
ABSTRACT
Active remodeling of the actin cytoskeleton in endothelial cells is necessary for allowing leukocytes to cross the barrier during the process of transendothelial migration (TEM). Involvement of RhoGTPases to regulate actin organization is inevitable, and we recently reported on the local function of RhoA in limiting vascular leakage during leukocyte TEM. As a follow-up we investigated here the possible involvement of two other closely-related GTPases; RhoB and RhoC, in regulating leukocyte TEM and vascular barrier maintenance. Physiological flow experiments showed no substantial involvement of either endothelial RhoB or RhoC in neutrophil adhesion and transmigration efficiency. Besides neutrophil TEM, we did not observe a role for endothelial RhoB or RhoC in limiting vascular leakage in both inflammatory conditions and during TEM. In conclusion, endothelial RhoB and RhoC are both dispensable for regulating leukocyte diapedesis and for maintaining vascular barrier function under inflammatory conditions and during leukocyte diapedesis.
Introduction
During immune surveillance and in case of inflammation, leukocytes leave the blood vessels and move towards the underlying tissues to clear pathogens and restore damaged tissues. In order to reach the distant sites of inflammation, the leukocytes have to cross the layer of endothelial cells (ECs) that functions as a tight barrier on the luminal site of blood vessels.Citation1,2 However, during inflammation this barrier becomes more permissive, resulting in local leakage of plasma proteins and finally edema formation. The adherens junctions consisting of vascular endothelial cadherin (VE-cadherin) between endothelial cells are indirectly linked to the actin cytoskeleton and involved in regulating vascular permeability.Citation3-Citation5 The passage of leukocytes through the EC layer and the regulation of vascular permeability have recently been shown to be uncoupled events.Citation4,6 Our work confirmed these findings and showed that the leakage is limited during leukocyte TEM by formation of a surrounding F-actin-rich ring, that is regulated via RhoA signaling and serves as an elastic strap.Citation7
The process of leukocyte transendothelial migration (TEM) is a tightly regulated process where spatiotemporal activation of RhoGTPases, to remodel the actin cytoskeleton, is necessary. Currently, only the involvement of RhoA in regulating leukocyte TEM and vascular permeability during TEM is described, and specific activation of RhoA during mid and late diapedesis is shown.Citation7 But it is very likely that during the other phases, i.e. the opening of the pore and the closure after leukocyte passage, which all involves actin cytoskeletal remodeling, require additional GTPase regulation. Therefore, we focused on the role of RhoB and RhoC, two GTPases with high similarity in amino acid sequence to RhoACitation8, in leukocyte TEM and vascular permeability.
Our data show that, in contrast to the essential function of RhoA in endothelial cells, neither RhoB nor RhoC are directly involved in leukocyte TEM and/or the limitation of vascular leakage during TEM.
Results
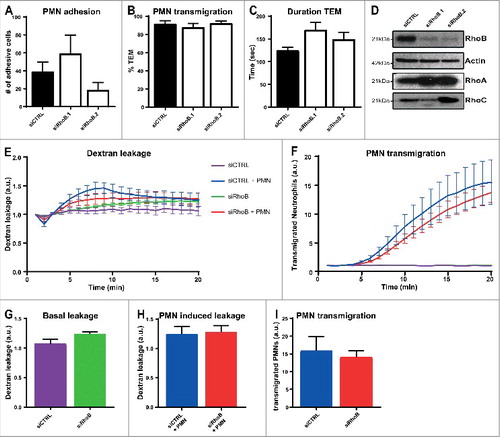
Depletion of endothelial RhoB has no effect on leukocyte TEM or vascular leakage. To study if RhoB plays a role in leukocyte transendothelial migration, HUVECs silenced with siRNA against RhoB or CTRL were cultured in specialized flow chambers. Cells were pretreated with TNFα 16 hours prior to the experiment. The results showed that neutrophil adhesion number and transmigration percentage under physiological flow conditions of 0.8 dyn/cm2 were unaltered in RhoB-depleted ECs using two different siRNA duplexes (). Besides the adhesion and transmigration, also the time from adhesion until completion of diapedesis, the duration of TEM, was not affected upon RhoB deficiency (). Successful RhoB depletion for both siRNAs was confirmed by Western blotting and showed also upregulation of RhoA and to a limited extend RhoC, in line with Marcos-Ramiro and colleagues ().Citation9 To further investigate the involvement of RhoB in vascular permeability, we measured simultaneous 70 kDa FITC-dextran leakage and neutrophil transmigration towards the chemokine C5a across TNFα-stimulated ECs for 20 minutes in a fluoroBlok-Transwell system. Silencing of endothelial RhoB showed no significant difference in basal leakage compared to control ECs under inflammatory conditions (, green and purple line, and ). Neutrophil transmigration across control ECs showed no significant increase in FITC-dextran leakage, and deficiency of RhoB had no additional effect on TEM-mediated permeability (, blue and red line, and ). Moreover, there was no significant effect observed in the number of neutrophils that transmigrated through RhoB-knockdown ECs, when compared to control-treated ECs (, blue and red line and ). Besides, no additional effects of RhoB depletion on both basal and leukocyte induced permeability were observed with 500 kDa FITC-dextran (data not shown). Thus, endothelial RhoB plays no essential role in neutrophil adhesion and transmigration, and is not involved in regulating basal vascular permeability or in maintaining the vascular barrier function during neutrophil TEM.
