
ABSTRACT
The loss of the von Hippel-Lindau (VHL) tumor-suppressor is a major driver of Clear Cell Renal Cell Carcinoma (CC-RCC) resulting in the stabilization and overactivation of hypoxia inducible factors (HIFs). ROCK1 is a well-known protein serine/threonine kinase which is recognized as having a role in cancer including alterations in cell motility, metastasis and angiogenesis. We recently investigated and identified a synthetic lethal interaction between VHL loss and ROCK1 inhibition in CC-RCC that is dependent on HIF overactivation. Increased expression and activity of both HIFs and ROCK1 occurs in many types of cancer supporting the potential therapeutic role of ROCK inhibitors beyond CC-RCC. We also discuss future research required to establish prognostic markers to predict tumor response to ROCK inhibitors.
Metastatic Clear Cell Renal Cell Carcinoma (CC-RCC) is a devastating disease with a 5-year survival 3 of 20%.Citation1 While surgical resection of the tumor is often curative for early stage disease, approximately one third of patients present with regional or metastatic CC-RCC.Citation2 Despite significant increases in T1a (< 4 cm) CC-RCC detection over the past few decades, with significant increases in surgical treatment, there has been no reduction in the number of patients presenting with more aggressive disease (regional or metastatic disease at time of diagnosis). As such, CC-RCC remains a major clinical challenge with great need for novel treatment strategies.
There are 2 types of FDA-approved therapies that are currently available for treating advanced-stage CC-RCC: immunotherapies (cytokines: interleukin-2 [IL2], interferon α [IFNα]; and programmed death-1 inhibitor [PD-1i]) and targeted therapies (receptor tyrosine kinase inhibitors [RTKis] and mammalian target of rapamycin inhibitors [mTORis]).
The first FDA-approved therapy for CC-RCC was IL2-based immunotherapy that has been shown to prolong overall patient survival to 17.5 months.Citation3 IL2 immunotherapy is both of limited efficacy and is associated with significant morbidity and some mortality. Since then, the first line RTKis − Sorafenib, Sunitinib, and Pazopanib − have been approved and prolong overall survival to 19.3 months,Citation4 29.3, and 28.4 months respectively.Citation5 Second line RTKi Axitinib has been shown to prolong overall survival to 13.6 months in Sorafenib-refractory patients and to 29.9 months in cytokine-refractory patients.Citation6 While immunotherapy and RTKis are used to treat advanced stage CC-RCC, mTORis are used to specifically treat metastatic CC-RCC. First line mTORi Temsirolimus is approved for poor-prognosis metastatic CC-RCC patients and prolongs overall survival to 10.9 months as compared with cytokine-based immunotherapy, which prolongs overall survival to 7.3 months.Citation7 Second line mTORi Everolimus increases overall survival of RTKi-refractory metastatic CC-RCC patients by 14.8 months.Citation8 While patients initially respond to FDA-approved therapies, the majority of patients develop drug resistance.Citation9
Recently, multiple next generation therapies, including 2 RTKis, Lenvatinib and Cabozantinib, and one immunotherapeutic, PD-1i Nivolumab, have been approved for CC-RCC. Along with inhibiting VEGF, both Lenvatinib and Cabozantinib also target additional RTKs that have been linked to RTKi drug resistance. Accordingly, upregulation of fibroblast growth factor receptor (FGFR) signaling has been shown to contribute to resistance to VEGFR inhibitors,Citation10 and Lenvatinib inhibits PDGFR, VEGFR, and FGFR. In RTKi refractory patients, Lenvatinib prolonged overall patient survival to 18.4 months in comparison to 17.5 months for Everolimus-treated patients. Combination treatment with both Lenvatinib and Everolimus was able to prolong overall patient survival to 25.5 months.Citation11 Overactivation of the hepatocyte growth factor receptor (HGFR, or MET) pathway has been also implicated in VEGFR inhibitor resistance,Citation12,13 and Cabozantinib inhibits both VEGFR and MET. Treatment with Cabozantinib prolonged overall survival to 21.4 months in RTKi refractory patients over Everolimus treatment, which only prolonged survival to 16.5 months. The approval of both Lenvatinib and Cabozantinib offers new options to overcome resistance to current RTKi, and further clinical trials are underway. Importantly, the therapeutic benefits of RTKis have been attributed to targeting of endothelial cells, and not direct targeting of cancer cells,Citation9,14 thus making the therapies, which directly target cancer cells, excellent candidates for combination treatment.
