
ABSTRACT
Oncogenic KRAS engages multiple effector pathways including the MAPK cascade to promote proliferation and survival of pancreatic cancer cells. KRAS-transformed cancer cells exhibit oncogene addiction to sustained activity of RAS for maintenance of malignant phenotypes. Previously, we have shown an essential role for the RHO guanine exchange factor ARHGEF2 for growth and survival of RAS-transformed pancreatic tumors. Here, we have determined that pancreatic cancer cells demonstrating KRAS addiction are significantly dependent on expression of ARHGEF2. Furthermore, enforced expression of ARHGEF2 desensitizes cells to pharmacological MEK inhibition and initiates a positive feedback loop which activates ERK phosphorylation and the downstream ARHGEF2 promoter. Therefore, targeting ARHGEF2 expression may increase the efficacy of MAPK inhibitors for treatment of RAS-dependent pancreatic cancers.
Introduction
The dependency on sustained flux through pro-proliferative signaling pathways leads to cellular vulnerabilities that present opportunity for therapeutic intervention. Constitutive activation of oncogenes such as RAS, can lead to cellular addiction to continued expression of the oncogene and its downstream activity.Citation47 Therefore, oncogenic signaling pathways are drug targets that can be exploited to halt mechanisms required for cancer cell growth and survival.
Small GTPases are molecular switches that cycle between active GTP-bound and inactive-GDP bound forms that regulate a variety of cellular signaling events including growth, cellular differentiation, cell motility and survival.Citation12,Citation23 Arguably the most common of the small GTPases, the Ras superfamily, include 5 main family members Ras, Rho, Ran, Rab, and Arf. These families can be further divided into subfamilies that share the common core G domain providing essential GTPase and nucleotide exchange activity to regulate discrete cellular processes.Citation14,Citation48
RAS is the prototypical member of the Ras family and is encoded by 3 proto-oncogenes, KRAS, HRAS and NRAS, which are frequently found mutated in human cancers.Citation13 Of these, KRAS mutations are most frequent, and are present in 33% of all tumors, including 95% of pancreas, 61% of colon and 17% of lung cancers. KRAS activation is coupled to transcription through the activation of multiple effector pathways including RAF-MAPK, PI3K, RALGDS, TIAM1-RAC and ARHGEF2-RHOA that underlie the phenotypes associated with pancreatic and other cancers.Citation9-Citation11,Citation15,Citation31,Citation37,Citation41 Finding therapeutically tractable targets in RAS effector pathways is critical for effective treatment of RAS driven cancers.
Pancreatic cancer is a highly metastatic deadly solid malignancy with a 5 y survival rate of less than 5%. Activating mutations in KRAS are nearly ubiquitous in pancreatic ductal adenocarcinoma (PDAC) occurring in 90–95% of cases.Citation1,Citation24,Citation42 Since mutation of KRAS is an important driver of PDAC often occurring with up regulation and hyperactivation of EGFR1/2, suggests pharmacological agents aimed at inhibition of the MAPK pathway offer promising treatments.Citation2,Citation7,Citation25,Citation46 However, combined treatment of MAPK inhibitors with current chemotherapy agents such as gemcitabine do not improve clinical outcome.Citation18,Citation30,Citation45 The effectiveness of pharmacological agents that target KRAS effectors often results in activation of compensatory pathways that limit their efficacy highlighting the importance of identifying mechanisms of adaptive response to MAPK inhibitory agents.
The RHO guanine exchange factor ARHGEF2 (also known as GEF-H1) is a microtubule associated guanine exchange factor and Dbl family member demonstrating exchange activity toward RHOA.Citation34 ARHGEF2 was found to contribute to cell survival and growth in KRAS and HRAS-transformed cells.Citation9 Furthermore, the oncogenic potential of ARHGEF2 has been demonstrated in NIH-3T3 fibroblast transformation assays in nude mice.Citation3 ARHGEF2 plays a critical role in supporting RAS transformation as depletion of ARHGEF2 hinders the growth of pancreatic xenografts in vivo.Citation9 We have found that ARHGEF2 is a transcriptional target of oncogenic KRAS and the ARHGEF2 promoter is transactivated downstream of multiple signaling pathways including MAPK and PI3K.Citation20
It is possible that RAS activation of the MAPK pathway is coupled to microtubule function through ARHGEF2 to promote and coordinate mitogenic signals with changes in cell shape, migration and morphogenesis. Indeed, cellular perturbation by mechanical strain has been shown to activate ERK in a RHOA dependent manner.Citation22,Citation39 Previously, we have demonstrated that ARHGEF2 contains negative regulatory sequences between amino acids 87–151 which sequester ARHGEF2 to the microtubules.Citation26 Lacking the microtubule binding domain, ARHGEF2Δ87–151 interacts with KSR-1, the best characterized scaffold for the MAPK pathway and was sufficient for activation of MEK1/2 and ERK1/2 phosphorylation in the absence of either PDGF or oncogenic RAS.Citation9
Expression of ARHGEF2 is correlated with KRAS dependency and growth of pancreatic cancer cell lines
Previous studies have described a spectrum of KRAS dependency for a panel of KRAS mutant cancer cell lines.Citation40 Since RNAi knockdown of KRAS in some cell lines activates apoptosis, the strict operational definition of “KRAS addiction” required that KRAS knockdown induced caspase-3 cleavage. This definition established a RAS dependency index (DI) for multiple lung and pancreatic cancer cell lines harboring KRAS mutations.