Figure 1. Depletion of endothelial RhoB has no effect on leukocyte TEM and on vascular leakage. (A) Quantification of neutrophil adhesion number and (B) transmigration percentage and (C) transmigration duration through TNFα-treated ECs under physiological flow conditions after 48 h of transfection with control siRNA or two different RhoB siRNAs. (D) Representative immunoblot of RhoB silencing in ECs with two different siRNAs (top panel). Actin was used as protein loading control (second panel). Increase in RhoA expression is shown in third panel and RhoC expression is slightly increased (bottom panel) (E) Extravasation kinetics of FITC-dextran and (F) calcein-red-labelled neutrophils through TNFα-treated ECs cultured on 3-µm pore FluoroBlok Transwell-filters. Neutrophils transmigrated towards C5a chemoattractant in the lower compartment. Four conditions were tested: control + neutrophils (blue line), endothelial RhoB deficiency + neutrophils (red line), control only (purple line) and endothelial RhoB depletion only (green line). (G) Quantification of FITC-dextran leakage after 20 minutes without neutrophil addition. (H) Quantification of FITC-dextran leakage after 20 minutes of neutrophil transmigration. (I) Quantification of calcein-red-labelled neutrophils after 20 minutes of transmigration. (A-I) Data are obtained from three independent experiments in duplicates for and triplicates for and show mean ± s.e.m. None of the quantifications show significant differences, tested with Mann-Whitney.
Endothelial RhoB depletion shows no visible effects on F-actin structure and VE-cadherin during leukocyte TEM. To examine the effect of RhoB depletion on endothelial F-actin architecture and endothelial junction integrity via VE-cadherin, immunofluorescent confocal microscopy was used. Control or RhoB siRNA-silenced HUVECs were transduced with Lifeact-GFP to visualize F-actin. In addition, a live staining of VE-cadherin, via a directly labelled fluorescent antibodyCitation10, was performed before addition and transmigration of neutrophils. Overall VE-cadherin distribution shows no defects upon silencing of RhoB, as the junctional protein is still present with comparable intensity in-between adjacent ECs (,). Thus, our data show the ability of RhoB-depleted ECs to form a monolayer with proper VE-cadherin expression and localization.
Figure 2. Endothelial RhoB depletion does not impair F-actin structure and VE-cadherin distribution during leukocyte TEM. (A) Confocal imaging at 40x magnification of control siRNA and RhoB deficiency in Lifeact-GFP expressing HUVECs that were stained live with VE-cadherin-ALEXA 647 to indicate endothelial cell-cell junctions after 4h TNFα stimulation. (B) Intensity plot of linescan perpendicular to cell-cell junction displays Intensity Value as measurement for the VE-cadherin along the red line indicated in . (C) Number of stress fibers present per cell in control and RhoB siRNA transfected cells. (D/E) Confocal imaging at 63x magnification of neutrophil transmigration through RhoB depleted or control cells that express Lifeact-GFP. HUVECs were treated with TNFα 4 h prior to administration of cell tracker labelled neutrophils that were allowed to transmigrate for 25 min. Arrows indicated formed pore in endothelial F-actin (green), showing a labeled neutrophil passing through (red). VE-cadherin (white) dots in RhoB depleted cells surround the nucleus, a more often observed effect with VE-cadherin staining.