While only 12.5% of CC-RCC patients enter partial remission with cytokine-based immunotherapy,Citation15 second line Nivolumab-based immunotherapy increases overall survival of RTKi-refractory metastatic CC-RCC patients by 25 months, as compared with Everolimus, which increases overall survival of RTKi-refractory metastatic CC-RCC patients by 19.6 months.Citation16 This represents a significant advance for immunotherapy, but there remains the need for novel targeted therapies to treat CC-RCC patients belonging to the group of poor-responders to immunotherapy.
The genetics of sporadic CC-RCC has been extensively studied and loss of function of the von Hippel-Lindau (VHL) tumor-suppressor occurs in up to 90% of CC-RCC patients.Citation17-Citation19 In addition, the VHL gene is affected by mutations/deletions in familial VHL disease, a syndrome, predisposing affected individuals to hemangioblastoma, pheochromocytoma, and CC-RCC.Citation20 This genetic alteration sets the stage for a synthetic lethality screen to find novel therapeutics specifically targeting VHL-deficient cancer cells and sparing VHL-expressing normal tissue. The principle underlying synthetic lethality screens is that cancer cells with a specific genetic alteration (e.g. VHL deficiency) will be more sensitive to targeted inhibition of a certain pathway than normal cells, where genetic alterations of tumor-suppressor genes are rare.Citation21 Thus, the resulting synthetic lethality compounds represent excellent candidates for therapies that target mutation-bearing cancer cells, but spare surrounding normal tissues.
We recently reported on a chemical library screen that revealed a synthetic lethal interaction of VHL deficiency with the Rho-associated protein kinase (ROCK) inhibitors – Y-27632, RKI 1447, and GSK 429286 – confirmed in several CC-RCC genetic backgrounds (see for VHL and ROCK pathway overview).Citation22 siRNA-mediated knockdown of ROCK1, but not ROCK2, replicated the synthetic lethality effect, suggesting that inhibition of ROCK1 by ROCK inhibitors is critical for targeting VHL-deficient CC-RCC. Importantly, we have shown that the hypoxia-inducible factor (HIF) pathway, which gets over-activated as a result of VHL loss, is critical for sensitivity to ROCK inhibitors. Based on these findings we expect that ROCK inhibitors would represent potential therapeutics not only for VHL-deficient CC-RCC, but also for CC-RCC with certain VHL mutations. In addition, ROCK inhibitors should target hemangioblastoma and pheochromocytoma with VHL deficiencies and certain mutations, as well as cancer types with overactivation of HIF pathway arising independent of VHL loss of function.
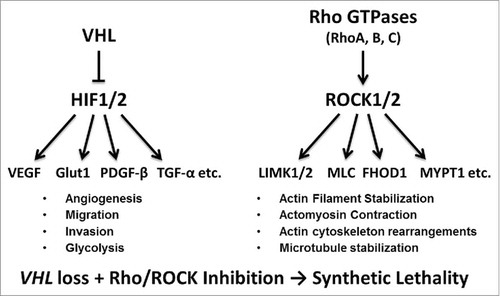
Figure 1. Overview of VHL/HIF and Rho/ROCK signaling pathways. VHL, left, is a part of an E3 ubiquitin ligase complex that targets HIF-1α and HIF-2α for degradation. The loss of VHL stabilizes HIFs, leading to elevated expression of a multitude of HIF-target genes, involved in angiogenesis, migration, invasion, glycolysis, etc. ROCK signaling, right, is dependent on activation by RhoGTPases that bind to ROCK1 and ROCK2. ROCK family kinases are major regulators of actin organization within the cell controlling actin filament stabilization, actomyosin contraction, actin cytoskeleton rearrangements, microtubule stabilization, etc. The combination of VHL loss leading to HIF overactivation and Rho/ROCK pathway inhibition triggers synthetic lethality.
VHL loss of function due to deletions, mutations, and promoter hypermethylation occurs in over 90% of sporadic CC-RCC cases.Citation23 The frequency of VHL mutations in CC-RCC tumors ranges from about 46% to 82% depending on the study,Citation17,24 and loss of heterozygosity occurs in up to 98% of cases.Citation25 In addition, VHL promoter hypermethylation occurs in about 10 to 20% of CC-RCC tumors.Citation19,26,27 Accordingly, VHL loss of function occurs by multiple mechanisms and is a hallmark of sporadic CC-RCC tumors, which has been shown to be a major driver of the disease.
