
ABSTRACT
Macrophages are innate immune cells that constantly patrol an organism to fulfill protective and homeostatic roles. Previous studies have shown that Rho GTPase activity is required for macrophage mobility, yet the roles of upstream regulatory proteins controlling Rho GTPase function in these cells are not well defined. Previously we have shown that the RhoA GEF Net1 is required for human breast cancer cell motility and extracellular matrix invasion. To assess the role of Net1 in macrophage motility, we isolated bone marrow macrophage (BMM) precursors from wild type and Net1 knockout mice. Loss of Net1 did not affect the ability of BMM precursors to differentiate into mature macrophages in vitro, as measured by CD68 and F4/80 staining. However, Net1 deletion significantly reduced RhoA activation, F-actin accumulation, adhesion, and motility in these cells. Nevertheless, similar to RhoA/RhoB double knockout macrophages, Net1 deletion did not impair macrophage recruitment to the peritoneum in a mouse model of sterile inflammation. These data demonstrate that Net1 is an important regulator of RhoA signaling and motility in mouse macrophages in vitro, but that its function may be dispensable for macrophage recruitment to inflammatory sites in vivo.
Introduction
Macrophages are monocyte-derived innate immune cells that patrol tissues for invading pathogens, contribute to tissue homeostasis, and aid in wound repair.Citation1 Thus, cell movement is an essential aspect of macrophage function.
Rho GTPases are fundamental regulators of actin cytoskeletal organization and cell motility, of which Cdc42, Rac1, and RhoA are the most highly investigated.Citation2,Citation3 A number of studies have demonstrated that their function is required for macrophage motility. For example, expression of dominant negative Cdc42 in Bac1.2F5 cells, which are a transformed mouse macrophage cell line, prevents cells from polarizing toward a CSF-1 gradient.Citation4 In primary mouse bone marrow macrophages (BMM), Rac1 deletion impairs cell spreading and membrane ruffling, but does not affect chemotaxis on glass or plastic.Citation5 Rac2 deletion in mouse BMMs inhibits haptotactic migration towards vitronectin or fibronectin, as well as cell spreading on these substrates.Citation6 Deletion of both Rac1 and Rac2 results in an increase in migration velocity due to a shift from classical lamellipodia driven motility to an alternating stellate and rounded cell motility. Rac1 was also shown to be required for efficient invasion through a Matrigel extracellular matrix.Citation7
RhoA and RhoB are also important for macrophage motility. RhoB deletion impairs mouse BMM adhesion and inhibits migration on select substrates due to a reduction in surface integrin expression.Citation8 RhoA deletion inhibits tail retraction, but surprisingly increases migration velocity. Deletion of both RhoA and RhoB dramatically alters BMM morphology in resting cells, producing a stellate morphology. During chemotactic migration these cells move with even greater velocity than single knockout cells.Citation9
Taken together, these studies indicate that Rho GTPases fulfill significant, non-overlapping roles in controlling macrophage motility. An important, unresolved question is the identity of Rho regulatory proteins controlling their activation. Rho GTPase activation is controlled by two families of enzymes known as guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs).Citation2 GEFs catalyze GDP release by Rho proteins, thereby promoting their GTP-bound, active state.Citation10 GAPs stimulate the intrinsic GTPase activity of Rho proteins to terminate signaling.Citation11 Most cells express multiple Rho GEFs and GAPs that control the spatial and temporal activation of Rho GTPases. Recently we created a mouse strain with a genetic deletion of Net1, which is a GEF for RhoA and RhoB. Net1 KO mice are viable and reproduce normally. However, they exhibit a delay in mammary gland development during puberty characterized by reduced invasion and branching of the ductal tree. Importantly, ductal epithelial cells lacking Net1 exhibited significantly reduced RhoA activation and myosin light chain phosphorylation, indicative of reduced actomyosin contractility.Citation12
Because macrophage function is important for mammary gland development and tumorigenesis,Citation13,Citation14 we examined the effects of Net1 deletion on RhoA signaling, cell adhesion, motility, and invasion in primary bone marrow macrophages. We observed that loss of Net1 significantly impaired RhoA activation, resulting in a decrease in F-actin content within cells. Net1 deletion also impaired macrophage motility and adhesion, but did not affect their ability to invade a Matrigel extracellular matrix in vitro, or to be recruited to the peritoneum in a sterile peritonitis mouse model. These data indicate that Net1 contributes to macrophage motility in vitro, but may be dispensable for macrophage recruitment to sites of inflammation in vivo.