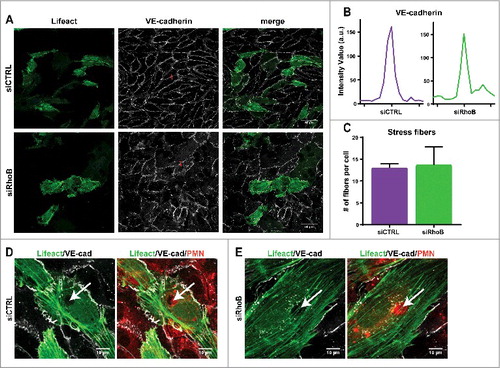
Also the formation and average number of actin stress fibers after TNFα stimulation is not impaired in RhoB-depleted ECs, compared to control cells (,). Actin cytoskeletal rearrangement is necessary upon leukocyte transmigration through the EC monolayer, therefore F-actin structure during neutrophil TEM was assessed, and both control and RhoB knockdown HUVECs show displacement of F-actin fibers at the site of neutrophil breaching, while the surrounding actin remains intact to secure maintenance of the endothelial barrier function ().
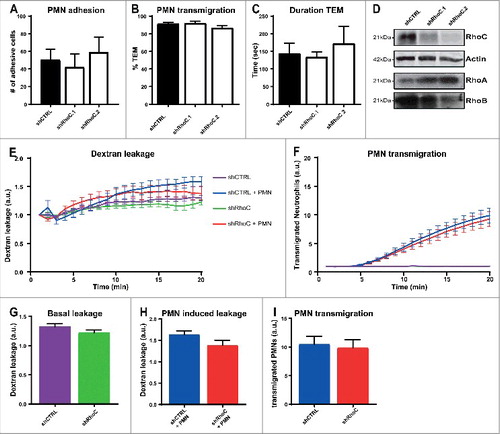
Depletion of endothelial RhoC has no effect on leukocyte TEM or vascular leakage. To study if the close-relative RhoC is involved in leukocyte TEM and regulation of vascular integrity, we performed physiological flow assays, as described before. The results showed no difference in the number of adhesive neutrophils and percentage of transmigrated neutrophils upon RhoC deficiency in ECs (). Besides the number of transmigrated neutrophils, the duration time from point of adhesion until complete diapedesis was also not affected in the absence of endothelial RhoC (). Western blotting showed successful RhoC knockdown with two different shRNAs, and similar as for RhoB depletion, an increase in RhoA expression was observed but not for RhoB (). Further investigation of RhoC involvement in vascular permeability during TEM was performed by simultaneous measurement of 70 kDa FITC-dextran leakage and calcein-red labelled neutrophil TEM to C5a chemokine across TNFα-stimulated ECs in a FluoroBlok-Transwell system setup. Basal leakage of FITC-dextran across RhoC-depleted ECs showed no significant difference compared to control situation (, green and purple line, and ). No additional effect on FITC-dextran leakage during neutrophil TEM was observed upon RhoC silencing (, red and blue line, and ). Also no effects on both basal and leukocyte induced permeability were observed with 500 kDa FITC-dextran (data not shown). Similar to the physiological flow experiment, the FluoroBlok-Transwell experiment also showed no significant difference in neutrophil TEM efficiency and kinetics between control and RhoC-depleted ECs (, red and blue line, and ).