Besides sporadic cases, CC-RCC frequently occurs in people affected by familial VHL disease. VHL disease is a heritable autosomal-dominant neoplastic syndrome with an incidence of 1 in 36,00028 that is associated with the development of renal cysts (60 to 70% of patients), with some cysts progressing to CC-RCC (40% of patients), spinal cord (60 to 80% of patients) and retinal (60% of patients) hemangioblastomas (tumors originating from the vasculature), and pheochromocytoma (adrenal gland tumors) (5% of patients).Citation29,30 Patients with VHL disease are born lacking one functional copy of the VHL gene, and during their lifetime lose a second functional copy due to mutation, deletion, or promoter hypermethylation in certain tissues, triggering cancer development. The disease is divided into distinct subtypes based on VHL status.Citation31 Type 1 VHL disease is associated with deletions and mutations in VHL that completely disrupt its function and are associated with high risk of CC-RCC and hemangioblastoma formation. Type 2 VHL disease is further split into 3 additional subsets, 2A-C, and is associated with VHL missense mutations.Citation32 Type 2A is associated with hemangioblastoma, pheochromocytoma, and a low risk for CC-RCC; type 2B is associated with hemangioblastoma, pheochromocytoma, and CC-RCC; whereas type 2C is associated with pheochromocytoma only.Citation23,31 Since our data indicate that VHL loss causing HIF stabilization sensitizes CC-RCC to ROCK inhibitors, we expect that tumors harboring VHL mutations which either completely disrupt VHL function (like those occurring in type 1 VHL disease, e.g., C162FCitation31), or specifically disrupt VHL's ability to regulate HIF activity (like those occurring in type 2A disease, e.g., Y98H, Y112H, A149T, T157I, and 2B disease, e.g., Y98N, Y112NCitation33,34 ) will be sensitive to ROCK inhibitors. The resulting overactivation of HIFs in these tumors will make them candidates for ROCK inhibitor-based therapies. Since VHL regulates many targets besides HIFα, including activated epidermal growth factor receptor, RNA Pol II subunits, protein kinase C, and othersCitation35 further investigation is needed to establish if, in addition to disruption of HIF regulation, disruption of any of these VHL functions by certain missense mutations is important for ROCK inhibitor sensitivity.
Overactivation of HIFs is a frequent event in cancer. Both HIF1α and HIF2α have been shown to be overexpressed in tumor samples compared with matched normal tissues in multiple cancer types besides CC-RCC including bladder, brain, breast, colon, ovarian, gastric, lung, melanoma, pancreatic, and prostate cancers.Citation36,37 While HIF activation often occurs in perinecrotic regions of solid tumors that lack adequate vasculature and oxygen supply,Citation38 there are multiple mechanisms by which HIFs are activated under normoxic conditions apart from VHL loss. For instance, the loss of p53 tumor suppressor leads to disruption of human homolog of mouse double minute 2 (HDM2)-mediated degradation of HIFα subunits resulting in HIF overactivation.Citation39 Similarly, the loss of phosphatase and tensin homolog (PTEN) leads to deregulation of phosphoinositide 3-kinase (PI3K) and protein kinase B (Akt) activity resulting in HIF overactivation.Citation40 Furthermore, disruption of prolyl hydroxylases (PHDs) by mutations blocks hydroxylation of the HIFα subunits and inhibits VHL-mediated HIFα degradation.Citation41 Multiple oncogenes that are commonly activated by mutations or overexpressed in cancer have also been shown to result in HIF overactivation, including Ha-rasCitation42 (via PI3K signaling), v-Src,Citation43 and c-Myc.Citation44 Although these findings suggest that HIF overactivation occurs frequently in multiple cancers, it is important to keep in mind that the magnitude of HIF activity is often less than in the case of VHL loss or hypoxic exposure, which are the main players of pathway controlling HIF activity.Citation37 Thus, additional experiments establishing the sensitivity of cancer cell lines with the genetic alterations listed above to ROCK inhibitors in normoxia and hypoxia are required to drive the conclusions on their sensitivity and utility of ROCK inhibitors for their targeting.