Using the validated shRNAs for KRAS described previously,Citation40 we subjected a panel of PDAC cell lines to shRNA mediated KRAS knockdown to establish a RAS DI for commonly used PDAC cell lines (). Most of the cell lines tested demonstrated exquisite sensitivity to KRAS ablation with the exception of KP4 which was relatively resistant to shRNA mediated KRAS depletion. These results demonstrate multiple cell lines are dependent on the sustained expression of KRAS for growth and viability.
Figure 1. ARHGEF2 expression correlates to KRAS dependency and influences survival. (A, B) Growth curves and western blots of the indicated cell lines expressing shRNA GFP control or one of 2 shRNAs targeting KRAS (A) or one of 2 shRNAs targeting ARHGEF2 (B). Growth rates were monitored over the indicted time course using the Essen Incucyte Zoom. Western analysis of KRAS and ARHGEF2 expression was examined 72 hours post infection. Quantification of KRAS (A) and ARHGEF2 (B) is indicated. (C) Correlation between the LOG2 normalized dependency index (DI) for ARHGEF2 and KRAS in the indicated pancreatic cancer cell lines. Cells were treated with KRAS or ARHGEF2 targeting shRNAs for 5 d and dependency index (DI) was calculated (see methods). Inset: β-actin normalized expression of ARHGEF2 mRNA in the indicated pancreatic cancer cell lines. The p-value indicates significant Pearson correlation between ARHGEF2 mRNA and LOG2 ARHGEF2 DI. (D) Western blot analysis of cleaved caspase-3 and p-ERK activation in Panc-1 and MiaPaCa-2 cell lines following acute knockdown of KRAS or ARHGEF2 with siRNA (siRNA 5 nM final). Lysates were probed with indicted antibodies 72 hours post transfection. Quantification of p-ERK is the average from 4 independent measurements.
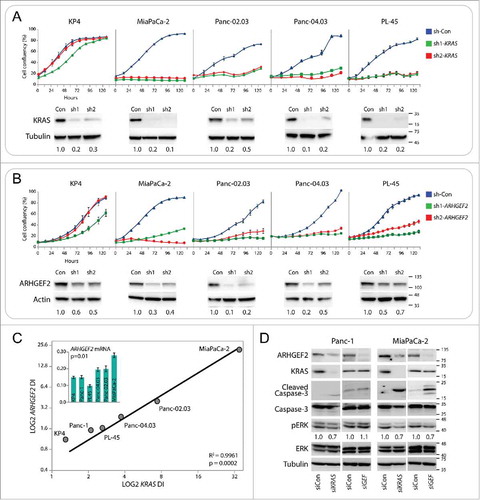
Since ARHGEF2 is required for pancreatic cancer cell growth and survival, we queried if the panel of PDAC cell lines were dependent on sustained ARHGEF2 expression. Using previously validated shRNAs for ARHGEF2,Citation9 we subjected the same panel of PDAC cell lines described above to shRNA mediated ARHGEF2 knockdown to establish dependency of cell lines on ARHGEF2 expression for growth (). Similar to KRAS knockdown, most of the cell lines tested demonstrated striking sensitivity to ARHGEF2 ablation with the exception of KP4 which was relatively insensitive to either shRNA targeting ARHGEF2. As we anticipated, these results mirrored the sensitivity to KRAS knockdown. When LOG2 transformed DI for ARHGEF2 was plotted versus the DI for KRAS we observed a linear relationship with an R2 Pearson correlation of 0.99 (; P = 2.0 × 10−4). In addition, we also observed a significant Pearson correlation of 0.85 between ARHGEF2 mRNA expression and the LOG2 ARHGEF2 DI in the panel of cell lines ( inset; p = 0.01). Collectively, these results show that PDAC cell lines which are KRAS dependent are also ARHGEF2 dependent.
