTGGGGGAGGATTGTG; Bcl2-R: CGTACAGTTCCACAAAGGCA; Bax-F: AGGTCTTTTTCCGAGTGGCA; Bax-R: CCCGGAGGAAGTCCAATGTC; GAPDH-F: CCACATCGCTCAGACACCAT; GAPDH-R: ACCAGAGTTAAAAGCAGCCCT; 18S-F: GCAATTATTCCCCATGAACG; and 18S-R: GGCCTCACTAAACCATCCAA. The PCR was initiated by 10 min at 95°C before 40 thermal cycles, each of 15 s at 95°C and 1 min at 60°C. Data were analyzed according to the comparative Ct method and were normalized by GAPDH and 18S rRNA expression in each sample.
Western Blot analysis
Western Blot analysis was conducted to measure the alterations in the protein expression of genes. MiaPaCa-2 and Panc-1 PC cells were treated with or without 500 nM KPT-9274 or KPT-9307 for 72 hours. In a separated experiment, MiaPaCa-2, BxPC-3, HPAC and Panc-1 PC cells were treated with KPT-9274 for 48 hours followed by 30 nM Calyculin A treatment of 40 min. After treatment, the cells were lysed in RIPA buffer, and protein concentration was measured using BCA protein assay (PIERCE, Rockford, IL). The proteins were subjected to 14% SDS-PAGE, and electrophoretically transferred to nitrocellulose membrane. The membranes were incubated with specific primary antibodies, and subsequently incubated with secondary antibody conjugated with peroxidase (Bio-rad, Hercules, CA). The signal was detected using the chemiluminescent detection system (PIERCE) and the signals in films were semi-quantified by AlphaEaseFC (Alpha Innotech, San Leandro, CA). The ratio of p-Bad/actin signals was calculated.
Growth inhibition assay
MiaPaCa-2 cells were treated with 250–4000 nM KPT-9274 and/or 150–600 nM Gemcitabine for 72 hours. Then, the cells were subjected to cell proliferation assay using MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide]. Thespectrophotometric absorbance of the samples was determinedby using a plate reader SynergyHT (BioTek, Winooski, WI) at 470 nm. The combination index value and isobologram were calculated and created by using CalcuSyn software (Biosoft, Cambridge, UK).
Quantification of apoptosis by Annexin V FITC assay
Cell apoptosis was detected using Annexin V FITC (Biovision Danvers MA) according to the manufacturer's protocol as we reported previously.Citation21 PDAC cells were treated with 500 nM KPT-9274 for 72 hrs. At the end of treatment, cells were trypsinized and equal numbers were stained with Annexin V and Propidum Iodide. The stained cells were analyzed using a Becton Dickinson flow cytometer at the Karmanos Cancer Institute Flow cytometry core.
Statistics
Wherever appropriate, the data were subjected to a Student's t-test using GraphPad Prism software (La Jolla, CA). p < 0.05 was considered statistically significant.
Disclosure of potential conflicts of interest
William Senapedis, Erkan Baloglu, Yosef Landesman, Michael Kauffman and Sharon Shacham are employees of Karyopharm Therapeutics Inc. hold patent, equity and stocks and has received both major and minor renumerations from Karyopharm. All other authors have no potential conflict of interests.
Funding
Work in the laboratory of Azmi AS is supported by NIH R21 grant 1R21CA188818–01A1. The authors thank the SKY Foundation, James H Thie foundation and Perri Foundation for supporting part of this study.