Another important factor that needs to be taken into consideration for the prediction of sensitivity to ROCK inhibitors is expression of the drug target, ROCK1, in cancers other than CC-RCC. In this respect, elevated ROCK1 expression was reported in breastCitation45 and prostateCitation46 cancers at the protein level and lung cancer at the mRNA level.Citation47 Activating ROCK1 somatic mutations have been also reported in breast and lung cancers,Citation48 affecting the autoinhibitory region of ROCK1, resulting in increased activity even though protein levels remain unchanged. Interestingly, ROCK1 has been reported to be a HIF-target gene in breast cancer,Citation49 although the regulation likely involves cell-type-specific components since we found ROCK1 expression to be similar in VHL-deficient CC-RCC and CC-RCC with re-introduced VHL.Citation22 In addition, overexpression at the mRNA and protein levels of upstream regulators of ROCK – Rho GTPases RhoA and RhoCCitation50 – occurs in breast, prostate, lung, bladder, colon, ovarian, gastric, melanoma, and pancreatic cancers.Citation51 While somatic mutations resulting in increased Rho activity are rare,Citation52 regulators of Rho, guanine-nucleotide exchange factors (GEFs), GTPase-activating proteins (GAPs) and guanine-nucleotide dissociation inhibitors (GDIs), have been shown to be deregulated in cancerCitation51 contributing to the activity of the Rho/ROCK pathway. Currently, a specific Rho GTPase inhibitor, Cethrin, was developed for the management of spinal cord injuries compatible with intrathecal delivery,Citation53 but feasibility of its systemic delivery needs evaluation. RhoGTPases can also be targeted indirectly, e.g., by statins (HMG-CoA Reductase inhibitors), which inhibit Rho GTPase isoprenylation and translocation to the plasma membrane,Citation54 although statins are far from being specific toward Rho, and also inhibit Ras and Rac GTPases dependent on isoprenylation.Citation54 Together, these data suggest that the Rho/ROCK pathway is active and can be targeted in multiple cancer types.
Overall, ROCK and HIF co-activation frequently occurs in several cancer types, suggesting that ROCK inhibitors should be effective for targeting those cancers. Accordingly, ROCK inhibitors have shown an anti-cancer effect in breast,Citation55 prostate,Citation46 ovarian,Citation56 and melanomaCitation57 cancers both in vitro and in vivo in mouse models. Recently, AT13148, a multi-kinase inhibitor targeting ROCK, has shown an anti-cancer effect in mouse models of breast,Citation58 prostate,Citation58 lung,Citation58 uterine,Citation58 gastric,Citation59 and melanomaCitation57 types of cancer. Currently, there is an ongoing phase I clinical trial (NCT01585701) of AT13148 administered to breast, prostate, and ovarian cancer patients, which will be evaluated for normal tissue toxicity and possible anti-tumor response. It is likely that the synthetic lethal interaction between ROCK inhibition and HIF overactivation contributes to sensitivity of these forms of cancer to ROCK inhibitors. Additional studies are required to develop reliable markers for prediction of ROCK inhibitor anti-tumor response.
In conclusion, the synthetic lethal interaction between ROCK inhibition and HIF overactivation is important since it justifies ROCK inhibitors as candidate therapeutics for multiple forms of cancer. While ROCK inhibitors represent good candidates for targeting hypoxic regions of solid tumors, where HIFs are stabilized due to the lack of adequate vasculature and oxygen supply, ROCK inhibitors are also expected to target cancers where HIFs are overactivated by mutations in their upstream regulators. It is also worth investigating which VHL mutations can confer sensitivity to ROCK inhibitors to a degree similar to VHL loss in CC-RCC, hemangioblastoma, and pheochromocytoma. Since ROCK1 is expressed in multiple cancers, it represents a druggable molecule. Further research is needed to evaluate the impact of the discovered synthetic lethal interaction on sensitivity of other forms of cancer besides CC-RCC to ROCK inhibitors; and develop the plan for patient stratification into ROCK inhibitor-sensitive and -insensitive groups.
