
ABSTRACT
Vascular resistance is a major determinant of BP and is controlled, in large part, by RhoA-dependent smooth muscle cell (SMC) contraction within small peripheral arterioles and previous studies from our lab indicate that GRAF3 is a critical regulator of RhoA in vascular SMC. The elevated contractile responses we observed in GRAF3 deficient vessels coupled with the hypertensive phenotype provided a mechanistic link for the hypertensive locus recently identified within the GRAF3 gene. On the basis of our previous findings that the RhoA signaling axis also controls SMC contractile gene expression and that GRAF3 expression was itself controlled by this pathway, we postulated that GRAF3 serves as an important counter-regulator of SMC phenotype. Indeed, our new findings presented herein indicate that GRAF3 expression acts as a pressure-sensitive rheostat to control vessel tone by both reducing calcium sensitivity and restraining expression of the SMC-specific contractile proteins that support this function. Collectively, these studies highlight the potential therapeutic value of GRAF3 in the control of human hypertension.
Introduction
Hypertension (or uncontrolled blood pressure) is a prevalent and debilitating disease that currently afflicts 74 million people in the US alone (approximately 1 in 3 adults).Citation1 Hypertension contributes to myriad diseases including myocardial infarction, peripheral vascular disease, end-stage renal disease, aortic aneurysm, and retinopathy, and continues to be the leading risk factor for global disease burden. Unfortunately, despite the fact that nearly 70 drugs are approved for hypertension management, reasonable BP control is achieved in only about half of treated patients. This insufficiency is due, in part, to our lack of understanding of the molecular and genetic underpinnings of this complex disease and the resultant use of empiric therapeutic regimens. A more complete understanding of the contributing factors should aid in the future discovery of more effective and perhaps personalized therapeutic strategies.
Blood pressure is tightly controlled by integrated signaling between multiple organs including the brain, heart, kidneys, GI tract, endocrine and vascular systems. The vasculature is a major target tissue as BP is defined as cardiac output multiplied by the total peripheral resistance imparted by peripheral vessel diameter which is controlled primarily by vascular smooth muscle cell (vSMC) contractility.Citation2–Citation6 Differentiated vSMC express a variety of SMC-specific contractile and contractile-associated proteins that contribute to this function including SM myosin heavy chain (SM MHC), SM22, calponin (CNN1), and SM α-actin. Unlike cardiac and skeletal muscle cells in which contractility is controlled by Ca2+-dependent troponin-actin interactions, excitation-contraction coupling in SMC is mediated by the Ca2+-calmodulin dependent activation of myosin light chain kinase (MLCK), and SMC tension is directly proportional to myosin light chain (MLC) phosphorylation as this enables myosins molecular interaction with actin.Citation7,8
Notably, active RhoA stimulates Rho-kinase (ROCK) and ROCK-dependent inhibition of myosin phosphatase results in elevated MLC phosphorylation and enhanced Ca2+ sensitization.Citation9–Citation12 Importantly, besides promoting an increase in intracellular Ca2+, many GPCR-coupled contractile agonists including angiotensin II (AII), norepinephrine, and endothelin-1 (ET1) also stimulate RhoA activity in SMC and in intact arteriesCitation9,13,14 and several studies in animal models and patients indicate that RhoA-dependent pathways are involved in the increased vascular resistance associated with hypertension.Citation9,13–16 The findings that increased ROCK activity has been observed in hypertensive patients and in spontaneously hypertensive rats and ROCK inhibitors like Y-27632, Fasudil, and SAR407899 have been shown to reduce BP in these settingsCitation9,13 indicates that targeting the Rho signaling pathway might provide a new therapeutic avenue for controlling hypertension and associated sequelae.
RhoA activity is regulated by guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs) that facilitate GDP-GTP exchange or RhoA's intrinsic GTPase activity respectively. The findings that germline deletion of the Rho-specific GEF, LARG or SMC-specific knockout of the related GEF, p115RhoGEF, in mice inhibited salt-Citation14 or AngII-Citation13 induced hypertension respectively, reinforced the notion that hypertensive agonists influence BP by elevating SMC RhoA activity. Interestingly, over the last several years, we have shown that GRAF3 is a SMC-selective GAP protein that imparts tight control of basal blood pressure homeostasis by limiting RhoA activity and modulating vascular resistance. Specifically, we showed that mice with gene-trap-mediated reductions in GRAF3 levels exhibited significant hypertension and elevated pressor responses to AngII and endothelin-1Citation17. Of clinical importance, three separate GWAS studies identified a blood-pressure associate allele within the GRAF3 locus and our recently published follow-up studies revealed that GRAF3 mRNA levels were approximately 3-fold higher in arteries from individuals homozygous for the protective allele and our human genetic data from well-characterized untreated patients confirmed that the protective allele was associated with a 5 mm Hg decrease in BP.Citation18–Citation21 Mechanistically, our data supported a model wherein GRAF3 controls blood pressure homeostasis by limiting RhoA-dependent MLC phosphorylation and Ca2+−mediated SMC contractility in resistance vessels.Citation17,19,22–24
Besides influencing pMLC-mediated contractility, RhoA has a major impact on the transcription of numerous contractile genes. Serum response factor (SRF) is a major driver of SMC contractile gene transcription and recent studies from our labs and others have shown that RhoA-dependent actin polymerization controls SRF activity. SRF binds CArG (CC(A/T)6GG) cis elements that are found in the promoters of numerous SMC contractile genes and SRF activity is regulated, to a large extent, by its interaction with other cell-type selective transcription factors or co-factorsCitation25–Citation29 including the Myocardin-Related Transcription Factors, MRTF-A and MRTF-B which physically interact with SRF and powerfully trans-activate SMC contractile genes.Citation30,31 Based upon a model originally described by the Treisman labCitation32,33 we previously demonstrated that the effects of RhoA on SMC contractile gene expression is mediated by actin polymerization-dependent regulation of MRTF-A and MRTF-B nuclear localization.Citation31,34,35 \ Under conditions of low RhoA activity (as in serum-starved cells), MRTF nuclear localization is strongly inhibited by G-actin binding to an N-terminal RPEL domain, an event that masks a nuclear localization sequence. The fall in cytoplasmic G-actin levels that occurs upon serum or agonist-induced RhoA-mediated actin polymerization promotes MRTF nuclear accumulation and promotes SRF/MRTF-dependent gene transcription. Interestingly, Esnault et. al. reported that the actin-polymerization induced SRF/MRTF gene signature includes several actin isoforms and dozens of genes involved in actin filament formation, contraction and force transmission.Citation36 These findings indicate that RhoA activates a feed-forward signaling cascade to promote a contractile phenotype. Coupled with our findings that GRAF3 expression was itself controlled by the SRF/MRTF pathway following cell stretch,Citation36 we postulated that GRAF3 may serve as an important counter-regulator of the contractile SMC phenotype. Indeed, our new findings presented herein indicate that GRAF3 expression acts as a pressure-sensitive rheostat to control vessel tone by not only reducing calcium sensitivity but also by restraining expression of the SMC-specific contractile proteins that support this function.