References
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017; 67:7-30; https://doi.org/10.3322/caac.21387
- Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med 2014; 371:1039-49; PMID:25207767; https://doi.org/10.1056/NEJMra1404198
- Azmi AS, Mohammad RM. p21-activated kinase 4: a druggable target in the elusive oncogenic KRAS pathway? Future Med Chem 2015; 7:5-7; PMID:25582328; https://doi.org/10.4155/fmc.14.144
- Alan JK, Lundquist EA. Mutationally activated Rho GTPases in cancer. Small GTPases 2013; 4:159-63; PMID:24088985; https://doi.org/10.4161/sgtp.26530
- Popoff MR, Geny B. Multifaceted role of Rho, Rac, Cdc42 and Ras in intercellular junctions, lessons from toxins. Biochim Biophys Acta 2009; 1788:797-812; PMID:19366594; https://doi.org/10.1016/j.bbamem.2009.01.011
- Yeo D, He H, Baldwin GS, Nikfarjam M. The role of p21-activated kinases in pancreatic cancer. Pancreas 2015; 44:363-9; PMID:25760284; https://doi.org/10.1097/mpa.0000000000000276
- Gnesutta N, Qu J, Minden A. The serine/threonine kinase PAK4 prevents caspase activation and protects cells from apoptosis. J Biol Chem 2001; 276:14414-9; PMID:11278822; https://doi.org/10.1074/jbc.M011046200
- Winter PS, Sarosiek KA, Lin KH, Meggendorfer M, Schnittger S, Letai A, Wood KC. RAS signaling promotes resistance to JAK inhibitors by suppressing BAD-mediated apoptosis. Sci Signal 2014; 7:ra122; PMID:25538080; https://doi.org/10.1126/scisignal.2005301
- Liu Y, Sun SY, Owonikoko TK, Sica GL, Curran WJ, Khuri FR, Deng X. Rapamycin induces Bad phosphorylation in association with its resistance to human lung cancer cells. Mol Cancer Ther 2012; 11:45-56; PMID:22057915; https://doi.org/10.1158/1535-7163.mct-11-0578
- Cannings E, Kirkegaard T, Tovey SM, Dunne B, Cooke TG, Bartlett JM. Bad expression predicts outcome in patients treated with tamoxifen. Breast Cancer Res Treat 2007; 102:173-9; PMID:17004114; https://doi.org/10.1007/s10549-006-9323-8
- Choudhry ZS, Tripathi V, Sutton M, Bao B, Mohammad RM, Azmi AS. Regulation of KRAS-PAK4 axis by microRNAs in cancer. Curr Pharm Des 2014; 20:5275-8; PMID:24479809; https://doi.org/10.2174/1381612820666140128203452
- Feng X, Jiang J, Shi S, Xie H, Zhou L, Zheng S. Knockdown of miR-25 increases the sensitivity of liver cancer stem cells to TRAIL-induced apoptosis via PTEN/PI3K/Akt/Bad signaling pathway. Int J Oncol 2016; 49:2600-10; PMID:27840896; https://doi.org/10.3892/ijo.2016.3751
- Subramani R, Gangwani L, Nandy SB, Arumugam A, Chattopadhyay M, Lakshmanaswamy R. Emerging roles of microRNAs in pancreatic cancer diagnosis, therapy and prognosis (Review). Int J Oncol 2015; 47:1203-10; PMID:26314882; https://doi.org/10.3892/ijo.2015.3129
- Ding W, Tan H, Zhao C, Li X, Li Z, Jiang C, Zhang Y, Wang L. MiR-145 suppresses cell proliferation and motility by inhibiting ROCK1 in hepatocellular carcinoma. Tumour Biol 2016; 37:6255-60; PMID:26615424; https://doi.org/10.1007/s13277-015-4462-3
- Zheng M, Sun X, Li Y, Zuo W. MicroRNA-145 inhibits growth and migration of breast cancer cells through targeting oncoprotein ROCK1. Tumour Biol 2016; 37:8189-96; PMID:26715279; https://doi.org/10.1007/s13277-015-4722-2