Abbreviations
| Akt | = | protein kinase B |
| CC-RCC | = | Clear Cell Renal Cell Carcinoma |
| GAPs | = | GTPase-activating proteins |
| GDIs | = | guanine-nucleotide dissociation inhibitors |
| FGFR | = | fibroblast growth factor receptor |
| GEFs | = | guanine-nucleotide exchange factors |
| HIFs | = | hypoxia inducible factors |
| IFNα | = | interferon α |
| IL-2 | = | interleukin-2 |
| mTORis | = | mammalian target of rapamycin inhibitors |
| PD-1i | = | programmed death-1 inhibitor |
| PHD | = | prolyl hydroxylases |
| PI3K | = | phosphoinositide 3-kinase |
| PTEN | = | phosphatase and tensin homolog |
| ROCK | = | Rho-associated protein kinase |
| RTKis | = | receptor tyrosine kinase inhibitors |
| VHL | = | von Hippel-Lindau |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Author contributions
JMT, JL, and OVR wrote the paper.
Funding
This commentary was funded by ACS-IRG-98–279–10 and NCI R03 CA202563–01 to OVR, and NIH T32 (2T32CA009054–36A1) to JMT.
References
- Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, Heng DY, Larkin J, Ficarra V. Renal cell carcinoma. Nat Rev Dis Prim 2017; 3:17009; PMID:28276433; https://doi.org/10.1038/nrdp.2017.9
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. Cancer J Clin 2016; 66:7-30; https://doi.org/10.3322/caac.21332
- McDermott DF, Regan MM, Clark JI, Flaherty LE, Weiss GR, Logan TF, Kirkwood JM, Gordon MS, Sosman JA, Ernstoff MS, et al. Randomized phase III trial of high-dose interleukin-2 versus subcutaneous interleukin-2 and interferon in patients with metastatic renal cell carcinoma. J Clin Oncol 2005; 23:133-41; PMID:15625368; https://doi.org/10.1200/JCO.2005.03.206
- Escudier B, Eisen T, Stadler W, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 2007; 456:125-34; https://doi.org/10.1056/NEJMoa060655
- Motzer RJ, Hutson TE, Cella D, Reeves J, Hawkins R, Guo J, Nathan P, Staehler M, de Souza P, Merchan JR, et al. Pazopanib versus Sunitinib in metastatic renal-cell carcinoma. N Engl J Med 2013; 369:722-31; PMID:23964934; https://doi.org/10.1056/NEJMoa1303989
- Escudier B, Gore M. Axitinib for the management of metastatic renal cell carcinoma. Drugs R D 2011; 11:113-26; PMID:21679004; https://doi.org/10.2165/11591240-000000000-00000
- McKay RR, Lin X, Albiges L, Fay AP, Kaymakcalan MD, Mickey SS, Ghoroghchian PP, Bhatt RS, Kaffenberger SD, Simantov R, et al. Statins and survival outcomes in patients with metastatic renal cell carcinoma. Eur J Cancer 2016; 52:155-62; PMID:26687834; https://doi.org/10.1016/j.ejca.2015.10.008
- Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grünwald V, Thompson JA, Figlin RA, Hollaender N, et al. Phase 3 trial of everolimus for metastatic renal cell carcinoma: Final results and analysis of prognostic factors. Cancer 2010; 116:4256-65; PMID:20549832; https://doi.org/10.1002/cncr.25219
- Buczek M, Escudier B, Bartnik E, Szczylik C, Czarnecka A. Resistance to tyrosine kinase inhibitors in clear cell renal cell carcinoma: From the patient's bed to molecular mechanisms. Biochim Biophys Acta 2014; 1845:31-41; PMID:24135488; https://doi.org/10.1016/j.bbcan.2013.10.001
- Criscitiello C, Esposito A, De Placido S, Curigliano G. Targeting fibroblast growth factor receptor pathway in breast cancer. Curr Opin Oncol 2015; 27:452-6; PMID:26397764; https://doi.org/10.1097/CCO.0000000000000224. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26397764