Consistent with previous results, when either Panc-1 or Miapaca-2 cells were treated with siRNA targeting KRAS a significant decrease in ARHGEF2 expression was observed (). The reduced ARHGEF2 expression observed with KRAS knockdown was of similar level as that seen by targeting ARHGEF2 with siRNA directly (). We hypothesized that ARHGEF2 knockdown would phenocopy KRAS knockdown in activation of apoptosis in PDAC cells with high DI. Therefore, we assessed levels of caspase-3 cleavage in Panc-1 and MiaPaCa-2 cells, cell lines at the opposite spectrum of RAS-ARHGEF2 DI treated with siRNA targeting either KRAS or ARHGEF2. Panc-1 cells were relatively KRAS and ARHGEF2 independent by the DI prediction () and demonstrated a relatively low apoptotic response when treated with siRNA targeting either KRAS or ARHGEF2 (). In contrast, MiaPaCa-2 cells were the most KRAS and ARHGEF2 dependent cell line by the DI prediction in the panel of cell lines tested (), and demonstrated a high apoptotic response as measured by caspase-3 cleavage when either KRAS or ARHGEF2 were knocked down (). We also observed a reproducible reduction in sustained activity of the MAPK pathway in MiaPaCa-2 cells treated with siRNAs targeting either KRAS or ARHGEF2 as measured by decreased ERK1/2 phosphorylation (). These results highlight an essential role for ARHGEF2 cooperativity with oncogenic KRAS required for optimal activation of the MAPK signaling pathway.
Activation of ARHGEF2 promotes an adaptive escape pathway from MEK inhibition
Previous studies have shown that cancer cells can activate adaptive response pathways downstream of PI3K, Wnt or TGF-β to maintain cell survival and proliferation following MEK1/2 inhibition.Citation36 We find these pathways also activate a minimal ARHGEF2 promoter reporter constructCitation20 suggesting that up regulation of ARHGEF2 could be an underlying mechanism for cellular escape from MEK inhibition. Therefore, we hypothesized that ectopic expression of ARHGEF2 would desensitize cells to MEK1/2 inhibition.
Since active ARHGEF2Δ87–151 was shown to induce ERK1/2 phosphorylation,Citation9 we challenged MiaPaCa-2 cells containing a doxycycline (dox) inducible ARHGEF2Δ87–151-GFP to grow in the presence of multiple doses of the MEK1/2 inhibitor AZD6244. Currently in clinical trials, AZD6244 is a potent MEK1/2 inhibitor with IC50 of 14 nM and blocks ERK1/2 activation in PDAC and colorectal cancer cell lines.Citation49 In the absence of dox, AZD6244 impaired MiaPaCa-2 cell proliferation with concomitant block in ERK1/2 phosphorylation ( and ; -dox). In contrast, induction of active ARHGEF2 expression by dox mitigated the effect of AZD6244 on MiaPaCa-2 cell proliferation even in the presence of high doses of drug (; +dox). The enforced expression of ARHGEF2Δ87–151-GFP potently activated ERK1/2 phosphorylation even at AZD6244 doses where MAPK signaling was inhibited in the absence of dox ().
Figure 2. Enforced ARHGEF2 expression desensitizes cells to AZD6244 treatment via activation of the MAPK pathway. (A) Growth curves of doxycycline inducible ARHGEF2Δ87–151-GFP-MiaPaCa-2 cells grown in the absence (open shapes, low ARHGEF2) or in the presence of doxycycline (filled shapes, high ARHGEF2) treated with the indicated doses of AZD6244. Growth rates were monitored over the indicted time course using the Essen Incucyte Zoom. (B) Western blot analysis of p-ERK activation in doxycycline inducible ARHGEF2Δ87–151-GFP MiaPaCa-2 cells grown in the absence [-dox] or in the presence of doxycycline [+dox]. Lysates were probed with the indicted antibodies 24 hours post induction of ARHGEF2Δ87–151-GFP. GAPDH served as a loading control. Quantification of p-ERK is indicated. (C) Normalized luciferase activity generated from the minimal ARHGEF2 promoter (AP-min) or pGL3-Basic empty vector control (EV) transfected in doxycycline inducible ARHGEF2Δ87–151-GFP MiaPaCa-2 cells grown in the absence [-] or in the presence of doxycycline [+]. Cells were treated with the indicated doses of AZD6244 8 hours after induction of ARHGEF2. Luciferase activity was normalized to renilla expression and data are plotted as the fold change over empty vector. Error bars represent standard deviations from 3 independent transfections.