Results
Using a CRE-lox strategy we previously created mice with whole body deletion of Net1, which eliminated the expression of all Net1 isoforms.Citation12 To assess the role of Net1 in mouse macrophage function, primary bone marrow macrophage (BMM) precursors were isolated from the femurs and tibia of wild type and Net1 KO mice, and then incubated in the presence of M-CSF for a week to promote macrophage differentiation. Using quantitative real time PCR we confirmed that Net1 KO BMMs lacked expression of Net1 mRNAs (). Using flow cytometry (Fig. S1), we observed that greater than 80% of the cells were CD45+CD11b+CD68+F4/80+ mature macrophages. Importantly, using these markers we did not detect a significant difference in macrophage differentiation between wild type and Net1 KO BMMs (). Thus, loss of Net1 does not appear to affect macrophage differentiation in vitro.
Figure 1. Net1 deletion impairs RhoA activation in mouse bone marrow macrophages. (A) Net1 mRNA expression in M-CSF differentiated mouse bone marrow macrophages (BMMs). (B, C) Representative examples of F4/80 and CD68 expression in wild type (B) and Net1 KO (C) BMMs. (D) Quantification of the percent of cells with CD68 and F4/80 expression in differentiated BMMs. (E) Representative example of RhoA activity in BMMs, as measured by GST-RBD pulldown. (F) Quantification of RhoA activity in BMMs. For all plots, bars = median values. n.s. = not significant. ** = p < 0.01. *** = p < 0.001.
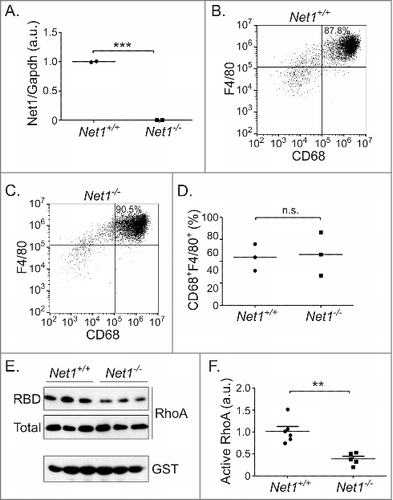
As Net1 is a GEF for RhoA, we then examined whether Net1 contributed to RhoA activation in primary macrophages. Whole cell lysates from serum-starved BMMs were isolated and RhoA activation was assessed using GST-RBD pulldown assays. We observed that Net1 deletion resulted in a significant decrease in RhoA activation (, ). This is consistent with the requirement for Net1 expression for full RhoA activation in mouse mammary epithelial cells and human breast cancer cells.Citation12,Citation15
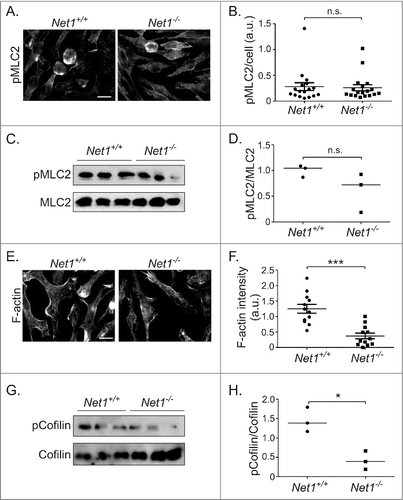
RhoA controls actomyosin contraction by stimulating ROCK-dependent phosphorylation of myosin phosphatase target subunit 1. This inhibits myosin phosphatase, thereby increasing phosphorylation of myosin regulatory light chain (MLC) and subsequent myosin activation.Citation2,Citation16 Thus, we examined whether loss of Net1 resulted in a change in MLC2 phosphorylation by immunofluorescence microscopy and Western blotting. Using these assays we did not observe a significant difference MLC2 phosphorylation between wild type and Net1 KO BMMs (). RhoA activation also increases F-actin content through a combination of actomyosin contraction, formin-stimulated actin polymerization, and inhibition of the F-actin severing activity of cofilin.Citation3 Thus, we tested for F-actin content in wild type and Net1 KO cells by phalloidin staining. We observed that loss of Net1 significantly reduced the amount of F-actin in these cells (, ). To determine whether an increase in cofilin activity contributed to the decrease in F-actin content, we tested for phosphorylation of cofilin on its inhibitory phosphorylation site serine 3. We observed that there was a significant decrease in cofilin Ser3 phosphorylation in Net1 KO BMMs, suggesting that cofilin activity was increased in response to Net1 deletion (, ). Taken together, these data indicate that Net1 is required for RhoA signaling to control F-actin content in BMMs, and that this may be due, in part, to control of cofilin activity.