Figure 3. Depletion of endothelial RhoC has no effect on leukocyte TEM and on vascular leakage. (A) Quantification of neutrophil adhesion number and (B) transmigration percentage and (C) transmigration duration through TNFα-treated ECs under physiological flow conditions after 48 h of transduction with lentiviral particles containing control shRNA or two different RhoC shRNAs. (D) Representative immunoblot of RhoC silencing with two different shRNAs (top panel). Actin was used as loading control (second panel) and increased RhoA expression is shown in third panel. Expression of RhoB was unaffected as shown in bottom panel. (E) Extravasation kinetics of FITC-dextran and (F) calcein-red-labelled neutrophils through TNFα-treated ECs cultured on 3-µm pore FluoroBlok Transwell-filters. Neutrophils transmigrated towards C5a chemoattractant in the lower compartment. Four conditions were tested: control + neutrophils (blue line), endothelial RhoC depletion + neutrophils (red line), control only (purple line) and endothelial RhoC deficiency only (green line). (G) Quantification of FITC-dextran leakage after 20 minutes without neutrophil addition. (H) Quantification of FITC-dextran leakage after 20 minutes of neutrophil transmigration. (I) Quantification of calcein-red-labelled neutrophils after 20 minutes of transmigration. (A-I) Data are obtained from three independent experiments in duplicates for and triplicates for and show mean ± s.e.m. None of the quantifications show significant differences, tested with Mann-Whitney.
To conclude, in line with the data for RhoB, also RhoC is not crucial in ECs for proper neutrophil adhesion and transmigration, and plays no essential role in maintaining the vascular barrier function, both in basal- and neutrophil-induced conditions.
Knockdown of endothelial RhoC shows no visible effects on F-actin structure and VE-cadherin during leukocyte TEM. Confocal microscopy was used to examine the effect of RhoC silencing on the endothelial F-actin architecture and endothelial junction integrity by means of VE-cadherin distribution. Transduction with Lifeact-GFP of control or RhoC shRNA-silenced HUVECs was used to visualize endothelial F-actin. In addition, a live staining of VE-cadherin, via a directly labelled fluorescent antibodyCitation10, was performed before freshly isolated neutrophils were added. Gray value intensity of VE-cadherin in-between adjacent ECs is still comparable between control and RhoC depleted cells, and IF images show no difference in overall VE-cadherin distribution between the EC in the monolayer (,). Thereby it shows the non-essential role of RhoC in the formation of a monolayer with proper VE-cadherin expression and localization.
Figure 4. Endothelial RhoC depletion does not impair F-actin structure and VE-cadherin distribution during leukocyte TEM. (A) Confocal imaging at 40x magnification of control and RhoC shRNA silencing in Lifeact-GFP expressing HUVECs that were stained live with VE-cadherin-ALEXA 647 to indicate endothelial cell-cell junctions after 4h TNFα stimulation. (B) Intensity plot of linescan perpendicular to cell-cell junction displays Intensity Value as measurement for the VE-cadherin along the red line indicated in . (C) Number of stress fibers present per cell in control and RhoC shRNA transduced cells. (D/E) Confocal imaging at 63x magnification of neutrophil transmigration through RhoC depleted or control cells that express Lifeact-GFP. HUVECs were treated with TNFα 4 h prior to administration of cell tracker labelled neutrophils that were allowed to transmigrate for 25 min. Arrows indicated formed pore in endothelial F-actin (green), showing a labeled neutrophil passing through (red) and VE-cadherin (white).
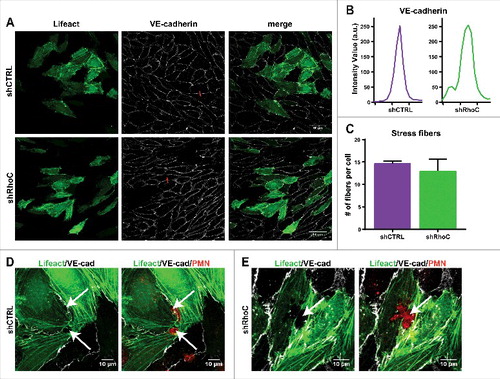
Also silencing of RhoC had no effect on the formation and average number of actin stress fibers after TNFα stimulation (,). The F-actin fiber displacement during neutrophil breaching through the EC monolayer is not impaired upon RhoC knockdown and neutrophil-surrounding actin remained intact in both shCTRL and shRhoC ECs to secure maintenance of the endothelial barrier function (). This, together with the RhoB silencing data () confirms the recent study that transmigrating neutrophils squeeze themselves through the ECs and bend surrounding endothelial F-actin fibers instead of using endothelial actomyosin contractility to open the pore.Citation11,12
Discussion
Our data show no significant effect of depletion of endothelial RhoB or RhoC on the number of adhesive neutrophils, the percentage of transmigrated neutrophils or the duration of TEM, indicating no substantial role for either endothelial RhoB or RhoC in regulating neutrophil TEM. These observations do fit with the recent publication that depletion or inhibition of endothelial RhoA shows no essential role in regulation of leukocyte transmigration efficiencyCitation7 and the publication where RhoB depletion has no effect on T-cell transmigration efficiency.Citation9 However, RhoA regulates a contractile F-actin ring that surrounds passing leukocytes to limit vascular leakage during TEM.Citation7 In contrast to RhoA, no function of RhoB or RhoC was found in maintaining vascular barrier function both in basal conditions and during leukocyte transmigration, and is partly reflected by the observation of unaltered VE-cadherin distribution at the endothelial cell-cell junctions upon RhoB knockdown.