- Garajova I, Le Large TY, Frampton AE, Rolfo C, Voortman J, Giovannetti E. Molecular mechanisms underlying the role of microRNAs in the chemoresistance of pancreatic cancer. Biomed Res Int 2014; 2014:678401; PMID:25250326; https://doi.org/10.1155/2014/678401
- Brunetti O, Russo A, Scarpa A, Santini D, Reni M, Bittoni A, Azzariti A, Aprile G, Delcuratolo S, Signorile M et al. MicroRNA in pancreatic adenocarcinoma: predictive/prognostic biomarkers or therapeutic targets? Oncotarget 2015; 6:23323-41; PMID:26259238; https://doi.org/10.18632/oncotarget.4492
- Lou E, Subramanian S, Steer CJ. Pancreatic cancer: modulation of KRAS, MicroRNAs, and intercellular communication in the setting of tumor heterogeneity. Pancreas 2013; 42:1218-26; PMID:24152947; https://doi.org/10.1097/mpa.0000000000000007
- Azmi AS, Senapedis W, Baloglu E, Landesman Y, Kauffman M, Shacham S, Wu J, Aboukameel A, Muqbil I, Mohammad RM. Abstract 4688: Overcoming drug resistance and stemness in oncogenic kras driven pancreatic ductal adenocarcinoma through PAK4 inhibition. Cancer Res 2015; 75:4688; https://doi.org/10.1158/1538-7445.am2015-4688
- Abu Aboud O, Chen CH, Senapedis W, Baloglu E, Argueta C, Weiss RH. Dual and Specific Inhibition of NAMPT and PAK4 By KPT-9274 Decreases Kidney Cancer Growth. Mol Cancer Ther 2016; 15:2119-29; PMID:27390344; https://doi.org/10.1158/1535-7163.mct-16-0197
- Aboukameel A, Muqbil I, Senapedis W, Baloglu E, Landesman Y, Shacham S, Kauffman M, Philip PA, Mohammad RM, Azmi AS. Novel p21-activated kinase 4 (PAK4) allosteric modulators overcome drug resistance and stemness in pancreatic ductal adenocarcinoma. Mol Cancer Ther 2017; 16:76-87; PMID:28062705; https://doi.org/10.1158/1535-7163.mct-16-0205
- Ju HQ, Zhuang ZN, Li H, Tian T, Lu YX, Fan XQ, Zhou HJ, Mo HY, Sheng H, Chiao PJ et al. Regulation of the Nampt-mediated NAD salvage pathway and its therapeutic implications in pancreatic cancer. Cancer Lett 2016; 379:1-11; PMID:27233476; https://doi.org/10.1016/j.canlet.2016.05.024
- King H, Thillai K, Whale A, Arumugam P, Eldaly H, Kocher HM, Wells CM. PAK4 interacts with p85 alpha: implications for pancreatic cancer cell migration. Sci Rep 2017; 7:42575; PMID:28205613; https://doi.org/10.1038/srep42575
- Bold RJ, Virudachalam S, McConkey DJ. BCL2 expression correlates with metastatic potential in pancreatic cancer cell lines. Cancer 2001; 92:1122-9; PMID:11571724; https://doi.org/10.1002/1097-0142(20010901)92:5<1122::AID-CNCR1429>3.0.CO;2-H
- Westphal S, Kalthoff H. Apoptosis: targets in pancreatic cancer. Mol Cancer 2003; 2:6; PMID:12605713; https://doi.org/10.1186/1476-4598-2-6
- Datta SR, Ranger AM, Lin MZ, Sturgill JF, Ma YC, Cowan CW, Dikkes P, Korsmeyer SJ, Greenberg ME. Survival factor-mediated BAD phosphorylation raises the mitochondrial threshold for apoptosis. Dev Cell 2002; 3:631-43; PMID:12431371; https://doi.org/10.1016/S1534-5807(02)00326-X