Results and discussion
GRAF3 controls RhoA/MRTFA-dependent SMC contractile gene expression
Based upon our results that ectopic expression of GRAF3 in SMC markedly attenuated RhoA-dependent actin stress fiber formation, while depletion of endogenous SMC GRAF3 led to more abundant actin-stress fibersCitation17, we reasoned that endogenous GRAF3 might play an important function in limiting SRF/MRTF-dependent SMC contractile gene expression. Consistent with this paradigm, we found that aortic SMC isolated from GRAF3 deficient mice (GRAF3gt/gt) had significantly higher levels of SM-MHC, SM α-actin, CNN1, and SM22 mRNA and protein levels when compared to aortic SMC isolated from control animals (,). Notably, GRAF3gt/gt mice were generated using embryonic stem cells that contained an inhibitory LacZ gene trap within the GRAF3 third intronCitation17 and since the gene-trap cassette in this model is flanked by loxP sites, normal GRAF3 expression in these cells can be permanently restored by Cre recombinase-dependent excision. This provides a powerful model system to define GRAF3 –dependent events because GRAF3-depleted and reconstituted cells are derived from the same population of primary SMC avoiding potential compensatory changes that are often problematic when establishing primary cultures from knock-out animals. Importantly, as shown in , Cre-dependent restoration of GRAF3 expression to endogenous levels in these cells normalized SMC contractile gene expression. We next used validated siRNAs to confirm a direct role for endogenous GRAF3 in modulating human SMC phenotypes. As shown in , siRNA-mediated depletion of GRAF3 from cultured human coronary SMC also resulted in a profound increase in contractile genes and similar findings were observed in human aortic SMC (data not shown). These data confirm that endogenous GRAF3 levels govern SMC contractile gene expression.
Figure 1. GRAF3 depletion promoted RhoA/MRTFA-dependent SMC contractile gene expression. a-c. Aortic SMC were isolated from wild type (wt) or GRAF3gt/gt mice (gt) and contractile gene expression was measured by RT-PCR or Western blot (panel a, representation; panel c shows quantification from 3 separate experiments and values are expressed as mean +/- SEM. *<0.05). Smooth muscle α-actin (SMA), calponin (CNN1), smooth muscle myosin heavy chain (MHC). In panel b, cell isolates were treated with LacZ (control) or Cre adenovirus to assess the capacity of GRAF3 re-expression to reverse changes in gene expression. d. Human coronary (HuCo) SMC were treated with control (Csi) or GRAF3 (G3si) siRNA for 72 hr. and marker expression was assessed as in panel a. Quantification is shown from 3 separate experiments and values are expressed as mean +/- SEM. † p < 0.1; *<0.05; **p < 0.01. e, f. GRAF3 depletion enhanced actin polymerization (F-actin, red) and nuclear accumulation of GFP-MRTFA in transfected 10T1/2 cells treated with or without 10% serum for 1 hr. f, quantification of MRTFA staining in the nucleus (nuc), cytoplasm (cyto.) or diffuse in 4 separate experiments with at least 50 cells per condition g. GRAF3 depletion (G3) enhanced nuclear accumulation of endogenous MRTFA in rat aortic SMC treated with or without sphingosine 1-phosphate (S1p) for 1 hr as assessed by nuclear fractionation followed by Western blotting. Histone H3 (H3) is shown as a control h,i. SMC were treated with Csi or G3si as in panel g. Cells were treated with latrunculin B (LB, 10 μM) or vehicle for the last 24 hr. prior to assessing marker gene expression. Data from 3 separate experiments were quantified and values are expressed as mean +/- SEM. *<0.05.
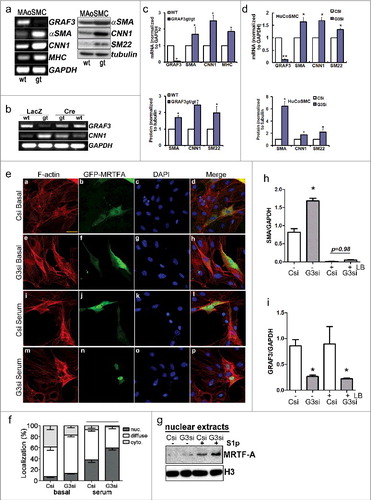
We next tested our hypothesis that GRAF3-mediated F- to G-actin cycling controls SRF activity by controlling the nucleo-cytoplasmic shuttling of MRTF. To this end, we first evaluated the subcellular locale of ectopically expressed green fluorescent protein (GFP)-MRTFA in 10T1/2 cells treated with control or GRAF3 specific siRNAs. As shown in and , GRAF3 depletion promoted actin polymerization (as assessed by phalloidin staining) and promoted serum-dependent nuclear translocation of MRTFA. Note that ectopic expression of MRTFA alone also modestly induced actin polymerization, in support of a positive feedback loop resulting from MRTFA-mediated induction of actin and contractile gene expression. Importantly, we also found that treatment of rat aortic SMC with GRAF3 siRNAs led to increased basal and S1P-stimulated nuclear accumulation of endogenous MRTFA as assessed by cell fractionation ().