The increase in RhoA expression levels upon depletion of either RhoB or RhoC shows the already described redundancy between these highly similar GTPases.Citation9,13,14 As the indispensable function for RhoA in limiting vascular leakage during leukocyte TEM has already been shownCitation7, it is difficult to completely exclude a RhoA compensatory mechanism in case of RhoB or RhoC deficiency. Therefore the results presented here may be an underestimation. However, in vivo inhibition with C3 transferase, that inhibits all three isoforms of the Rho family, showed the same effects as specific RhoA depletion in vitro.Citation7 Simultaneous knockdown of all three RhoGTPases in HUVECs did not increase neutrophil transmigration efficiency compared to single knockdowns.Citation9 These findings point to a more exclusive role for RhoA in limiting vascular leakage during leukocyte TEM.
Whether or not redundancy of RhoA is involved, this does not exclude involvement of two other well-known GTPases Rac1 and Cdc42. Small microvilli-like protrusions that surround a transmigrating leukocyte, formed by actin polymerization, are driven by RhoG and Rac1.Citation15-Citation17 Although so far these protrusions are thought to provide a platform for endothelial adhesion receptors and pro-inflammatory cytokinesCitation18,19 to mediate adhesion, no possible role in regulating vascular permeability has been proposed yet. Next to these apical protrusions, Rac1 is also involved in formation of ventral lamellipodia that rapidly restore the endothelial barrier upon injuryCitation20 and is thought to close the gap immediately after leukocyte transmigration, although direct evidence is still lacking.
Besides Rho and Rac1, Cdc42 is a well-known actin regulator, mainly in extension of filopodia.Citation21 Cdc42 was shown not to be involved in regulating endothelial permeability, however, this is only studied under basal conditions upon thrombin- and histamine-induced permeabilityCitation22, and not in an inflammatory background together with a leukocyte-induced vascular leakage context. Thus, a role for Cdc42 in leukocyte extravasation still needs to be established.
Even though many efforts have been made during the last couple of years to study the contribution of the small GTPases in the process of leukocyte transmigration, still relatively few details are known. Where most studies focused on the function of RhoA and Rac1, this report excludes direct involvement of RhoB and RhoC in leukocyte diapedesis and endothelial barrier maintenance.
Material and methods
DNA and RNA constructs. pLenti-Lifeact-GFP was a kind gift of Stephan Huveneers (AMC, Amsterdam, the Netherlands). shRNA in pLKO.1 targeting RhoC (TRCN47864 and TRCN47866), and control shRNA (shC002) were purchased from sigma Aldrich mission library. siRNA (working concentration 20nM) targeting RhoB (sc-29472, sc-29472A, sc-29472B), and scrambled non-silencing siRNA (sc-37007) were purchased from Santa Cruz.
Antibodies. Rabbit antibody against RhoA (Cat #2117X) and RhoC (Cat #3430) both Cell signaling. Polyclonal rabbit anti-RhoB (Cat#sc-180) was obtained from Santa Cruz and mouse-anti-Actin (Cat#A3853) from Sigma. Directly labelled VE-cadherin AF647 (Cat#561567) was acquired from BD Biosciences. Secondary donkey antibody against Rabbit-IR800 (Cat#926–32213) and Mouse-IR800 (Cat#926–32212) were both from LI-COR Biosciences. Secondary HRP-conjugated goat anti-mouse (Cat#P0447), swine anti-rabbit (Cat#P0399) antibodies were purchased from Dako. All antibodies were used according to manufacturer's protocol.