![Figure 2. Enforced ARHGEF2 expression desensitizes cells to AZD6244 treatment via activation of the MAPK pathway. (A) Growth curves of doxycycline inducible ARHGEF2Δ87–151-GFP-MiaPaCa-2 cells grown in the absence (open shapes, low ARHGEF2) or in the presence of doxycycline (filled shapes, high ARHGEF2) treated with the indicated doses of AZD6244. Growth rates were monitored over the indicted time course using the Essen Incucyte Zoom. (B) Western blot analysis of p-ERK activation in doxycycline inducible ARHGEF2Δ87–151-GFP MiaPaCa-2 cells grown in the absence [-dox] or in the presence of doxycycline [+dox]. Lysates were probed with the indicted antibodies 24 hours post induction of ARHGEF2Δ87–151-GFP. GAPDH served as a loading control. Quantification of p-ERK is indicated. (C) Normalized luciferase activity generated from the minimal ARHGEF2 promoter (AP-min) or pGL3-Basic empty vector control (EV) transfected in doxycycline inducible ARHGEF2Δ87–151-GFP MiaPaCa-2 cells grown in the absence [-] or in the presence of doxycycline [+]. Cells were treated with the indicated doses of AZD6244 8 hours after induction of ARHGEF2. Luciferase activity was normalized to renilla expression and data are plotted as the fold change over empty vector. Error bars represent standard deviations from 3 independent transfections.](/cms/asset/5db2cc85-0ac2-405a-9787-1eb5b5184b1a/ksgt_a_1337545_f0002_c.jpg)
We previously demonstrated that expression of ARHGEF2 is activated through the MAPK pathway via a minimal RAS-responsive ARHGEF2 promoter (AP-min). We envision that the AP-min construct can be used as a reporter for MAPK activity or signaling pathways that are able to activate expression of ARHGEF2. Using MiaPaCa-2 cells with dox inducible ARHGEF2Δ87–151-GFP, we find luciferase expression from the AP-min construct was potently enhanced in cells treated with dox compared with empty vector control (). Furthermore, dox induced ARHGEF2Δ87–151-GFP can active the AP-min promoter in the presence of AZD6244 (). These results demonstrate that expression of ARHGEF2 establishes a positive feedback loop that activates the expression of ARHGEF2 through activation of the MAPK pathway and provides a mechanism for cellular escape from pharmacological MEK inhibition. We are currently conducting small molecule screens to uncover the determinants of ARHGEF2 promoter regulation that will in combination with existing treatment regimes, increase the efficacy of MEK1/2 inhibitors in clinical trials for treatment of PDAC and other RAS dependent cancers.
Discussion
The multistage process of cancer development is driven by the acquisition of genetic lesions that initiate and maintain cancer cell proliferation and survival.Citation29,Citation47 In some cases cancer cells can become dependent on or addicted to one or several oncogenes driving tumor initiation and/or tumor growth.Citation16 The concept of “oncogene addiction” is also supported by preclinical and clinical data and suggests many cancers are sensitive to the inhibition of a single oncogene.Citation29 In the present study, we show that a panel of PDAC cell lines demonstrates a continuous distribution of KRAS dependency which correlates highly to ARHGEF2 dependency. These data suggest that one feature of KRAS dependency is coupled to the expression of ARHGEF2. Interestingly, circulating pancreatic tumor cells following surgery express a cell motility gene signature that includes increased levels of ARHGEF2 which was found to negatively predict overall survival.Citation38 Taken together, targeting ARHGEF2 may be a novel therapeutic approach for treatment of RAS-dependent cancers.