We next sought to test the extent that GRAF3-dependent regulation of contractile gene expression was dependent on F-actin mediated SRF/MRTFA signaling. To this end, we treated control- and GRAF3-siRNA transfected cells with the actin de-polymerization drug, latrunculin B (LatB) -which promotes MRTF cytoplasmic sequestration and blocks expression of MRTF-dependent genes.Citation37 As shown in , LatB treatment fully attenuated the increase in SMA expression induced by GRAF3 depletion. Basal SMA expression was also dramatically reduced LatB treatment, consistent with the paradigm put forth by Esnault et al that basal and serum-induced actin transcription requires inducible SRF binding which is controlled by MRTF-dependent nucleosome displacement.Citation36 On the other hand, LatB treatment did not influence basal GRAF3 mRNA levels (), which is consistent with our previous studies showing that while GRAF3 expression could be induced by RhoA signaling in an SRF/MRTF dependent fashion, basal GRAF3 levels were not affected by inhibition of this pathway.Citation19 Collectively, these data indicate that GRAF3 limits SRF/MRTFA-dependent expression of SMC contractile genes by a homeostatic mechanism that involves actin de-polymerization.
GRAF3 is a blood-volume dependent rheostat
Physical forces such as cell stretch and mechanical tension promote robust activation of the RhoA signaling cascade and are known to be increased in the vessel wall under hypertensive conditions.Citation38
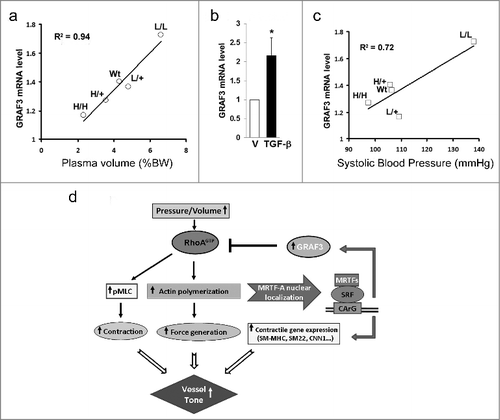
Since we previously showed that GRAF3 mRNA was significantly upregulated in SMC cultures subjected to cyclic stretch and in isolated portal vein segments subjected to static stretch and that these effects were mediated via the MRTF/SRF pathway,Citation19 we postulated that GRAF3 might serve as a transcriptionally mediated negative feedback loop of the RhoA signaling axis. In support of this possibility, arterial GRAF3 mRNA levels were significantly increased in mice made hypertensive by L-NAME or DOCA-salt regimens.Citation17 However, since both of these treatments have been reported to directly activate the RhoA pathway in SMC, the extent to which GRAF3 expression was induced in response to mechanical strain associated with the high BP was unclear from these studies. To gain further insight into the possibility that GRAF3 expression was induced by intraluminal pressure, we took advantage of an elegant mouse model developed by the Smithies lab in which plasma volumes of mice range from ∼50% below normal to ∼50% above normal.Citation39 In this model, a gene targeting-mediated procedure that altered the 3'UTR of the Tgfb1 gene generated four types of mice with Tgfb1 mRNA expression of ∼10%, 50%, 200%, and 300% of normal values and resulted in inversely progressive changes in aldosterone levels and plasma volume (i.e. the lowest TGFβ expressing mice (L/L) had the highest plasma volume). Remarkably, as shown in , GRAF3 expression increased in parallel to and was strongly correlated with plasma volume in tissues (r2 = 0.94). The finding that GRAF3 was inversely correlated with TGF-β levels in spite of the fact that TGF-β treatment of isolated SMC induces GRAF3 expression (Fig2b) indicates that the response in vivo is not due to direct effects of altered TGF-β levels. Interestingly, GRAF3 message was, in fact, much more strongly correlated with plasma volume than systolic pressure (r2 = 0.94 versus r2 = 0.72; ). This finding is relevant because it is well known that systolic BP reflects a compensated state due to activation of various counter-regulatory mechanisms to maintain normal blood pressure in the face of abnormally low or high plasma volumes. Indeed, in this particular model, it has been shown that significant compensatory changes in the renin–angiotensin system accompany the changes in plasma volumes, and are sufficient to maintain normal systolic blood pressure in the H/H, H/+, and L/+ mice, even though plasma volumes are abnormal39. Moreover, even the L/L mice that develop severe hypertension are in a compensated state as expression of plasma and kidney renin mRNA levels are 70% less than levels observed in normal mice.39 Taken together, these findings indicate GRAF3 serves as a mechanical strain-sensitive rheostat that acts to prevent excessive feed-forward activation of the RhoA signaling axis in order to control SMC tone and BP (see ).
Figure 2. GRAF3 expression directly correlates with plasma volume in TGFβ1 hypo/hypermorphic mice. a, c. GRAF3 (relative to GAPDH) mRNA levels in kidneys from Tgfb1L/L (L/L), Tgfb1L/+ (L/+), Tgfb1+/+ (WT), Tgfb1H/+ (H/+), and Tgfb1H/H (H/H) male mice with genetically graded expression of TGFβ1 from 10% (L/L) to 300% (H/H) normal was measured by qRT-PCR. GRAF3 (relative to GAPDH) mRNA levels were plotted against plasma volume as a % of BW. L (low); H (high).(a) or systolic blood pressure in mmHg (c). b. Rat aortic SMC were treated with TGF-β for 72 hr and GRAF3 mRNA was quantified and normalized to GAPDH. (d) Schematic representation of GRAF3-dependent counter-regulation of vessel tone.