Figure 2. Net1 deletion reduces F-actin content in BMMs. (A) Representative images of pMLC2 staining of BMMs from wild type and Net1 KO mice. Scale bar is 10 µm. (B) Quantification of pMLC2 staining in multiple fields from three different BMM isolates for each genotype. (C) Western blot for pS19-MLC2 and total MLC2 in three different isolates of wild type and Net1 knockout BMMs. (D) Quantification of pS19-MLC2/MLC2 Western blots. (E) Representative images of F-actin staining in BMMs from wild type and Net1 KO mice. Scale bar is 10 µm. (F) Quantification of F-actin staining in multiple fields from three different BMM isolates for each genotype. (G) Western blot for pS3 cofilin and total cofilin in three different isolates of wild type and Net1 knockout BMMs. (H) Quantification of pS3-cofilin/cofilin Western blots. Bars are median values for all plots. n.s. = not significant. * = p < 0.05. *** = p < 0.001.
Because RhoA activation is required for cell adhesion and motility, we examined whether loss of Net1 affected either of these functions. To measure effects on adhesion, wild type and Net1 KO macrophages were trypsinized and replated on glass coverslips coated with fibronectin. After 30 minutes, unattached cells were washed off and the attached cells were quantified after staining with DAPI. These experiments demonstrated that Net1 deletion significantly impaired macrophage adhesion to fibronectin (). We then tested whether loss of Net1 affected macrophage motility. Wild type and Net1 KO macrophages were serum-starved and then placed in the upper well of a transwell apparatus. Macrophage chemoattractant protein-1 (MCP-1) was added to the bottom well to provide a motility stimulus. We found that loss of Net1 significantly impaired macrophage movement towards MCP-1 (). We then assessed whether Net1 was required for extracellular matrix (ECM) invasion by testing cell movement through transwells coated with Matrigel. We observed that Net1 was not required for ECM invasion (). This is consistent with the work of others demonstrating a role for Rac1 but not RhoB in Matrigel invasion by BMMs.Citation7,Citation8
Figure 3. Net1 deletion impairs BMM adhesion and motility, but does not affect Matrigel invasion or peritoneal recruitment. (A) Adhesion of wild type and Net1 KO BMMs to fibronectin 30 minutes after plating. (B) Migration of wild type and Net1 KO BMMs in transwell inserts with MCP-1 in the bottom well. (C) Invasion of wild type and Net1 KO BMMs through Matrigel coated transwell inserts with MCP-1 in the bottom well. For all experiments, three different BMM isolates were assayed in triplicate for each genotype. (D) Representative examples of F4/80 and CD68 expressing cells in peritoneal isolates from wild type and Net1 KO mice three days after thioglycollate injection. (E, F, G) Quantification of F4/80+CD68+ (E), CD45+ (F), and CD45+CD11b+ cells in peritoneal isolates from thioglycollate injected wild type and Net1 KO mice. For all plots, bars = median values. n.s. = not significant; ** = p < 0.01; *** = p < 0.001.