- Motzer RJ, Hutson TE, Glen H, Michaelson MD, Molina A, Eisen T, Jassem J, Zolnierek J, Maroto JP, Mellado B, et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: A randomised, phase 2, open-label, multicentre trial. Lancet Oncol 2015; 16:1473-82; PMID:26482279; https://doi.org/10.1016/S1470-2045(15)00290-9
- Xie Z, Lee YH, Boeke M, Jilaveanu LB, Liu Z, Bottaro DP, Kluger HM, Shuch B. MET inhibition in clear cell renal cell carcinoma. J Cancer 2016; 7:1205-14; PMID:27390595; https://doi.org/10.7150/jca.14604
- Choueiri TK, Escudier B, Powles T, Tannir NM, Mainwaring PN, Rini BI, Hammers HJ, Donskov F, Roth BJ, Peltola K, et al. Cabozantinib versus everolimus in advanced renal cell carcinoma (METEOR): Final results from a randomised, open-label, phase 3 trial. Lancet Oncol 2016; 17:917-27; PMID:27279544; https://doi.org/10.1016/S1470-2045(16)30107-3
- Rini BI, Atkins MB. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol 2009; 10:992-1000; PMID:19796751; https://doi.org/10.1016/S1470-2045(09)70240-2
- Coppin C, Porzsolt F, Autenrieth M, Kumpf J, Coldman A, Wilt T. Immunotherapy for advanced renal cell cancer. Cochrane Database Syst Rev 2004; 3:CD001425; https://doi.org/10.1002/14651858.CD001425.pub2
- Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 2015; 373:1803-13; PMID:26406148; https://doi.org/10.1056/NEJMoa1510665
- Nickerson ML, Jaeger E, Shi Y, Durocher Ja, Mahurkar S, Zaridze D, Matveev V, Janout V, Kollarova H, Bencko V, et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res 2008; 14:4726-34; PMID:18676741; https://doi.org/10.1158/1078-0432.CCR-07-4921
- Chen F, Zhang Y, enbabao lu Y, Ciriello G, Yang L, Reznik E, Shuch B, Micevic G, De Velasco G, Shinbrot E, et al. Multilevel genomic-based taxonomy of renal cell carcinoma. Cell Rep 2016; 14:2476-89; PMID:26947078; https://doi.org/10.1016/j.celrep.2016.02.024
- Network CGAR. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013; 499:43-9; PMID:23792563; https://doi.org/10.1038/nature12222
- Latif F, Tory K, Gnarra J, Yao M, Duh F-M, Orcutt M, Stackhouse T, Kuzmin I, Modi W, Geil L, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Source Sci New Ser 1993; 260:1317-20.
- Chan Da, Giaccia AJ. Harnessing synthetic lethal interactions in anticancer drug discovery. Nat Rev Drug Discov 2011; 10:351-64; PMID:21532565; https://doi.org/10.1038/nrd3374
- Thompson JM, Nguyen QH, Singh M, Pavesic MW, Nesterenko I, Nelson LJ, Liao AC, Razorenova OV. Rho-associated kinase 1 inhibition is synthetically lethal with von Hippel-Lindau deficiency in clear cell renal cell carcinoma. Oncogene 2016; 36:1080-9; https://doi.org/10.1038/onc.2016.272
- Cowey CL, Rathmell WK. VHL gene mutations in renal cell carcinoma: Role as a biomarker of disease outcome and drug efficacy. Curr Oncol Rep 2009; 11:94-101; PMID:19216840; https://doi.org/10.1007/s11912-009-0015-5
- Shuin T, Kondo K, Torigoe S, Kishida T, Kubota Y, Hosaka M, Nagashima Y, Kitamura H, Latif F, Zbar B, et al. Frequent somatic mutations and loss of heterozygosity of the von Hippel-Lindau tumor suppressor gene in primary human renal cell carcinomas. Cancer Res 1994; 54:2852-5; PMID:8187067
- Gnarra JR, Tory K, Weng Y, Schmidt L, Wei MH, Li H, Latif F, Liu S, Chen F, Duh FM, et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet 1994; 7:85-90; PMID:7915601; https://doi.org/10.1038/ng0594-85
- Marshall FF. Promoter hypermethylation profile of kidney cancer. J Urol 2005; 174:484-5; PMID:16006871; https://doi.org/10.1016/S0022-5347(01)68286-7
- Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, Samid D, Duan DS, Gnarra JR, Linehan WM, et al. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci U S A 1994; 91:9700-4; PMID:7937876; https://doi.org/10.1073/pnas.91.21.9700
- Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH. Von Hippel-Lindau disease. Lancet 2003; 361:2059-67; https://doi.org/10.1016/S0140-6736(03)13643-4
- Gossage L, Eisen T, Maher ER. VHL, the story of a tumour suppressor gene. Nat Rev Cancer 2014; 15:55-64; https://doi.org/10.1038/nrc3844
- Maher ER, Neumann HP, Richard S. von Hippel-Lindau disease: A clinical and scientific review. Eur J Hum Genet 2011; 19:617-23; PMID:21386872; https://doi.org/10.1038/ejhg.2010.175
- Knauth K, Cartwright E, Freund S, Bycroft M, Buchberger A. VHL mutations linked to type 2C von Hippel-Lindau disease cause extensive structural perturbations in pVHL. J Biol Chem 2009; 284:10514-22; PMID:19228690; https://doi.org/10.1074/jbc.M809056200
- Kaelin WG. Molecular basis of the VHL hereditary cancer syndrome. Nat Rev Cancer 2002; 2:673-82; PMID:12209156; https://doi.org/10.1038/nrc885
- Li L, Zhang L, Zhang X, Yan Q, Minamishima YA, Olumi AF, Mao M, Bartz S, Kaelin WG Jr. Hypoxia-inducible factor linked to differential kidney cancer risk seen with type 2A and type 2B VHL mutations. Mol Cell Biol 2007; 27:5381-92; PMID:17526729; https://doi.org/10.1128/MCB.00282-07
- Knauth K, Bex C, Jemth P, Buchberger a. Renal cell carcinoma risk in type 2 von Hippel-Lindau disease correlates with defects in pVHL stability and HIF-1alpha interactions. Oncogene 2006; 25:370-7; PMID:16261165; https://doi.org/10.1038/sj.onc.1209062
- Zhang Q, Yang H. The roles of VHL-dependent ubiquitination in signaling and cancer. Front Oncol 2012; 2:1-7; PMID:22655263; https://doi.org/10.3389/fonc.2012.00035
- Talks KL, Turley H, Gatter KC, Maxwell PH, Pugh CW, Ratcliffe PJ, Harris AL. The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol 2000; 157:411-21; PMID:10934146; https://doi.org/10.1016/S0002-9440(10)64554-3
- Zhong H, De Marzo AM, Laughner E, Lim M, Hilton DA, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW. Overexpression of hypoxia-inducible factor 1alpha in common human cancers and their metastases. Cancer Res 1999; 59:5830-5; PMID:10582706
- Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer 2003; 3:721-32; PMID:13130303; https://doi.org/10.1038/nrc1187
- Sermeus A, Michiels C. Reciprocal influence of the p53 and the hypoxic pathways. Cell Death Dis 2011; 2:e164; PMID:21614094; https://doi.org/10.1038/cddis.2011.48
- Zundel W, Schindler C, Haas-Kogan D, Koong A, Kaper F, Chen E, Gottschalk AR, Ryan HE, Johnson RS, Jefferson AB, et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev 2000; 14:391-6; PMID:10691731
- Chan DA, Sutphin PD, Denko NC, Giaccia AJ. Role of prolyl hydroxylation in oncogenically stabilized hypoxia-inducible factor-1α. J Biol Chem 2002; 277:40112-7; PMID:12186875; https://doi.org/10.1074/jbc.M206922200
- Mazure NM, Chen EY, Laderoute KR, Giaccia AJ. Induction of vascular endothelial growth factor by Hypoxia is modulated by a phosphatidylinositol 3-kinase/Akt signaling pathway in Ha-ras-transformed cells through a hypoxia inducible factor-1 transcriptional element. Blood 1997; 90:3322-31; PMID:9345014
- Takacova M, Holotnakova T, Barathova M, Pastorekova S, Kopacek J, Pastorek J. Src induces expression of carbonic anhydrase IX via hypoxia-inducible factor 1. Oncol Rep 2010; 23:869-74; PMID:20127031
- Doe MR, Ascano JM, Kaur M, Cole MD. Myc post-transcriptionally induces HIF1 protein and target gene expression in normal and cancer cells. Cancer Res 2012; 72:949-57; PMID:22186139; https://doi.org/10.1158/0008-5472.CAN-11-2371