Conclusion
Vascular resistance is a major determinant of BP and is controlled, in large part, by RhoA-dependent smooth muscle cell (SMC) contraction within small peripheral arteriolesCitation2–Citation6 and studies from our lab indicate that GRAF3 is the major regulator that governs RhoA's on/off state in vascular SMC. Our previous studies indicated that GRAF3 limits acute SMC tone by limiting RhoA-mediated pMLC-dependent contractility. The current study demonstrates that GRAF3 also imparts tight control over the expression of SMC differentiation genes that support contractility and as such indicate that the levels of GRAF3 can have a long-lasting impact on vessel tone.
The Treisman lab and others have demonstrated that SRF/MRTF-mediated gene transcription is a prime target of actin treadmilling.Citation33,35,40,41 Indeed, G-actin is important for sequestration of MRTFA in the cytoplasm and for the nuclear export of MRTFA. Thus, depletion of the G-actin pool by Rho-A dependent F-actin assembly promotes MRTF nuclear accumulation, and enhances MRTF-recruitment to SRF-bound CArG elements, which ultimately leads to nucleosome displacement and transcriptional induction of SRF-controlled cytoskeletal genes. One of the key evolutionarily conserved targets of SRF is actin itself- indeed knockdown of MRTF or SRF in various cell types leads to a 50–100 fold reduction in basal actin expression.Citation42 Thus, it has been speculated that the RhoA/MRTF/SRF pathway is part of a robust and critical homeostatic feedback loop that ensures sufficient levels of G-actin are available to support complex cell behaviors such as motility.Citation43 Kalo et al, confirmed and extended this hypothesis using a cell system in which β-actin transcriptional output was quantified in real time through the use of an fluorophore stably integrated into the 3’ UTR of the endogenous gene.Citation44 Using single-molecule mRNA fluorescence in situ hybridization, the authors showed that a reduction in β-actin protein levels positively feeds back to the β-actin gene to induce a strong and long lasting transcriptional response that was highly synchronized between the β-actin alleles.Citation44
Interestingly, Esnault et. al. reported that the SRF/MRTFA transcriptional network in fibroblasts comprises over 600 genes and, remarkably, gene ontology analysis revealed hundreds of such genes aligned with actin-linked processes.Citation36 Indeed dozens of genes from each stage of mechano-transduction were identified from force generators to force transducers. Indeed, from the outside of the cell inward, 14 extracellular matrix (ECM) remodeling genes were identified as SRF/MRTF targets including collagens, matrix metalloproteases and matrix modifiers such as Lox, which promotes FAK-dependent matrix stiffening. Thirty five focal adhesion proteins were identified including major force sensors including talin, vinculin, p130Cas, and hic 5. Thirty Rho signaling molecules were identified including the guanine nucleotide exchange factors Vav3 and Trio as well as the RhoA effector ROCK2. Finally, 10 forms of actin and myosin genes were identified as well as 40 different actomyosin regulators including tropomyosin, Arp 2/3, and various actin capping proteins. Collectively, these data indicate that mechanical stress dependent Rho A activation promotes a robust positive feedback circuit thus enabling prolonged actomyosin-dependent functions.
Based on our studies, we posit that in SMC, RhoA-dependent GRAF3 expression provides an important break on this robust feed-forward signaling pathway. While basal GRAF3 expression was not markedly effected by MRTFA/SRF-mediated gene transcription (as indicated by lack of effect of Lat B treatment), GRAF3 message was dramatically up-regulated by inducers of RhoA activity in cells or intact vessels (i.e. S1P or mechanical tension respectively). Depletion of endogenous GRAF3 increased pMLC, promoted F-actin polymerization and focal adhesion formationCitation17,19 enhanced MRTFA nuclear accumulation, and stimulated transcription of SM α-actin, SM-myosin heavy chain, calponin and SM-22. Thus GRAF3 can impart tight control of SMC tone by reducing SMC calcium sensitivity and restraining expression of the SMC-specific contractile proteins that support this function.
It is well recognized that arterioles constrict and dilate in response to changes in perfusion pressure. This so-called ‘myogenic’ response of arterial SMC is controlled in large part by a pressure-dependent increase in RhoA activity.Citation45 Indeed, several studies revealed that graded increases in intraluminal perfusion pressure in isolated arterioles lead to a progressive rise in RhoA-dependent phosphorylation of MYPT1 (leading to increased pMLC and calcium sensitivity) and a progressive RhoA-dependent increase in the F- to G-actin ratio; the latter being the most critical for mediating the myogenic responses at high pressures (80–100 mm Hg).Citation45–Citation48 Such increased contraction of SMC in response to increased perfusion pressure is crucial for cardiovascular homeostasis as it shields downstream capillaries from the damaging effects of high intravascular pressure. However, counter-regulatory means to control the myogenic response are also important for cardiovascular homeostasis. Abnormal myogenic constriction (and decreased arterial diameter) can lead to tissue ischemia and high blood pressure and such dysfunction has been observed in several pathological conditions including hypertension, stroke, diabetes and Alzheimer's disease.Citation45 Our finding that GRAF3 expression directly correlates with plasma volume (and intraluminal pressure) indicates that GRAF3 likely serves as a critical counter-regulator of the myogenic response.