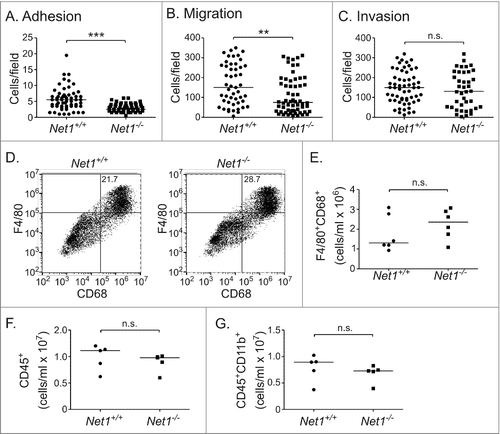
To determine whether Net1 was required for macrophage recruitment in vivo, we tested for peritoneal recruitment of macrophages in a sterile peritonitis model.Citation17 Wild type and Net1 KO mice were injected in the peritoneum with a sterile, 3% thioglycollate solution. Three days later the peritoneal fluid was isolated and tested for lymphocyte, monocyte, and macrophage content by flow cytometry. Three days was tested as this is the peak time for macrophage accumulation in this model. We observed that Net1 deletion did not adversely affect the recruitment of F4/80+CD68+ macrophages to the peritoneum (, ). Similarly, it did not affect the overall populations of CD45+ cells or CD45+CD11b+ monocytes (, ). Taken together, these data demonstrate that Net1 contributes significantly to mouse BMM adhesion and motility in vitro, but is dispensable for Matrigel invasion. These data also suggest that Net1 may be dispensable for macrophage recruitment to sites of inflammation.
Discussion
In this work we have shown that the RhoGEF Net1 contributes to RhoA activation, F-actin content, adhesion, and motility in primary mouse BMMs. However, it was dispensable for Matrigel invasion in vitro and peritoneal recruitment following an inflammatory stimulus in vivo. Although Net1 expression has been reported to increase during osteoclastogenesis and dendritic cell maturation,Citation18,Citation19 both of which are monocyte-derived cells, to our knowledge this is the first time that Net1 has been functionally tested for regulation of innate immune cell behaviors.
Net1 is a GEF for both RhoA and RhoB, but not Rac1 or Cdc42.Citation20-Citation22 Surprisingly, our results do not represent an exact synthesis of prior work examining RhoA and RhoB KO BMMs.Citation8,Citation9 For example, Konigs et al demonstrated that RhoA deletion increased BMM migration speed on fibronectin, even though the cells had significant difficulty retracting their trailing edges. Similarly, deletion of both RhoA and RhoB further increased migration speed.Citation9 In an earlier study by Wheeler et al, RhoB deletion did not affect BMM adhesion and migration on fibronectin, but did reduce BMM adhesion and motility on glass surfaces.Citation8 Our data indicate that Net1 contributes significantly to BMM motility and adhesion. These differences may reflect divergent experimental approaches, as the prior studies used modified Dunn chambers to measure motility. Alternatively, it is likely that deletion of an upstream GEF such as Net1 is not equivalent to deletion of the Rho genes themselves. Most cells express multiple RhoGEFs, so loss of any one GEF would be expected to only partially impair Rho protein activation, as we observed for RhoA activation in Net1 deleted BMMs (, ).
The roles of different RhoGEFs in controlling BMM motility have not been extensively studied. Vav1, Vav2, and Vav3, which are GEFs for the Rac1 and RhoA subfamilies in cells, have been shown to contribute to Rac1 and RhoA activation downstream of adhesion signaling, but not in response to the chemoattractant CSF-1.Citation23,Citation24 The Cdc42 GEF DOCK8 forms a complex with LRAP35a and MRCK to stimulate MLC2 phosphorylation and BMM motility.Citation25 Deletion of the Rac GAPs Abr and Bcr resulted in an increase in Rac1 activation and BMM motility towards CSF-1.Citation26 Although this is only a small sampling of the RhoGEFs and RhoGAPs that may be expressed in BMMs, these data strongly suggest that each family of regulatory proteins will function in spatially and temporally regulated manners to coordinate Rho GTPase activity to allow directed cell movement.
Because we observed a significant impairment of BMM motility in vitro, it was surprising to find that Net1 did not contribute to peritoneal macrophage recruitment in vivo. A possible explanation for these divergent effects is the timing of the assays. Our in vitro transwell assays were conducted over the span of two hours, while macrophage recruitment in the sterile peritonitis model was measured after three days. It is conceivable that if we had measured macrophage recruitment sooner we would have observed an effect of Net1 deletion, but there is a limit to this approach as macrophages are only beginning to accumulate in the peritoneum 24 hours after thioglycollate injection. Moreover, others have observed that deletion of both RhoA and RhoB actually increased macrophage recruitment 2 days after thioglycollate injection, indicating that these Rho proteins are dispensable is this assay.Citation9 Thus, it seems more likely that loss of Net1 or RhoA/RhoB will result in more subtle defects in vivo than can be assessed with this type of assay. It is also possible that Net1 and RhoA subfamily proteins make more important contributions to other macrophage functions, such as phagocytosis of pathogens or cell debris.Citation27 We also cannot rule out a role for Net1 in other types of monocytes. For example, RhoA function has been reported to be important for the survival of dendritic cell subtypes, as well as for migration from the skin to the lymph node.Citation28,Citation29 In the future it will be important to assess possible contributions of Net1 to these immune cell phenotypes.