Despite the high frequency of oncogenic RAS mutations in cancer,Citation32 there are no clinically successful drugs that target RAS proteins directly. RAS has been viewed as a challenging target due to an apparent lack of druggable pockets.Citation8,Citation28 In fact, the NCI launched a ‘RAS Project’ in 2013 aimed at renewing efforts to find clinical inhibitors of RAS signaling. A large body of evidence suggests that RAS is a clinically relevant target since continued expression of mutant RAS is necessary for tumor initiation and maintenance. For example, RNAi knockdown of RAS has been shown to impair the in vitro and in vivo growth of PDAC cell lines.Citation4 Similarly, in mouse models of PDAC driven by an inducible mutant RAS, lesions depended on continuous RAS expression for maintenance.Citation6,Citation50 However, after prolonged KRAS inactivation renewed tumor growth upon KRAS reactivation rapidly led to acquisition of resistance.Citation6
RHO has a demonstrated role in the RAS-transformation programCitation5,Citation33,Citation35 and can affect tumorigenic processes in multiple cancers.Citation43 Unlike RAS however, mutations of RHO in cancers are rareCitation44 suggesting RHO hyperactivation likely occurs through dysregulation of RHOGEFs and/or RHOGAPs downstream of oncogenes such as RAS. Although our results show that ARHGEF2 is essential for the survival of PDAC cells likely through regulation of the MAPK pathway, whether the dependency on ARHGEF2 requires its RHOGEF activity remains to be determined. The enforced expression of ARHGFE2 could promote signal diversification to potentiate positive feedback through the MAPK pathway and increase RHOA-GTP signaling.Citation20 Whether these 2 events are mechanistically linked is unclear. In HRAS transformed MEFs however, ERK1/2 phosphorylation by ARHGEF2 did not require its RHOGEF activity.Citation9 In addition, previous studies have found that RHOA activation by oncogenic RAS may depend on cytosolic p190-RHOGAP activityCitation5 rather than overexpression of a RHOGEF.
In the present study, we have used a constitutively active ARHGEF2 construct unable to associate with microtubules to uncouple localization and function. Microtubule repressed RHO signaling has been attributed to the microtubule sequestration of ARHGEF2 in an inactive state.Citation21 Previously, we have found that pharmacological disruption of microtubules by nocodazole induces microtubule depolymerization and released ARHGEF2 from the microtubules resulting in a spatially defined activation of RHOA.Citation26 Furthermore, we have found that wild type MEFs treated with nocodazole potently activate ERK whereas ARHGEF2−/−MEFs do not (Sandi and Rottapel, unpublished). These data establish a link between microtubule stability and the activation of RHO and RAS signaling pathways dependent on the cellular localization and therefore activation of ARHGEF2.
While RAS-effectors offer potential for therapeutic intervention, the efficacy of RAF and MEK inhibitors is primarily limited by the development of drug resistance. For example, the initial clinical response to Vemurafenib (a BRAF inhibitor) can be impressive in certain individuals but tumor resistance usually occurs after several months of treatment. Mechanisms of resistance include mutational activation of NRAS or receptor tyrosine kinase (RTK)-mediated activation of RAS, both leading to CRAF-dependent activation of MEK-ERK signaling.Citation19,Citation27 MEK has also become an attractive drug target and offers the promise of a novel therapeutic approach for RAS-mutant tumors.Citation36 AZD6244 is an allosteric MEK1/2 inhibitor that is highly selective, but has been found to exhibit modest clinical activity as a single agent and may show greater efficacy in combination therapies.Citation36 We have shown that enforced expression of active ARHGEF2 in MiaPaCa-2 cells can activate ERK1/2 phosphorylation in the presence of relatively high doses of AZD6244. Interestingly, AZD6244 in combination with docetaxol, a microtubule stabilizing agent and hence deactivator of ARHGEF226 results in regression in human tumor xenograft models and is currently being investigated in clinical trials.Citation17 Other reported mechanisms of MEK inhibition have been described including increased RAF dimerization, down regulation of the RASGAP NF1, and up regulation of RTKs which promote increased flux through the ERK signaling pathway.Citation36
In summary, we have determined that PDAC cells demonstrating KRAS addiction are significantly dependent on expression of ARHGEF2. The results presented here and in our previous studies, show ARHGEF2 signaling activates a positive feedback loop amplifying the MAPK pathway and is required for growth and survival of RAS-transformed cancer cells. Targeting ARHGEF2 expression and signaling may increase the efficacy of MAPK pathway inhibitors and inhibitors of other RAS effectors for treatment of pancreatic cancer.
Methods
Cell culture
All cell lines used in the study (ATCC) were cultured as described previously.Citation9 For siRNA mediated knockdown, cells (5.0 × 105) were transfected with siRNA against KRAS (siGenome) or ARHGEF2 (Silencer select, Invitrogen) at 5 nM final concentration using RNAiMax (Invitrogen) according to the manufacturer's protocol. Knockdown was confirmed by western blot. Stable ARHGEF2Δ87–151-GFP inducible cells were established as described previously.Citation20 To induce ARHGEF2Δ87–151-GFP expression, cells were grown in media supplemented with 0.1μg/mL doxycycline.