Collectively, our experiments presented herein and elsewhere, demonstrate that modulating GRAF3 levels has a major impact on vessel tone, and BP and that GRAF3 may serve as a rheostat to limit the range of BP changes that are imparted by a wide-variety of signals.Citation17,19 With respect to human hypertension, we and others have shown that a common GRAF3 polymorphism (rs604723) can influence GRAF3 expression and basal BP.Citation18–Citation21 Moreover, a more recent GWAS study determined that this locus interacts with sodium to influence human hypertension and we previously established causality between these pathways using our pre-clinical mouse model.Citation19,49 Interestingly, hypertension attributable to adrenal gland-dependent volume expansion is one of the more common forms of secondary hypertension, and our studies herein indicate that the ability to modulate GRAF3 levels in this context is likely important for controlling the outcome of this clinical problem. Our new finding that GRAF3 levels markedly impact the expression of SM contractile genes may explain how dampened GRAF3 gene expression could contribute to sustained SMC contractility and elevated vessel tone. Moreover, given that the intrinsic mechanical properties of VSMC play a critical role in modulating vessel stiffness- a valuable predictor of end organ failure,Citation50,51 future studies directed towards determining the influence of GRAF3 expression on vessel compliance are warranted.
Materials and methods
Reagents
The SM22α antibody was a kind gift from Mario Gimona (University of Salzburg, Salzburg, Austria). The anti-MRTF-A antibody was generated by our group and has been previously described.Citation31 All other antibodies were purchased commercially: Smooth muscle (SM) α-actin (A5228, Sigma), GAPDH (Santa Cruz), SM-MHC (ab53219, Abcam), Calponin/CNN1 (LS-B7497, LifeSpan Bio), SM22 (sc-271719, Santa Cruz), Histone H3 (ab1791, Abcam). Sphingosine-1-phosphate (S1P) was from Cayman Chemicals.
Mouse models
GRAF3 gene trap ES cells were developed by Genome Research Limited (Sanger Institute, SIGTR ES CE0477) and were distributed by the Mutant Mouse Regional Resource Center at the University of California, Davis and chimeric mice were produced in house by injection of GRAF3+/gt ES cells into the blastocoel cavity of mouse blastocysts by standard procedures and mice were genotyped as previously reportedCitation17. TGFβ1 hyper- (H) and hypo-morphic (L) mice were previously generated by replacing the 3′-UTR of the Tgfb1 gene with the stable 3′-UTR from the bovine growth hormone gene (bGH) or the unstable 3′-UTR of the FBJ osteosarcoma oncogene (Fos;Citation39). Mice were bred to obtain the following five genotypes: Tgfb1L/L (L/L), Tgfb1L/+ (L/+), Tgfb1+/+ (WT), Tgfb1H/+ (H/+), and Tgfb1H/H (H/H) mice and plasma volumes and BP were obtained and previously reportedCitation39. Animal husbandry was provided by staff within the University of North Carolina Division of Laboratory and Animal Medicine and all animal procedures were approved by our accredited American Association for Accreditation of Laboratory Animal Care committee.
Cell culture and transfections
Human coronary SMC (huCoSMC; Cambrex) and human aortic SMC (huAoSMC; Cambrex) were maintained in Clonectics SmGM media containing 5% FBS, insulin, hEGF, hFGF, and gentamicin and were used between passage 3 and 6. Primary aortic vascular SMC were isolated from Wistar rats or Wt or GRAF3gt/gt mice as describedCitation52. In brief, thoracic aortas were stripped of the endothelial and adventitial layers by micro-dissection. The SMC in the media were isolated by enzymatic digestion in buffer containing trypsin and collagenase. Cells were maintained in Dulbecco's modified eagle medium with F12 supplemented with 10% fetal bovine serum and 0.5% penicillin/streptomycin and used from passage 8–16. Each of our preparations is routinely tested for expression of smooth muscle-specific markers (by immunohistochemistry) and the ability to drive smooth muscle specific expression of reporter constructs. Only the cell lines that are deemed at least 85% pure by these measurements are utilized for further experimentation. Cells were infected with LacZ control adenovirus or Cre adenovirus at 1000MOI and harvested after 72 hours for mRNA analysis. Multipotential10T1/2 cells were maintained and transfected as previously described.Citation34 In brief, cells were plated on CC2 chamber slides, transfected with GFP-MRTFA 24 hours after plating at 70% to 80% confluence using the transfection reagent, TransIT-LT1 (Mirus) according to manufacturer's protocol. GRAF3 was depleted using short interfering RNA (siRNA) (Ambion) with the following sequences: sense, 5′-GCCUGAUAGUGACUCCUAUtt; antisense, 5′-AUAGGAGUCACUAUCAGGCtc (Human); sense, 5′-CUACUGUACUUACGACAAAtt; antisense, 5′-UUUGUCGUAAGUACAGUAGtg (Mouse and Rat). Cells were transfected with gene-specific siRNA (100 nmol/L) or non-targeted control (NTC) siRNA designed against green fluorescent protein using DharmaFECT reagent 1 for 72–96 hr.
Cell fractionation
Cell fractionation was conducted as previously describedCitation53. In short, RAoSMC cells were gently scraped into cytoplasmic lysis buffer and incubated on ice for 15 min. Nuclei were pelleted by centrifugation, washed three times and re-suspended in cytoplasmic lysis buffer containing 0.5 M NaCl.
Western blotting
Cleared cell lysates or nuclear fractions were separated by SDS-PAGE, transferred to nitrocellulose, and then probed with the indicated antibodies. Blots were visualized by chemiluminescence.