Cell cultures and treatments. Pooled human umbilical vein endothelial cells (HUVECs) perchased from Lonza (P1012, Cat # C2419A), were cultured until passage 7 on fibronectin (FN)-coated dished in EGM-2 medium, supplemented with singlequots (Lonza). Human Embryonic Kidney (HEK)-293T cells were maintained in DMEM (Invitrogen), containing 10% (v/v) heat-inactivated fetal calf serum, 100 U/ml penicillin and streptomycin and 1x sodium pyruvate (all Invitrogen). All cells were cultured at 37°C and 5% CO2. Pretreatment with 10 ng/ml recombinant TNF-α (Peprotech) for 24 h before each leukocyte TEM experiment. Lentiviral constructs were packaged into lentivirus in Human Embryonic Kidney (HEK)-293T cells by means of third generation lentiviral packaging plasmids (Dull et al., 1998; Hope et al 1990). Lentivirus containing supernatant was harvested on day 2 and 3 after transfection. Lentivirus was concentrated by Lenti-X concentrator (Clontech, Cat# 631232). Transduced target cells were used for assays after 72 hours. Cells were transfected with the expression vectors according to manufacturer's protocol with Trans- IT-LT1 (Myrus) and siRNA transfections were performed according to manufacturer's protocol using INTERFERin (Polyplus).
Neutrophil isolation. Polymorphonuclear (PMN) neutrophils were isolated from whole blood derived from healthy donors. Whole blood was diluted (1:1) with 5% (v/v) TNC in PBS. Diluted whole blood was pipetted carefully on 12,5 ml Percoll (room temperature) 1.076 g/ml. Tubes were centrifuged (Rotanta 96R) at 2000 rpm, slow start, low brake for 20 minutes. After erythrocyte lysis in an ice-cold isotonic lysis buffer (155 mM NH4CL, 10 mM KHCO3, 0.1 mM EDTA, pH7.4 in Milli-Q (Millipore), neutrophils were centrifuged at 1500 rpm for five minutes at 4°C, incubated once with lysis buffer for 5 minutes on ice, centrifuged again at 1500 rpm for five minutes at 4°C, washed once with PBS, centrifuged again at 1500 rpm for five minutes at 4°C and resuspended in HEPES medium pH7.4 (20 mM HEPES, 132 mM NaCl, 6 mM KCL, 1 mM CaCL2, 1 mM MgSO4, 1.2 mM K2HPO4, 5 mM glucose (all from Sigma-Aldrich), and 0.4% (w/v) human serum albumin (Sanquin Reagents) and kept at room temperature for not longer than four hours until use. Neutrophil counts were determined by cell counter (Casey).
FITC-dextran permeability assay. 200,000 ECs were cultured in (FN) treated 24 well cell culture inserts (Corning FluoroBlok, Falcon, 3.0 μm pore size Cat# 351151) and treated with TNF-α overnight. 30 μg FITC-dextran (70 kDa) (Sigma) in HEPES medium pH7.4 was added to the upper and 0.1 nM C5a (Sigma C-5788) in HEPES medium pH7.4 was added to the lower compartment. FITC-dextran and calcein red-orange (Molecular probes C34851) labeled neutrophil (200,000 cells) extravasation was monitored simultaneously for a period of 60 minutes with an interval of 1 minute by an Infinite F200 pro plate reader (TECAN) at 37 °C. EX BP 485/9 and EM BP 535/20 was used to measure FITC-dextran kinetics. EX BP 570/9 and EM BP 595/20 was used to measure neutrophil (calcein red-orange) transmigration kinetics.
Neutrophil TEM under physiological flow. 150.000 HUVECs were cultured per channel in a FN-coated ibidi µ-slide VI0.Citation4 (Ibidi) the day before the experiment was executed and stimulated overnight with 10 ng/ml TNFα (Peprotech). Freshly isolated neutrophils were resuspended at 1*106 cells/ml in HEPES medium pH7.4 and were incubated for 30 minutes at 37ºC. Cultured HUVECs in Ibidi flow chambers were connected to a perfusion system and exposed to 0.5 ml/minute HEPES medium pH7.4 shear flow for 5 minutes (0.8 dyne/cm2). Neutrophils were subsequently injected into the perfusion system and real-time leukocyte-endothelial interactions were recorded for 20 minutes by a Zeiss Observer Z1 microscope. All live imaging was performed at 37°C in the presence of 5% CO2. Transmigrated neutrophils were distinguished from those adhering to the apical surface of the endothelium by their transition from bright to phase-dark morphology. Percentage adherent or transmigrated neutrophils were manually quantified using the ImageJ plug-in Cell Counter (type 1, adherent cells, type 2, transmigrated cells).