- Hayakawa J, Ohmichi M, Kurachi H, Kanda Y, Hisamoto K, Nishio Y, Adachi K, Tasaka K, Kanzaki T, Murata Y. Inhibition of BAD phosphorylation either at serine 112 via extracellular signal-regulated protein kinase cascade or at serine 136 via Akt cascade sensitizes human ovarian cancer cells to cisplatin. Cancer Res 2000; 60:5988-94; PMID:11085518
- Schurmann A, Mooney AF, Sanders LC, Sells MA, Wang HG, Reed JC, Bokoch GM. p21-activated kinase 1 phosphorylates the death agonist bad and protects cells from apoptosis. Mol Cell Biol 2000; 20:453-61; PMID:10611223; https://doi.org/10.1128/MCB.20.2.453-461.2000
- Fang X, Yu S, Eder A, Mao M, Bast RC, Jr., Boyd D, Mills GB. Regulation of BAD phosphorylation at serine 112 by the Ras-mitogen-activated protein kinase pathway. Oncogene 1999; 18:6635-40; PMID:10597268; https://doi.org/10.1038/sj.onc.1203076
- Harada H, Becknell B, Wilm M, Mann M, Huang LJ, Taylor SS, Scott JD, Korsmeyer SJ. Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol Cell 1999; 3:413-22; PMID:10230394; https://doi.org/10.1016/S1097-2765(00)80469-4
- Tran NH, Frost JA. Phosphorylation of Raf-1 by p21-activated kinase 1 and Src regulates Raf-1 autoinhibition. J Biol Chem 2003; 278:11221-6; PMID:12551923; https://doi.org/10.1074/jbc.M210318200
- Wu X, Carr HS, Dan I, Ruvolo PP, Frost JA. p21 activated kinase 5 activates Raf-1 and targets it to mitochondria. J Cell Biochem 2008; 105:167-75; PMID:18465753; https://doi.org/10.1002/jcb.21809
- Bradshaw-Pierce EL, Pitts TM, Tan AC, McPhillips K, West M, Gustafson DL, Halsey C, Nguyen L, Lee NV, Kan JL et al. Tumor P-Glycoprotein Correlates with Efficacy of PF-3758309 in in vitro and in vivo Models of Colorectal Cancer. Front Pharmacol 2013; 4:22; PMID:23524533; https://doi.org/10.3389/fphar.2013.00022
- Kimmelman AC, Hezel AF, Aguirre AJ, Zheng H, Paik JH, Ying H, Chu GC, Zhang JX, Sahin E, Yeo G et al. Genomic alterations link Rho family of GTPases to the highly invasive phenotype of pancreas cancer. Proc Natl Acad Sci U S A 2008; 105:19372-7; PMID:19050074; https://doi.org/10.1073/pnas.0809966105
- Radu M, Semenova G, Kosoff R, Chernoff J. PAK signalling during the development and progression of cancer. Nat Rev Cancer 2014; 14:13-25; PMID:24505617; https://doi.org/10.1038/nrc3645
- Zheng M, Wu Z, Wu A, Huang Z, He N, Xie X. MiR-145 promotes TNF-alpha-induced apoptosis by facilitating the formation of RIP1-FADDcaspase-8 complex in triple-negative breast cancer. Tumour Biol 2016; 37:8599-607; PMID:26733177; https://doi.org/10.1007/s13277-015-4631-4
- Blick C, Ramachandran A, McCormick R, Wigfield S, Cranston D, Catto J, Harris AL. Identification of a hypoxia-regulated miRNA signature in bladder cancer and a role for miR-145 in hypoxia-dependent apoptosis. Br J Cancer 2015; 113:634-44; PMID:26196183; https://doi.org/10.1038/bjc.2015.203
- Liu RL, Dong Y, Deng YZ, Wang WJ, Li WD. Tumor suppressor miR-145 reverses drug resistance by directly targeting DNA damage-related gene RAD18 in colorectal cancer. Tumour Biol 2015; 36:5011-9; PMID:25913620; https://doi.org/10.1007/s13277-015-3152-5
- Zhan M, Zhao X, Wang H, Chen W, Xu S, Wang W, Shen H, Huang S, Wang J. miR-145 sensitizes gallbladder cancer to cisplatin by regulating multidrug resistance associated protein 1. Tumour Biol 2016; 37:10553-62; PMID:26852750; https://doi.org/10.1007/s13277-016-4957-6