Semi quantitative RT-PCR
Cells were homogenized, and RNA was extracted using Zymo or Qiagen RNA isolation kit. Semi quantitative RT-PCR was performed with the following primers: Human SM α-actin 5’- TAAAGCTTCCCAGACTTCCGC, 3’-TGTCCCATTCCCACCATCAC;
Human SM22 5’- GTGCATTTCAGGCAGGCTCTC, 3’- GCACTATGATCCACTCCACCA;
Human Calponin 5’- AACAACTTCATGGACGGCCT, 3’- TCTCCAGCTGGTGCCAATTT;
Mouse Myosin Heavy Chain 5’-GTTCATTCGCATCAACTTCG,
3’-CGGCGAGCAGGTAGTAGAAG;
Mouse Calponin 5’-CTCTGCTTTGCCAGAGCCCCC, 3’-TGACGCCGTGTACCGTAGGC;
Mouse SM α-actin 5’-CGCTGTCAGGAACCCTGAGA, 3’-CGAAGCCGGCCTTACAGAG;
GRAF3 5’-CTGCCCACTCTGGAGTTCAGCG, 3’-GCTGCACCGATCTGTTCTTTTCG;
GAPDH 5’-ATGGGTGTGAACCACGAGAA, 3’-GGCATGGACTGTGGTCATGA as described previously.Citation54
Cytofluorescence
10T1/2 cells were plated and transfected in 4-well chamber slides and maintained overnight in media containing 10% serum. Cells were transfected with SiRNA and then GFP-MRTFA plasmid the next day. The third day, cells were serum starved overnight, and then treated with or without serum for 1 hr. Cells were washed in PBS, fixed in 3.7% paraformaldehyde/phosphate-buffered saline for 20 minutes prior to imaging. GFP fluorescence was scored into three separate categories: nuclear, cytoplasmic, or diffuse (a nuclear:cytoplasmic ratio from approximately 0.75 to 1.25). Localization results are presented as averages of at least three separate experiments in which at least 100 cells per condition were scored.
Statistical analysis
All data represent at least 3 separate experiments presented as means±SEM. Means were compared by Student's t test or ANOVA followed by Chi squared analysis (where indicated) and statistical significant is reported as p values. All other data, including Western blot analysis and RT-PCR, are representative of at least 3 individual experiments. Sample sizes were chosen based on an extensive literature search.
Acknowledgements
These studies were supported by NIH National Heart, Lung, and Blood Institute (NHLBI) R01 HL130367 (awarded to JMT and CPM) and NIH-NHLBI R01 HL109607 (to CPM) as well as American Heart Fellowships to XB (14POST20380117) and KDM (15PRE25340001).
Additional information
Funding
References
- Carretero OA, Oparil S. Essential hypertension. Part I: definition and etiology. Circulation. 2000;101:329–335. doi:10.1161/01.CIR.101.3.329. PMID:10645931.
- Cowley AW, Jr. . The genetic dissection of essential hypertension. Nat Rev Genet. 2006;7:829–40. doi:10.1038/nrg1967. PMID:17033627.
- Davis MJ, Wu X, Nurkiewicz TR, Kawasaki J, Davis GE, Hill MA, Meininger GA. Integrins and mechanotransduction of the vascular myogenic response. Am J Physiol Heart Circ Physiol. 2001;280:H1427–1433. PMID:11247750.
- Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. 1999;79:387–423. PMID:10221985.
- Hall JE. The kidney, hypertension, and obesity. Hypertension. 2003;41:625–33. doi:10.1161/01.HYP.0000052314.95497.78. PMID:12623970.
- Lifton RP, Gharavi AG, Geller DS. Molecular mechanisms of human hypertension. Cell. 2001;104:545–56. doi:10.1016/S0092-8674(01)00241-0. PMID:11239411.
- Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–35. doi:10.1038/nature01148. PMID:12478284.
- Budzyn K, Marley PD, Sobey CG. Targeting Rho and Rho-kinase in the treatment of cardiovascular disease. Trends Pharmacol Sci. 2006;27:97–104. doi:10.1016/j.tips.2005.12.002. PMID:16376997.
- Loirand G, Pacaud P. The role of Rho protein signaling in hypertension. Nat Rev Cardiol. 2010;7:637–47. doi:10.1038/nrcardio.2010.136. PMID:20808285.
- Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y, Kaibuchi K. Phosphorylation and activation of myosin by Rho-associated kinase (Rho- kinase). J Biol Chem. 1996;271:20246–9. doi:10.1074/jbc.271.34.20246. PMID:8702756.
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho- kinase). Science. 1996;273:245–8. doi:10.1126/science.273.5272.245. PMID:8662509.
- Mueller BK, Mack H, Teusch N. Rho kinase, a promising drug target for neurological disorders. Nat Rev Drug Discov. 2005;4:387–98. doi:10.1038/nrd1719. PMID:15864268.
- Wirth A, Benyo Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, Orsy P, Horvath B, Maser-Gluth C, Greiner E, et al. G12-G13-LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med. 2008;14:64–8. doi:10.1038/nm1666 10.1038/nm0208-222. PMID:18084302.
- Guilluy C, Bregeon J, Toumaniantz G, Rolli-Derkinderen M, Retailleau K, Loufrani L, Henrion D, Scalbert E, Bril A, Torres RM, et al. The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat Med. 2010;16:183–190. doi:10.1038/nm.2079. PMID:20098430.
- Masumoto A, Hirooka Y, Shimokawa H, Hironaga K, Setoguchi S, Takeshita A. Possible involvement of Rho-kinase in the pathogenesis of hypertension in humans. Hypertension. 2001;38:1307–10. doi:10.1161/hy1201.096541. PMID:11751708.
- Guilluy C, Eddahibi S, Agard C, Guignabert C, Izikki M, Tu L, Savale L, Humbert M, Fadel E, Adnot S, et al. RhoA and Rho kinase activation in human pulmonary hypertension: role of 5-HT signaling. Am J Respir Crit Care Med. 2009;179:1151–8. doi:10.1164/rccm.200805-691OC. PMID:19299501.