Western blotting. Cells were washed once with PBS+/+ (1mM CaCl and 0.5 mM MgCl), and lysed with 95°C SDS-sample buffer containing 4% β-mecapto-ethanol. Samples were boiled at 95°C for 5–10 minutes to denature proteins. Proteins were separated on 12,5% SDS running gel in running buffer (200 mM Glycine, 25 mM Tris, 0.1% SDS (pH8.6)), transferred to nitrocellulose membrane (Thermo Scientific Cat#26619) in blot buffer (48 nM Tris, 39 nM Glycine, 0.04% SDS, 20% MeOH) and subsequently blocked with 5% (w/v) milk (Campina) in Tris-buffered saline with Tween 20 (TBST) for 45 minutes. The immunoblots were analyzed using primary antibodies incubated overnight at 4°C and secondary antibodies linked to horseradish peroxidase (HRP) (Dako, Aligent Tochnologies) or IR800 (LI-COR biosciences), after each step immunoblots were washed 4x with TBST. HRP signals were visualized by enhanced chemiluminescence (ECL) (Thermo Scientific) and light sensitive films. IR800 signals were scanned with Odyssey.
Confocal laser scanning microscopy. HUVECs were cultured on FN-coated 12 mm glass coverslips and transfected/transduced as indicated and stimulated with TNFα 4 h and stained for VE-cadherin 30 min prior to the experiment. Freshly isolated neutrophils were labelled with 1 µM cell tracker blue (Life Technologies) for 30 min at 37°C and 250.000 cells in 500 µl HEPES medium pH7.4 were added to the HUVECs. PMNs were allowed to transmigrate for 25 minutes at 37°C, 5% CO2 before fixation with 3,7% PFA (Merck). Coverslips were washed with PBS+/+ and subsequently mounted with mowiol (Merck). Z-stack image acquisition was performed on a confocal laser scanning microscope (Leica SP8) using a 40x NA 1.3 or 63x NA 1.4 oil immersion objective.
Additional information
Funding
References
- Schimmel L, Heemskerk N, van Buul JD. Leukocyte transendothelial migration: A local affair. Small GTPases. 2017;8:1–15. doi:10.1080/21541248.2016.1197872. PMID:27715453.
- Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol. 2015;15:692–704. doi:10.1038/nri3908. PMID:26471775
- Vestweber D, Winderlich M, Cagna G, Nottebaum AF. Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends Cell Biol. 2009;19:8–15. doi:10.1016/j.tcb.2008.10.001. PMID:19010680
- Wessel F, Winderlich M, Holm M, Frye M, Rivera-Galdos R, Vockel M, Linnepe R, Ipe U, Stadtmann A, Zarbock A, et al. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat Immunol. 2014;15:223–30. doi:10.1038/ni.2824. PMID:24487320
- Trani M, Dejana E. New insights in the control of vascular permeability: vascular endothelial-cadherin and other players. Curr Opin Hematol. 2015;22:267–72. doi:10.1097/MOH.0000000000000137. PMID:25767951
- Vestweber D, Wessel F, Nottebaum AF. Similarities and differences in the regulation of leukocyte extravasation and vascular permeability. Semin Immunopathol. 2014;36:177–92. doi:10.1007/s00281-014-0419-7. PMID:24638889
- Heemskerk N, Schimmel L, Oort C, Van Rijssel J, Yin T, Ma B, van Unen J, Pitter B, Huveneers S, Goedhart J, et al. F-actin-rich contractile endothelial pores prevent vascular leakage during leukocyte diapedesis through local RhoA signalling. Nat Commun. 2016;7:10493. doi:10.1038/ncomms10493. PMID:26814335
- Reinhard NR, van Helden SF, Anthony EC, Yin T, Wu YI, Goedhart J, Gadella TWJ, Hordijk PL. Spatiotemporal analysis of RhoA/B/C activation in primary human endothelial cells. Sci Rep. 2016;6:25502. doi:10.1038/srep25502. PMID:27147504