- Lane J, Martin T, Watkins G, Mansel R, Jiang W. The expression and prognostic value of ROCK I and ROCK II and their role in human breast cancer. Int J Oncol 2008; 33:585-93; PMID:18695890
- Zhang C, Zhang S, Zhang Z, He J, Xu Y, Liu S. ROCK has a crucial role in regulating prostate tumor growth through interaction with c-Myc. Oncogene 2013; 33:5582-91; https://doi.org/10.1038/onc.2013.505
- Nakagawa O, Fujisawa K, Ishizaki T, Saito Y, Nakao K, Narumiya S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett 1996; 392:189-93; PMID:8772201; https://doi.org/10.1016/0014-5793(96)00811-3
- Lochhead Pa, Wickman G, Mezna M, Olson MF. Activating ROCK1 somatic mutations in human cancer. Oncogene 2010; 29:2591-8; PMID:20140017; https://doi.org/10.1038/onc.2010.3
- Gilkes DM, Xiang L, Lee SJ, Chaturvedi P, Hubbi ME, Wirtz D, Semenza GL. Hypoxia-inducible factors mediate coordinated RhoA-ROCK1 expression and signaling in breast cancer cells. Proc Natl Acad Sci U S A 2013; 111:E384-93; https://doi.org/10.1073/pnas.1321510111
- Morgan-Fisher M, Wewer UM, Yoneda A. Regulation of ROCK activity in cancer. J Histochem Cytochem 2013; 61:185-98; PMID:23204112; https://doi.org/10.1369/0022155412470834
- Orgaz JL, Herraiz C, Sanz-Moreno V. Rho GTPases modulate malignant transformation of tumor cells. Small GTPases 2014; 5:2154-56; https://doi.org/10.4161/sgtp.29019
- Lin Y, Zheng Y. Approaches of targeting Rho GTPases in cancer drug discovery. Expert Opin Drug Discov 2015; 10:1-20; https://doi.org/10.1517/17460441.2015.1058775
- Lord-Fontaine S, Yang F, Diep Q, Dergham P, Munzer S, Tremblay P, McKerracher L. Local inhibition of Rho signaling by cell-permeable recombinant protein BA-210 prevents secondary damage and promotes functional recovery following acute spinal cord injury. J Neurotrauma 2008; 25:1309-22; PMID:19061375; https://doi.org/10.1089/neu.2008.0613
- Rikitake Y, Liao JK. Rho GTPases, statins, and nitric oxide. Circ Res 2005; 97:1232-5; PMID:16339495; https://doi.org/10.1161/01.RES.0000196564.18314.23
- Patel Ra, Forinash KD, Pireddu R, Sun Y, Sun N, Martin MP, Schönbrunn E, Lawrence NJ, Sebti SM. RKI-1447 is a potent inhibitor of the Rho-associated ROCK kinases with anti-invasive and antitumor activities in breast cancer. Cancer Res 2012; 72:5025-34; PMID:22846914; https://doi.org/10.1158/0008-5472.CAN-12-0954
- Ogata S, Morishige K-I, Sawada K, Hashimoto K, Mabuchi S, Kawase C, Ooyagi C, Sakata M, Kimura T. Fasudil inhibits lysophosphatidic acid-induced invasiveness of human ovarian cancer cells. Int J Gynecol Cancer 2009; 19:1142-9; https://doi.org/10.1111/IGC.0b013e3181c03909
- Sadok A, McCarthy A, Caldwell J, Collins I, Garrett MD, Yeo M, Hooper S, Sahai E, Kuemper S, Mardakheh FK, et al. Rho kinase inhibitors block melanoma cell migration and inhibit metastasis. Cancer Res 2015; 75:2272-84; PMID:25840982; https://doi.org/10.1158/0008-5472.CAN-14-2156
- Yap TA, Walton MI, Grimshaw KM, Te Poele RH, Eve PD, Valenti MR, de Haven Brandon AK, Martins V, Zetterlund A, Heaton SP, et al. AT13148 is a novel, oral multi-AGC kinase inhibitor with potent pharmacodynamic and antitumor activity. Clin Cancer Res 2012; 18:3912-23; PMID:22781553; https://doi.org/10.1158/1078-0432.CCR-11-3313
- Xi Y, Niu J, Shen Y, Li D, Peng X, Wu X. AT13148, a first-in-class multi-AGC kinase inhibitor, potently inhibits gastric cancer cells both in vitro and in vivo. Biochem Biophys Res Commun 2016; 478:330-6; PMID:26828267; https://doi.org/10.1016/j.bbrc.2016.01.167