ARHGEF2 expression analysis
Cells were transfected with siRNA against KRAS (siGenome) or ARHGEF2 (Silencer select, Invitrogen) at 1–5 nM final concentration using RNAiMax (Invitrogen) according to the manufacturer's protocol. Total RNA was isolated from cells with TRIZOL (Invitrogen). cDNAs were made using the QuantiTect kit (Qiagen). QPCR was performed using an ABI7900 system with the Fast-SYBR Green PCR core reagent (Applied Biosystems). Expression of ARHGEF2 normalized to β-actin. Primer sequences as described previously.Citation20
Promoter assays
Luciferase reporter assays were conducted using the Dual-Luciferase Reporter Assay System (Promega) and reading on a Glo-Max dual injector luminometer (Promega) as described previously. The ARHGEF2 promoter (AP-min) was defined as −264 to +23 relative to the TSS and cloned into pGL3-Basic (Invitrogen) as described previously.Citation20
Relative dependency index
Transient shRNA knockdown of ARHGEF2 and KRAS was accomplished with lentivirus as described previously.Citation9 Cell lines were infected with hairpin control (shGFP) or ARHGEF2 targeting shRNAs (shARHGEF2–1 and shARHGEF2–2) in parallel at an MOI of 5. After 48 hours of puromycin selection, infected cells were counted and replated at equal cell densities in 96 well plates in quadruplicate. Relative cellular viabilities were measured by quantitating optical densities (ODs) using AlamarBlue (Thermo-Fisher Scientific) when shGFP control-expressing cells reached confluence. ODs were used to calculate the relative dependency index using the formula: RDI at MOI of 5 = (1/OD shARHGEF2–1 + 1/OD shARHGEF2–2) x ODshGFP (in each cell line tested). Proliferation rates were measured by plating 5000 cells/well in a 96-well plate and monitoring growth with Essen Incucyte Zoom (Essen Biosciences).
Statistical analysis
Statistical analysis was done using Student's t-test, assuming equal variance, and p-values were calculated based on 2-tailed test.
Western blot
Cells were lysed with 2X sample buffer and boiled to denature proteins. Standard protocols for western blots were followed and blots were imaged and quantified with BIO-RAD Quantity One. Antibodies used are as described previously.Citation20
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Dr. Helen Burston, Dr. Mauricio Medrano and Dr. Melany Wagner for careful reading of the manuscript and helpful suggestions.
Funding
This work was supported by grants from the Canadian Cancer Society and the Canadian Institute for Health Research.
References
- Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell 1988; 53:549-54; PMID:2453289; https://doi.org/10.1016/0092-8674(88)90571-5
- Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, Delgiorno KE, Carpenter ES, Halbrook CJ, Hall JC, et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell 2012; 22:304-17; PMID:22975374; https://doi.org/10.1016/j.ccr.2012.07.024
- Brecht M, Steenvoorden AC, Collard JG, Luf S, Erz D, Bartram CR, Janssen JW. Activation of gef-h1, a guanine nucleotide exchange factor for RhoA, by DNA transfection. Int J Cancer 2005; 113:533-40; PMID:15455375; https://doi.org/10.1002/ijc.20626
- Brummelkamp TR, Bernards R, Agami R. Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell 2002; 2:243-7; PMID:12242156; https://doi.org/10.1016/S1535-6108(02)00122-8
- Chen JC, Zhuang S, Nguyen TH, Boss GR, Pilz RB. Oncogenic Ras leads to Rho activation by activating the mitogen-activated protein kinase pathway and decreasing Rho-GTPase-activating protein activity. J Biol Chem 2003; 278:2807-18; PMID:12429740; https://doi.org/10.1074/jbc.M207943200
- Collins MA, Brisset JC, Zhang Y, Bednar F, Pierre J, Heist KA, Galban CJ, Galban S, di Magliano MP. Metastatic pancreatic cancer is dependent on oncogenic Kras in mice. PLoS One 2012; 7:e49707; PMID:23226501; https://doi.org/10.1371/journal.pone.0049707
- Collisson EA, Trejo CL, Silva JM, Gu S, Korkola JE, Heiser LM, Charles RP, Rabinovich BA, Hann B, Dankort D, et al. A central role for RAF–>MEK–>ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer Discov 2012; 2:685-93; PMID:22628411; https://doi.org/10.1158/2159-8290.CD-11-0347
- Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov 2014; 13:828-51; PMID:25323927; https://doi.org/10.1038/nrd4389
- Cullis J, Meiri D, Sandi MJ, Radulovich N, Kent OA, Medrano M, Mokady D, Normand J, Larose J, Marcotte R, et al. The RhoGEF GEF-H1 is required for oncogenic RAS signaling via KSR-1. Cancer Cell 2014; 25:181-95; PMID:24525234; https://doi.org/10.1016/j.ccr.2014.01.025
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417:949-54; PMID:12068308; https://doi.org/10.1038/nature00766
- Engers R, Zwaka TP, Gohr L, Weber A, Gerharz CD, Gabbert HE. Tiam1 mutations in human renal-cell carcinomas. Int J Cancer 2000; 88:369-76; PMID:11054665; https://doi.org/10.1002/1097-0215(20001101)88:3%3c369::AID-IJC8%3e3.0.CO;2-K
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature 2002; 420:629-35; PMID:12478284; https://doi.org/10.1038/nature01148
- Fernandez-Medarde A, Santos E. Ras in cancer and developmental diseases. Genes Cancer 2011; 2:344-58; PMID:21779504; https://doi.org/10.1177/1947601911411084
- Goitre L, Trapani E, Trabalzini L, Retta SF. The Ras superfamily of small GTPases: the unlocked secrets. Methods Mol Biol 2014; 1120:1-18; PMID:24470015
- Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, et al. Patterns of somatic mutation in human cancer genomes. Nature 2007; 446:153-8; PMID:17344846; https://doi.org/10.1038/nature05610
- Hahn WC, Weinberg RA. Modelling the molecular circuitry of cancer. Nat Rev Cancer 2002; 2:331-41; PMID:12044009; https://doi.org/10.1038/nrc795
- Holt SV, Logie A, Odedra R, Heier A, Heaton SP, Alferez D, Davies BR, Wilkinson RW, Smith PD. The MEK1/2 inhibitor, selumetinib (AZD6244; ARRY-142886), enhances anti-tumour efficacy when combined with conventional chemotherapeutic agents in human tumour xenograft models. Br J Cancer 2012; 106:858-66; PMID:22343622; https://doi.org/10.1038/bjc.2012.8
- Infante JR, Somer BG, Park JO, Li CP, Scheulen ME, Kasubhai SM, Oh DY, Liu Y, Redhu S, Steplewski K, et al. A randomised, double-blind, placebo-controlled trial of trametinib, an oral MEK inhibitor, in combination with gemcitabine for patients with untreated metastatic adenocarcinoma of the pancreas. Eur J Cancer 2014; 50:2072-81; PMID:24915778; https://doi.org/10.1016/j.ejca.2014.04.024
- Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 2010; 468:968-72; PMID:21107320; https://doi.org/10.1038/nature09627
- Kent OA, Sandi MJ, Burston HE, Brown KR, Rottapel R. An oncogenic KRAS transcription program activates the RHOGEF ARHGEF2 to mediate transformed phenotypes in pancreatic cancer. Oncotarget 2017; 17:4484-500.
- Krendel M, Zenke FT, Bokoch GM. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat Cell Biol 2002; 4:294-301; PMID:11912491; https://doi.org/10.1038/ncb773
- Laboureau J, Dubertret L, Lebreton-De Coster C, Coulomb B. ERK activation by mechanical strain is regulated by the small G proteins rac-1 and rhoA. Exp Dermatol 2004; 13:70-7; PMID:15009099; https://doi.org/10.1111/j.0906-6705.2004.00117.x
- Macara IG, Lounsbury KM, Richards SA, McKiernan C, Bar-Sagi D. The Ras superfamily of GTPases. FASEB J 1996; 10:625-30; PMID:8621061
- Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol 2008; 3:157-88; PMID:18039136; https://doi.org/10.1146/annurev.pathmechdis.3.121806.154305
- Matsuoka T, Yashiro M. Molecular targets for the treatment of pancreatic cancer: Clinical and experimental studies. World J Gastroenterol 2016; 22:776-89; PMID:26811624; https://doi.org/10.3748/wjg.v22.i2.776
- Meiri D, Marshall CB, Greeve MA, Kim B, Balan M, Suarez F, Bakal C, Wu C, Larose J, Fine N, et al. Mechanistic insight into the microtubule and actin cytoskeleton coupling through dynein-dependent RhoGEF inhibition. Mol Cell 2012; 45:642-55; PMID:22405273; https://doi.org/10.1016/j.molcel.2012.01.027
- Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010; 468:973-7; PMID:21107323; https://doi.org/10.1038/nature09626
- Ostrem JM, Shokat KM. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat Rev Drug Discov 2016; 15:771-85; PMID:27469033