- Marcos-Ramiro B, García-Weber D, Barroso S, Feito J, Ortega MC, Cernuda-Morollón E, Reglero-Real N, Fernández-Martín L, Durán MC, Alonso MA, et al. RhoB controls endothelial barrier recovery by inhibiting Rac1 trafficking to the cell border. J Cell Biol. 2016;213:385–402. doi:10.1083/jcb.201504038. PMID:27138256
- Kroon J, Daniel AE, Hoogenboezem M, van Buul JD. Real-time Imaging of Endothelial Cell-cell Junctions During Neutrophil Transmigration Under Physiological Flow. J Vis Exp. 2014;90:e51766. PMID:25146919
- Barzilai S, Yadav SK, Morrell S, Roncato F, Klein E, Stoler-Barak L, Golani O, Feigelson SW, Zemel A, Nourshargh S, et al. Leukocytes Breach Endothelial Barriers by Insertion of Nuclear Lobes and Disassembly of Endothelial Actin Filaments. Cell Rep. 2017;18:685–99. doi:10.1016/j.celrep.2016.12.076. PMID:28099847
- Alon R, van Buul JD. Leukocyte Breaching of Endothelial Barriers: The Actin Link. Trends Immunol. 2017;xx:1–10.
- Wang L, Zheng Y. Cell type-specific functions of Rho GTPases revealed by gene targeting in mice. Trends Cell Biol. 2007;17:58–64. doi:10.1016/j.tcb.2006.11.009. PMID:17161947
- Narumiya S, Yasuda S. Rho GTPases in animal cell mitosis. Curr Opin Cell Biol. 2006;18:199–205. doi:10.1016/j.ceb.2006.02.002. PMID:16487696
- Schnoor M, Lai FPL, Zarbock A, Kläver R, Polaschegg C, Schulte D, Weich HA, Oelkers JM, Rottner K, Vestweber D. Cortactin deficiency is associated with reduced neutrophil recruitment but increased vascular permeability in vivo. J Exp Med. 2011;208:1721–35. doi:10.1084/jem.20101920. PMID:21788407
- Van Buul JD, Allingham MJ, Samson T, Meller J, Boulter E, Garcia-Mata R, Burridge K. RhoG regulates endothelial apical cup assembly downstream from ICAM1 engagement and is involved in leukocyte trans-endothelial migration. J Cell Biol. 2007;178:1279–93. doi:10.1083/jcb.200612053. PMID:17875742
- van Rijssel J, Kroon J, Hoogenboezem M, van Alphen FPJ, de Jong RJ, Kostadinova E, Geerts D, Hordijk PL, van Buul JD. The Rho-guanine nucleotide exchange factor Trio controls leukocyte transendothelial migration by promoting docking structure formation. Mol Biol Cell. 2012;23:2831–44. doi:10.1091/mbc.E11-11-0907. PMID:22696684
- Middleton J, Neil S, Wintle J, Clark-Lewis I, Moore H, Charles L, Auer M, Elin H, Antal R. Transcytosis and surface presentation of IL-8 by venular endothelial cells. Cell. 1997;91:385–95. doi:10.1016/S0092-8674(00)80422-5. PMID:9363947
- Whittall C, Kehoe O, King S, Rot A, Patterson A. Europe PMC Funders Group Europe PMC Funders Author Manuscripts A chemokine self-presentation mechanism involving formation of endothelial surface microstructures.. 2013;190:1725–36.
- Martinelli R, Kamei M, Sage PT, Massol R, Varghese L, Sciuto T, Toporsian M, Dvorak AM, Kirchhausen T, Springer T a., et al. Release of cellular tension signals self-restorative ventral lamellipodia to heal barrier micro-wounds. J Cell Biol. 2013;201:449–65. doi:10.1083/jcb.201209077. PMID:23629967
- Nobes CD, Hall A. Rho, Rac, and Cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi:10.1016/0092-8674(95)90370-4. PMID:7536630
- Wojciak-Stothard B, Potempa S, Eichholtz T, Ridley AJ. Rho and Rac but not Cdc42 regulate endothelial cell permeability. JCell Sci. 2001;114:1343–55.