- Bai X, Lenhart KC, Bird KE, Suen AA, Rojas M, Kakoki M, Li F, Smithies O, Mack CP, Taylor JM. The smooth muscle-selective RhoGAP GRAF3 is a critical regulator of vascular tone and hypertension. Nat Commun. 2013;4:2910. doi:10.1038/ncomms3910. PMID:24335996.
- Wain LV, Verwoert GC, O'Reilly PF, Shi G, Johnson T, Johnson AD, Bochud M, Rice KM, Henneman P, Smith AV, et al. Genome-wide association study identifies six new loci influencing pulse pressure and mean arterial pressure. Nat Genet. 43:1005–11. doi:10.1038/ng.922. PMID:21909110.
- Bai X, Mangum KD, Dee RA, Stouffer GA, Lee CR, Oni-Orisan A, Patterson C, Schisler JC, Viera AJ, Taylor JM, et al. Blood pressure-associated polymorphism controls ARHGAP42 expression via serum response factor DNA binding. J Clin Invest. 2017;127:670–80. doi:10.1172/JCI88899. PMID:28112683.
- International Consortium for Blood Pressure Genome-Wide Association, S., Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, et. al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478:103–09.
- Kato N, Loh M, Takeuchi F, Verweij N, Wang X, Zhang W, Kelly TN, Saleheen D, Lehne B, Leach IM, et al. Trans-ancestry genome-wide association study identifies 12 genetic loci influencing blood pressure and implicates a role for DNA methylation. Nat Genet. 2016;47:1282–93. doi:10.1038/ng.3405.
- Hildebrand JD, Taylor JM, Parsons JT. An SH3 domain-containing GTPase-activating protein for Rho and Cdc42 associates with focal adhesion kinase. Mol Cell Biol. 1996;16:3169–78. doi:10.1128/MCB.16.6.3169. PMID:8649427.
- Taylor JM, Macklem MM, Parsons JT. Cytoskeletal changes induced by GRAF, the GTPase regulator associated with focal adhesion kinase, are mediated by Rho. J Cell Sci. 1999;112 (Pt 2):231–42. PMID:9858476.
- Taylor JM, Hildebrand JD, Mack CP, Cox ME, Parsons JT. Characterization of graf, the GTPase-activating protein for rho associated with focal adhesion kinase. Phosphorylation and possible regulation by mitogen-activated protein kinase. J Biol Chem. 1998;273:8063–70. doi:10.1074/jbc.273.14.8063. PMID:9525907.
- Mack CP, Thompson MM, Lawrenz-Smith S, Owens GK. Smooth muscle alpha-actin CArG elements coordinate formation of a smooth muscle cell-selective, serum response factor-containing activation complex. Circ Res. 2000;86:221–32. doi:10.1161/01.RES.86.2.221. PMID:10666419.
- Dalton S, Treisman R. Characterization of SAP-1, a protein recruited by serum response factor to the c-fos serum response element. Cell. 1992;68:597–612. doi:10.1016/0092-8674(92)90194-H. PMID:1339307.
- Hill CS, Treisman R. Differential activation of c-fos promoter elements by serum, lysophosphatidic acid, G proteins and polypeptide growth factors. Embo J. 1995;14:5037–47. PMID:7588632.
- Chen CY, Schwartz RJ. Recruitment of the tinman homolog Nkx-2.5 by serum response factor activates cardiac alpha-actin gene transcription. Mol Cell Biol. 1996;16:6372–84. doi:10.1128/MCB.16.11.6372. PMID:8887666.
- Chang DF, Belaguli NS, Iyer D, Roberts WB, Wu SP, Dong XR, Marx JG, Moore MS, Beckerle MC, Majesky MW, et al. Cysteine-rich LIM-only proteins CRP1 and CRP2 are potent smooth muscle differentiation cofactors. Dev Cell. 2003;4:107–18. doi:10.1016/S1534-5807(02)00396-9. PMID:12530967.
- Wang DZ, Olson EN. Control of smooth muscle development by the myocardin family of transcriptional coactivators. Curr Opin Genet Dev. 2004;14:558–66. doi:10.1016/j.gde.2004.08.003. PMID:15380248.
- Hinson JS, Medlin MD, Lockman K, Taylor JM, Mack CP. Smooth muscle cell-specific transcription is regulated by nuclear localization of the myocardin-related transcription factors. Am J Physiol Heart Circ Physiol. 2007;292:H1170–1180. doi:10.1152/ajpheart.00864.2006. PMID:16997888.
- Sotiropoulos A, Gineitis D, Copeland J, Treisman R. Signal-regulated activation of serum response factor is mediated by changes in actin dynamics. Cell. 1999;98:159–69. doi:10.1016/S0092-8674(00)81011-9. PMID:10428028.
- Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003;113:329–42. doi:10.1016/S0092-8674(03)00278-2. PMID:12732141.
- Staus DP, Blaker AL, Taylor JM, Mack CP. Diaphanous 1 and 2 regulate smooth muscle cell differentiation by activating the myocardin-related transcription factors. Arterioscler Thromb Vasc Biol. 2007;27:478–86. doi:10.1161/01.ATV.0000255559.77687.c1. PMID:17170370.
- Lockman K, Hinson JS, Medlin MD, Morris D, Taylor JM, Mack CP. . Sphingosine 1-phosphate stimulates smooth muscle cell differentiation and proliferation by activating separate serum response factor co-factors. J Biol Chem. 2004;279:42422–30. doi:10.1074/jbc.M405432200. PMID:15292266.
- Esnault C, Stewart A, Gualdrini F, East P, Horswell S, Matthews N, Treisman R. Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev. 2014;28:943–58. doi:10.1101/gad.239327.114. PMID:24732378.
- Descot A, Hoffmann R, Shaposhnikov D, Reschke M, Ullrich A, Posern G. Negative regulation of the EGFR-MAPK cascade by actin-MAL-mediated Mig6/Errfi-1 induction. Mol Cell. 2009;35:291–304. doi:10.1016/j.molcel.2009.07.015. PMID:19683494.