Materials and methods
Mouse husbandry and care
Mice were housed in the Center for Laboratory Animal Medicine and Care within the Medical School at the University of Texas Health Science Center at Houston. All studies were approved by the Institutional Animal Care and Use Committee (protocol AWC 14-007) and were conducted in accordance with the guidelines of the U.S. Public Health Service Policy for Humane Care and Use of Laboratory Animals.
Mouse strains used and genotyping
Net1−/− mice in a C57BL/6 background were as previously described.Citation12 These mice were backcrossed to FVB/J mice for 10 generations to create a congenic FVB/J strain. Primers for genotyping Net1 deficient mice were as follows: forward primer 5GF3, 5′-TGCTATGCTATTGCTGCTT-3′, and the reverse primer 3GR1, 5′-AGAACACCACCAAGTAACAA-3′ (amplifies the wild type Net1 allele); forward primer 5GF1, 5′-TTGTTACTTGGTGGTGTTCT-3′ and the reverse primer TV3–1R, 5′-AAGTGCTAACCTTCCTGC-3′ (amplifies the Net−/− allele).Citation12
Isolation, culture, and differentiation of bone marrow macrophage precursors
Mouse femurs and tibias were carefully separated from the surrounding hair and skin, and the muscles were removed. After a quick soaking in 30% ethanol, the bones were transferred to sterile phosphate buffered saline (PBS) in a tissue culture hood, and then further cleaned by removing all connective tissue and remaining muscle. Using a 27-gauge needle, the marrow was gently flushed from the bone with warm Dulbecco's modified Eagle's medium (DMEM)/F12 plus 100 U/ml Penicillin/100 µg/ml Streptomycin (P/S) (Hyclone) and 10% fetal bovine serum (FBS)(Sigma Aldrich) into a 50 ml tube until the bones appeared white. Clumps were broken up by trituration and washed twice with 20 ml of complete DMEM/F12 medium (Hyclone). Bone marrow cells were re-suspended in DMEM/F12 with P/S and 10% FBS, plus 10 ng/ml macrophage colony stimulating factor (M-CSF) (R&D Systems), and seeded in two 6-well plates per mouse. The cells were cultured at 37°C in a 5% CO2 incubator. Every two days, 2 ml complete medium with M-CSF was added to the cells. Bone marrow macrophages were considered fully differentiated after Day 7.
Isolation of mouse peritoneal macrophages
Mice were injected into the peritoneal cavity with 5 ml of 3% sterile thioglycollate medium in PBSCitation17 using a 25-G needle. After 3 days, mice were euthanized and the outer skin was gently peeled off to expose the peritoneum. The peritoneal cavity was injected with 10 ml of cold PBS using a 27-G needle. After massaging the peritoneum, the peritoneal fluid was gently collected with a 25-G needle attached to a 10 ml syringe. A small incision was then made in peritoneum and the remaining fluid was collected with a plastic Pasteur pipette. Samples were discarded if visible blood contamination was detected. The cell suspension was pelleted at 600 × g for 5 minutes at 4 °C and the cells were re-suspended in PBS for counting and flow cytometry analysis.