- Pagliarini R, Shao W, Sellers WR. Oncogene addiction: pathways of therapeutic response, resistance, and road maps toward a cure. EMBO Rep 2015; 16:280-96; PMID:25680965
- Philip PA, Goldman B, Ramanathan RK, Lenz HJ, Lowy AM, Whitehead RP, Wakatsuki T, Iqbal S, Gaur R, Benedetti JK, et al. Dual blockade of epidermal growth factor receptor and insulin-like growth factor receptor-1 signaling in metastatic pancreatic cancer: phase Ib and randomized phase II trial of gemcitabine, erlotinib, and cixutumumab versus gemcitabine plus erlotinib (SWOG S0727). Cancer 2014; 120:2980-5; PMID:25041791
- Philp AJ, Campbell IG, Leet C, Vincan E, Rockman SP, Whitehead RH, Thomas RJ, Phillips WA. The phosphatidylinositol 3′-kinase p85alpha gene is an oncogene in human ovarian and colon tumors. Cancer Res 2001; 61:7426-9; PMID:11606375
- Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res 2012; 72:2457-67; PMID:22589270
- Qiu RG, Chen J, McCormick F, Symons M. A role for Rho in Ras transformation. Proc Natl Acad Sci U S A 1995; 92:11781-5; PMID:8524848
- Ren Y, Li R, Zheng Y, Busch H. Cloning and characterization of GEF-H1, a microtubule-associated guanine nucleotide exchange factor for Rac and Rho GTPases. J Biol Chem 1998; 273:34954-60; PMID:9857026
- Sahai E, Olson MF, Marshall CJ. Cross-talk between Ras and Rho signalling pathways in transformation favours proliferation and increased motility. EMBO J 2001; 20:755-66; PMID:11179220
- Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov 2014; 13:928-42; PMID:25435214
- Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle 2004; 3:1221-4; PMID:15467468; https://doi.org/10.4161/cc.3.10.1164
- Sergeant G, van Eijsden R, Roskams T, Van Duppen V, Topal B. Pancreatic cancer circulating tumour cells express a cell motility gene signature that predicts survival after surgery. BMC Cancer 2012; 12:527; PMID:23157946; https://doi.org/10.1186/1471-2407-12-527
- Silbert O, Wang Y, Maciejewski BS, Lee HS, Shaw SK, Sanchez-Esteban J. Roles of RhoA and Rac1 on actin remodeling and cell alignment and differentiation in fetal type II epithelial cells exposed to cyclic mechanical stretch. Exp Lung Res 2008; 34:663-80; PMID:19085564; https://doi.org/10.1080/01902140802339615
- Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N, Settleman J. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 2009; 15:489-500; PMID:19477428; https://doi.org/10.1016/j.ccr.2009.03.022
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006; 314:268-74; PMID:16959974; https://doi.org/10.1126/science.1133427
- Smit VT, Boot AJ, Smits AM, Fleuren GJ, Cornelisse CJ, Bos JL. KRAS codon 12 mutations occur very frequently in pancreatic adenocarcinomas. Nucleic Acids Res 1988; 16:7773-82; PMID:3047672; https://doi.org/10.1093/nar/16.16.7773
- Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett 2008; 582:2093-101; PMID:18460342; https://doi.org/10.1016/j.febslet.2008.04.039
- Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer 2010; 10:842-57; PMID:21102635; https://doi.org/10.1038/nrc2960
- Walker EJ, Ko AH. Beyond first-line chemotherapy for advanced pancreatic cancer: an expanding array of therapeutic options? World J Gastroenterol 2014; 20:2224-36; PMID:24605022; https://doi.org/10.3748/wjg.v20.i9.2224
- Walters DM, Lindberg JM, Adair SJ, Newhook TE, Cowan CR, Stokes JB, Borgman CA, Stelow EB, Lowrey BT, Chopivsky ME, et al. Inhibition of the growth of patient-derived pancreatic cancer xenografts with the MEK inhibitor trametinib is augmented by combined treatment with the epidermal growth factor receptor/HER2 inhibitor lapatinib. Neoplasia 2013; 15:143-55; PMID:23441129
- Weinstein IB. Cancer. Addiction to oncogenes–the Achilles heal of cancer. Science 2002; 297:63-4; PMID:12098689
- Wennerberg K, Rossman KL, Der CJ. The Ras superfamily at a glance. J Cell Sci 2005; 118:843-6; PMID:15731001
- Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, Parry J, Smith D, Brandhuber BJ, Gross S, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res 2007; 13:1576-83; PMID:17332304
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012; 149:656-70; PMID:22541435