- Yan S, Li X, Jin Q, Yuan J. MicroRNA-145 sensitizes cervical cancer cells to low-dose irradiation by downregulating OCT4 expression. Exp Ther Med 2016; 12:3130-6; PMID:27882128; https://doi.org/10.3892/etm.2016.3731
- Lin Y, Ge X, Wen Y, Shi ZM, Chen QD, Wang M, Liu LZ, Jiang BH, Lu Y. MiRNA-145 increases therapeutic sensibility to gemcitabine treatment of pancreatic adenocarcinoma cells. Oncotarget 2016; 7:70857-68; PMID:27765914; https://doi.org/10.18632/oncotarget.12268
- Zhu X, Wu L, Yao J, Jiang H, Wang Q, Yang Z, Wu F. MicroRNA let-7c Inhibits Cell Proliferation and Induces Cell Cycle Arrest by Targeting CDC25A in Human Hepatocellular Carcinoma. PLoS One 2015; 10:e0124266; PMID:25909324; https://doi.org/10.1371/journal.pone.0124266
- Zhou Z, Lu X, Wang J, Xiao J, Liu J, Xing F. microRNA let-7c is essential for the anisomycin-elicited apoptosis in Jurkat T cells by linking JNK1/2 to AP-1/STAT1/STAT3 signaling. Sci Rep 2016; 6:24434; PMID:27087117; https://doi.org/10.1038/srep24434
- Sugimura K, Miyata H, Tanaka K, Hamano R, Takahashi T, Kurokawa Y, Yamasaki M, Nakajima K, Takiguchi S, Mori M et al. Let-7 expression is a significant determinant of response to chemotherapy through the regulation of IL-6/STAT3 pathway in esophageal squamous cell carcinoma. Clin Cancer Res 2012; 18:5144-53; PMID:22847808; https://doi.org/10.1158/1078-0432.ccr-12-0701
- Cui SY, Huang JY, Chen YT, Song HZ, Feng B, Huang GC, Wang R, Chen LB, De W. Let-7c governs the acquisition of chemo- or radioresistance and epithelial-to-mesenchymal transition phenotypes in docetaxel-resistant lung adenocarcinoma. Mol Cancer Res 2013; 11:699-713; PMID:23562878; https://doi.org/10.1158/1541-7786.mcr-13-0019-t
- Chang CJ, Hsu CC, Chang CH, Tsai LL, Chang YC, Lu SW, Yu CH, Huang HS, Wang JJ, Tsai CH et al. Let-7d functions as novel regulator of epithelial-mesenchymal transition and chemoresistant property in oral cancer. Oncol Rep 2011; 26:1003-10; PMID:21725603; https://doi.org/10.3892/or.2011.1360
- Zhang B, Zhao R, He Y, Fu X, Fu L, Zhu Z, Fu L, Dong JT. MicroRNA 100 sensitizes luminal A breast cancer cells to paclitaxel treatment in part by targeting mTOR. Oncotarget 2016; 7:5702-14; PMID:26744318; https://doi.org/10.18632/oncotarget.6790
- Xu C, Zeng Q, Xu W, Jiao L, Chen Y, Zhang Z, Wu C, Jin T, Pan A, Wei R et al. miRNA-100 inhibits human bladder urothelial carcinogenesis by directly targeting mTOR. Mol Cancer Ther 2013; 12:207-19; PMID:23270926; https://doi.org/10.1158/1535-7163.mct-12-0273
- Lu Y, Wu D, Wang J, Li Y, Chai X, Kang Q. miR-320a regulates cell proliferation and apoptosis in multiple myeloma by targeting pre-B-cell leukemia transcription factor 3. Biochem Biophys Res Commun 2016; 473:1315-20; PMID:27086852; https://doi.org/10.1016/j.bbrc.2016.04.069
- Liang X, Zhou D, Wei C, Luo H, Liu J, Fu R, Cui S. MicroRNA-34c enhances murine male germ cell apoptosis through targeting ATF1. PLoS One 2012; 7:e33861; PMID:22479460; https://doi.org/10.1371/journal.pone.0033861