Flow cytometry analysis
Cell suspensions were counted and 2–5 × 106 cells were used for flow cytometry analysis. Cells were rinsed with ice cold fluorescence activated cell sorting (FACS) buffer (PBS, 2% FBS, 0.1 mM EDTA). Each sample was then incubated with blocking solution (1 µl mouse FC block in 50 µl FACS) (eBioscience) for 10 minutes at room temperature. After blocking, 500 µl cold FACS buffer was added and samples were spun at 600 × g for 5 minutes at 4°C. Supernatants were carefully removed and the sample tubes were respectively incubated with 50 µl primary antibody-conjugated fluorophores (anti-mouse CD45 efluor 450, anti-mouse CD11b APC-efluor 780, anti-mouse F4/80 PE/cy5, 1:50 in FACS) and FMOs (fluorescence minus one) for 15 minutes at room temperature. After staining, samples and FMO tubes were rinsed with 1 ml PBS and single control tubes were rinsed with 1 ml FACS buffer. Sample and FMO tubes were incubated with 1 ml of Live/dead fixable Aqua stain kit (L34957, ThermoFisher Scientific) for 10 minutes at room temperature in the dark. After rinsing with 1 ml of FACS buffer, sample and FMO tubes were incubated with 250 µl permeabilization and fixation solutions (554715, BD Biosciences) for 20 minutes at 4°C in the dark. Sample and FMO tubes were rinsed with 1 ml of 1x permeabilization and wash buffer (554715, BD Biosciences) twice. Samples and FMO tubes were incubated with 50 µl anti-mouse APC-conjugated CD68 antibody (1:50 in wash buffer) for 20 minutes at 4 °C in the dark. Samples tubes were rinsed with 1 ml of wash buffer and 1 ml of FACS buffer subsequently. Data were acquired on a CytoFLEX flow cytometer (Beckman Coulter) or LSRFortessa flow cytometer (BD Biosciences) and analyzed using FlowJo software (Treestar Inc.). No less than 2× 106 events were recorded for each sample. Cell specific fluorescence minus one controls were used to confirm individual antibody specificity. For single color controls, one drop of compensation beads (01-1111-42, eBioscience) dissolved in 50 µl FACS was incubated with single antibody, respectively.
Quantitative real time PCR
Total RNA was extracted from bone marrow macrophages with on-column DNAse digestion (E.Z.N.A.® Total RNA Kit I) and cDNA synthesized using Moloney Murine Leukemia Virus reverse transcriptase (Life Technologies) and random primers. To quantify mRNA expression of both isoforms, Net1-specific qPCR primers were designed towards a region encoded by exons 9 and 10 that is located in the loxP flanked deletion region. The primers were 5′-ACATTCTCGTGAACTGGTTA-3′ (forward) and 5′-GCTGGAGGAAGTCTTGGA-3′ (reverse). The primers for mouse GAPDH were 5′-AGGTCGGTGTGAACGGATTTG-3′ (forward) and 5′-TGTAGACCATGTAGTTGAGGTCA-3′ (reverse). Relative mRNA abundance was determined by real-time PCR (Roche LightCycler 480) with SYBR green master mix (Sigma) and normalized to Gapdh. Amplification conditions included a preincubation at 95°C for 3 min followed by amplification of the target DNA for 45 cycles (95°C for 45 s, 57°C for 45 s and 72°C for 45 s) and fluorescence collection at 60°C.
Antibodies
The following antibodies were used. Anti-pSer19-MLC2 (3675); anti-MLC2 (8505); anti-pSer3-cofilin (3313); anti-cofilin (5175) (Cell Signaling Technology). Anti-CD68 (AB125212) (Abcam). Anti-GST (sc-138) (Santa Cruz Biotechnology). Anti-RhoA (ARH03) (Cytoskeleton, Inc.). For flow cytometry, anti-CD45 (48-0451-82) (eBioscience); anti-CD11b (47-0112-82) (eBioscience); anti-F4/80 (123111) (Biolegend); anti-CD68 (137007) (Biolegend).
GST-RBD assays
6× 106 bone marrow macrophages were lysed in 0.6 ml Triton lysis buffer with 10 mM MgCl2. The cell lysates were centrifuged at 14,000 rpm at 4°C for 15 min to pellet cell membranes and insoluble material. Equal amounts of soluble lysate were incubated with 30 µg of Glutathione-S-transferase-Rhotekin Rho binding domain (GST-RBD) protein beads (Cytoskeleton, Inc.) for 1 h at 4°C. Precipitates were washed three times with wash buffer (25 mM Tris-HCl [pH 7.5], 30 mM MgCl2, 40 mM NaCl), and resuspended in 50 µl of 2× Laemmli sample buffer. Precipitates and cell lysates were resolved by Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene difluoride (PVDF) membranes, and analyzed by Western blotting for RhoA and GST.