- Albinsson S, Nordstrom I, Hellstrand P. Stretch of the vascular wall induces smooth muscle differentiation by promoting actin polymerization. J Biol Chem. 2004;279:34849–55. doi:10.1074/jbc.M403370200. PMID:15184395.
- Kakoki M, Pochynyuk OM, Hathaway CM, Tomita H, Hagaman JR, Kim HS, Zaika OL, Mamenko M, Kayashima Y, Matsuki K, et al. Primary aldosteronism and impaired natriuresis in mice underexpressing TGFbeta1. Proc Natl Acad Sci U S A. 2013;110:5600–05. doi:10.1073/pnas.1302641110. PMID:23503843.
- Miralles F, Visa N. Actin in transcription and transcription regulation. Curr Opin Cell Biol. 2006;18:261–6. doi:10.1016/j.ceb.2006.04.009. PMID:16687246.
- Vartiainen MK, Guettler S, Larijani B, Treisman R. Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science. 2007;316:1749–52. doi:10.1126/science.1141084. PMID:17588931.
- Knoll B. Actin-mediated gene expression in neurons: the MRTF-SRF connection. Biol Chem. 2010;391:591–7. doi:10.1515/bc.2010.061. PMID:20370316.
- Salvany L, Muller J, Guccione E, Rorth P. The core and conserved role of MAL is homeostatic regulation of actin levels. Genes Dev. 2014;28:1048–53. doi:10.1101/gad.237743.114. PMID:24831700.
- Kalo A, Kanter I, Shraga A, Sheinberger J, Tzemach H, Kinor N, Singer RH, Lionnet T, Shav-Tal Y. Cellular Levels of Signaling Factors Are Sensed by beta-actin Alleles to Modulate Transcriptional Pulse Intensity. Cell Rep. 2015;11:419–432. doi:10.1016/j.celrep.2015.03.039. PMID:25865891.
- El-Yazbi AF, Abd-Elrahman KS. ROK and Arteriolar Myogenic Tone Generation: Molecular Evidence in Health and Disease. Front Pharmacol. 2017;8:87. doi:10.3389/fphar.2017.00087. PMID:28280468.
- Moreno-Dominguez A, El-Yazbi AF, Zhu HL, Colinas O, Zhong XZ, Walsh EJ, Cole DM, Kargacin GJ, Walsh MP, Cole WC. Cytoskeletal reorganization evoked by Rho-associated kinase- and protein kinase C-catalyzed phosphorylation of cofilin and heat shock protein 27, respectively, contributes to myogenic constriction of rat cerebral arteries. J Biol Chem. 2014;289:20939–52. doi:10.1074/jbc.M114.553743. PMID:24914207.
- El-Yazbi AF, Johnson RP, Walsh EJ, Takeya K, Walsh MP, Cole WC. Pressure-dependent contribution of Rho kinase-mediated calcium sensitization in serotonin-evoked vasoconstriction of rat cerebral arteries. J Physiol. 2010;588:1747–62. doi:10.1113/jphysiol.2010.187146. PMID:20351047.
- Moreno-Dominguez A, Colinas O, El-Yazbi A, Walsh EJ, Hill MA, Walsh MP, Cole WC. Ca2+ sensitization due to myosin light chain phosphatase inhibition and cytoskeletal reorganization in the myogenic response of skeletal muscle resistance arteries. J Physiol. 2013;591:1235–50. doi:10.1113/jphysiol.2012.243576. PMID:23230233.
- Li C, He J, Chen J, Zhao J, Gu D, Hixson JE, Rao DC, Jaquish CE, Gu CC, Chen J, Huang J, Chen S, Kelly TN. Genome-Wide Gene-Sodium Interaction Analyses on Blood Pressure: The Genetic Epidemiology Network of Salt-Sensitivity Study. Hypertension. 2016;68:348–55. doi:10.1161/HYPERTENSIONAHA.115.06765. PMID:27271309.
- Galmiche G, Labat C, Mericskay M, Aissa KA, Blanc J, Retailleau K, Bourhim M, Coletti D, Loufrani L, Gao-Li J, et al. Inactivation of serum response factor contributes to decrease vascular muscular tone and arterial stiffness in mice. Circ Res. 2013;112:1035–1045. doi:10.1161/CIRCRESAHA.113.301076. PMID:23426017.
- Qiu H, Zhu Y, Sun Z, Trzeciakowski JP, Gansner M, Depre C, Resuello RR, Natividad FF, Hunter WC, Genin GM, et al. Short communication: vascular smooth muscle cell stiffness as a mechanism for increased aortic stiffness with aging. Circ Res. 2010;107:615–9. doi:10.1161/CIRCRESAHA.110.221846. PMID:20634486.
- Sundberg-Smith LJ, DiMichele LA, Sayers RL, Mack CP, Taylor JM. The LIM protein leupaxin is enriched in smooth muscle and functions as an serum response factor cofactor to induce smooth muscle cell gene transcription. Circ Res. 2008;102:1502–11. doi:10.1161/CIRCRESAHA.107.170357. PMID:18497331.
- Staus DP, Weise-Cross L, Mangum KD, Medlin MD, Mangiante L, Taylor JM, Mack CP. Nuclear RhoA signaling regulates MRTF-dependent SMC-specific transcription. Am J Physiol Heart Circ Physiol. 2014;307:H379–390. doi:10.1152/ajpheart.01002.2013. PMID:24906914.
- Lockman K, Taylor JM, Mack CP. The histone demethylase, Jmjd1a, interacts with the myocardin factors to regulate SMC differentiation marker gene expression. Circ Res. 2007;101:e115–123. doi:10.1161/CIRCRESAHA.107.164178. PMID:17991879.