Immunofluorescent staining and microscopy
Bone marrow macrophages cells were plated on acid washed glass coverslips, fixed in 4% paraformaldehyde in PBS, and permeabilized with 0.2% Triton X-100 in PBS + 0.05% Tween 20 (PBST). Cells were incubated with the relevant primary antibodies diluted in PBST plus 1% bovine serum albumin fraction V (BSA) for 1 h at 37°C. For anti-pMLC2 staining, Tween 20 was omitted for primary antibody incubation. After washing in PBST, cells were incubated for 30 min at 37°C with secondary antibodies diluted 1:1000 in PBST, plus 4′, 6-diamidino-2-phenylindole (DAPI) (1 µg/ml) (Sigma-Aldrich). Secondary antibodies were anti-mouse Alexa Fluor-488, and anti-rabbit Alexa Fluor-594 (Life Technologies). For F-actin staining, Acti-stain 670-Phalloidin (Cytoskeleton) was used. Cells were then washed in PBST and coverslips were mounted on glass slides with Fluormount reagent (EMD Millipore Chemicals). Fluorescent staining was visualized using a Zeiss Axiophot epifluorescence microscope, and image acquisition was performed using Axiovision software. Intensity of fluorescent signals was quantified using Image J software (NIH).
Western blot analyses
For Western blotting, cells were washed with PBS and harvested in SDS lysis buffer (2.0% SDS, 20 mM Tris-HCl [pH 8.0], 100 mM NaCl, 80 mM β-glycerophosphate, 1 mM Na3VO4, 20 mM NaF, 10 μg/ml leupeptin, 10 μg/ml pepstatin A, 10 μg/ml aprotinin, 1 mM PMSF and 1 mM DTT). Harvested cells were sonicated and boiled for 5 min in the presence of Laemmli sample buffer. Equal volumes of cell lysates were resolved by SDS-PAGE. Proteins were transferred to PVDF membranes and blocked in 5% non-fat dried milk in TBST for 1h at room temperature. Membranes were incubated with the relevant antibodies diluted in TBST + 0.05% milk, or 2% BSA, at 4°C overnight.
Cell motility and adherence assays
Bone marrow macrophages were starved in DMEM/F12 plus 0.5% FBS for 16 h prior to trypsinization. 1× 105 cells were placed in the upper chamber of a Transwell insert with 5 µm pores (BD Biosciences). Media in the bottom well was supplemented with macrophage chemotactic protein 1 (50 ng/ml, R&D Systems). Cells were allowed to migrate for 2 h, and then the cells in the upper well were removed using a cotton swab. Cells on the bottom of the membrane were fixed and stained with DAPI (1 µg/ml; Sigma-Aldrich). Cells that had traversed the membrane were counted in 10 random fields using a 20× objective and a Zeiss Axiophot microscope. Ten images per membrane were captured with an Axiocam MRm camera and Axiovision software. Cell numbers were quantified using ImageJ software.
Statistical analysis
Comparisons were made using unpaired Student's t-tests. P < 0.05 was considered statistically significant. GraphPad Prism 5 software was employed for statistical analyses. All experiments were repeated independently at least three times. For immunofluorescence experiments at least 20 cells per condition per experiment were analyzed.
supp_data_1405772.zip
Download Zip (145 KB)Acknowledgments
We would like to thank members of the Frost, Denicourt, and Du labs for helpful suggestions.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunolg. 2011;11:723–737.
- Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269.
- Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690–701.
- Allen WE, Zicha D, Ridley AJ, et al. A role for Cdc42 in macrophage chemotaxis. J Cell Biol. 1998;141:1147–1157.
- Wells CM, Walmsley M, Ooi S, et al. Rac1-deficient macrophages exhibit defects in cell spreading and membrane ruffling but not migration. J Cell Sci. 2004;117:1259–1268.
- Pradip D, Peng X, Durden DL. Rac2 specificity in macrophage integrin signaling: potential role for Syk kinase. J Biol Chem. 2003;278:41661–41669.
- Wheeler AP, Wells CM, Smith SD, et al. Rac1 and Rac2 regulate macrophage morphology but are not essential for migration. J Cell Sci. 2006;119:2749–2757.
- Wheeler AP, Ridley AJ. RhoB affects macrophage adhesion, integrin expression and migration. Exp Cell Res. 2007;313:3505–3516.
- Konigs V, Jennings R, Vogl T, et al. Mouse macrophages completely lacking Rho subfamily GTPases (RhoA, RhoB, and RhoC) have severe lamellipodial retraction defects, but robust chemotactic navigation and altered motility. J Biol Chem. 2014;289:30772–30784.
- Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180.
- Tcherkezian J, Lamarche-Vane N. Current knowledge of the large RhoGAP family of proteins. Biol Cell. 2007;99:67–86.
- Zuo Y, Berdeaux R, Frost JA. The RhoGEF Net1 Is Required for Normal Mammary Gland Development. Mol Endocrinol. 2014;28:1948–1960.
- Schwertfeger KL, Rosen JM, Cohen DA. Mammary gland macrophages: pleiotropic functions in mammary development. J Mammary Gland Biol Neoplasia. 2006;11:229–238.
- Coussens LM, Pollard JW. Leukocytes in mammary development and cancer. Cold Spring Harb Perspect Biol. 2011;3:a003285.
- Carr HS, Zuo Y, Oh W, et al. Regulation of FAK activation, breast cancer cell motility and amoeboid invasion by the RhoA GEF Net1. Mol Cell Biol. 2013;33:2773–2786.
- Narumiya S, Tanji M, Ishizaki T. Rho signaling, ROCK and mDia1, in transformation, metastasis and invasion. Cancer Metastasis Rev. 2009;28:65–76.
- Hoover DL, Nacy CA. Macrophage activation to kill Leishmania tropica: defective intracellular killing of amastigotes by macrophages elicited with sterile inflammatory agents. J Immunol. 1984;132:1487–1493.
- Brazier H, Stephens S, Ory S, et al. Expression profile of RhoGTPases and RhoGEFs during RANKL-stimulated osteoclastogenesis: identification of essential genes in osteoclasts. J Bone Miner Res. 2006;21:1387–1398.
- Tureci O, Bian H, Nestle FO, et al. Cascades of transcriptional induction during dendritic cell maturation revealed by genome-wide expression analysis. FASEB J. 2003;17:836–847.
- Alberts AS, Treisman R. Activation of RhoA and SAPK/JNK signalling pathways by the RhoA-specific exchange factor mNET1. EMBO J. 1998;17:4075–4085.
- Qin H, Carr HS, Wu X, et al. Characterization of the biochemical and transforming properties of the neuroepithelial transforming protein 1. J Biol Chem. 2005;280:7603–7613.
- Srougi MC, Burridge K. The nuclear guanine nucleotide exchange factors Ect2 and Net1 regulate RhoB-mediated cell death after DNA damage. PLoS ONE. 2011;6:e17108.
- Wells CM, Bhavsar PJ, Evans IR, et al. Vav1 and Vav2 play different roles in macrophage migration and cytoskeletal organization. Exp Cell Res. 2005;310:303–310.
- Bhavsar PJ, Vigorito E, Turner M, et al. Vav GEFs regulate macrophage morphology and adhesion-induced Rac and Rho activation. Exp Cell Res. 2009;315:3345–3358.
- Shiraishi A, Uruno T, Sanematsu F, et al. DOCK8 Protein Regulates Macrophage Migration through Cdc42 Protein Activation and LRAP35a Protein Interaction. J Biol Chem. 2017;292:2191–2202.
- Cho YJ, Cunnick JM, Yi SJ, et al. Abr and Bcr, two homologous Rac GTPase-activating proteins, control multiple cellular functions of murine macrophages. Mol Cell Biol. 2007;27:899–911.
- Mulloy JC, Cancelas JA, Filippi MD, et al. Rho GTPases in hematopoiesis and hemopathies. Blood. 2010;115:936–947.
- Li S, Dislich B, Brakebusch CH, et al. Control of Homeostasis and Dendritic Cell Survival by the GTPase RhoA. J Immunol. 2015;195:4244–4256.
- Vargas P, Maiuri P, Bretou M, et al. Innate control of actin nucleation determines two distinct migration behaviours in dendritic cells. Nat Cell Biol. 2016;18:43–53.