
ABSTRACT
Activating somatic K-Ras mutations are associated with >15% all human tumors and up to 90% of specific tumor types such as pancreatic cancer. Successfully inhibiting abnormal K-Ras signaling would therefore be a game changer in cancer therapy. However, K-Ras has long been considered an undruggable target for various reasons. This view is now changing by the discovery of allosteric inhibitors that directly target K-Ras and inhibit its functions, and by the identification of new mechanisms to dislodge it from the plasma membrane and thereby abrogate its cellular activities. In this review, we will discuss recent progresses and challenges to inhibiting aberrant K-Ras functions by these two approaches. We will also provide a broad overview of other approaches such as inhibition of K-Ras effectors, and offer a brief perspective on the way forward.
Introduction
Ras proteins are plasma membrane (PM)-associated molecular switches that oscillate between GDP-bound inactive and GTP-bound active conformational states [Citation1] to regulate a variety of signaling pathways crucial for cell growth and differentiation [Citation2]. The three major Ras isoforms in humans (H-Ras, N-Ras and K-Ras) share 95% sequence identity at their catalytic domain (residues 1–166) but diverge at their C-terminal 21/22 residues that harbor a lipid-modified membrane targeting motif [Citation3–6]. The catalytic domain consists of lobe1 (residues 1–86) and lobe2 (residues 87–166) [Citation6]. The nucleotide binding and effector-interacting switch 1 (residues 30–38) and switch 2 (residues 59–76) regions are located in lobe1 and undergo major conformational changes upon GDP/GTP exchange and GTP hydrolysis. Nucleotide exchange is facilitated by guanine nucleotide exchange factors (GEFs) such as son of sevenless and GTP hydrolysis is catalysed by GTPase activating proteins (GAPs) such as neurofibromin. Defective GTPase activity of Ras due to somatic mutations, typically at codons 12, 13 and 61, is associated with 15–20% of all human cancers, and K-Ras mutations account for 85% of all Ras mutations [Citation4,Citation7,Citation8]. It is thus clear that a drug that selectively inhibits K-Ras would be a game changer in cancer therapy.
Ras proteins primarily localize to the inner leaflet of the PM in order to transduce extracellular signals to the nucleus [Citation9,Citation10]. For high affinity PM binding, Ras proteins undergo a series of post-translational modifications at the C-terminal CAAX motif (where C = Cys, A = aliphatic amino acids, and X = Met or Ser). First, a cytosolic farnesyltransferase attaches a farnesyl group to the Cys, which allows Ras to attach to the cytosolic leaflet of the ER [Citation11,Citation12]. RCE1 (Ras converting CAAX endopeptidase 1) then removes the AAX tripeptide, followed by the methylation of the now C-terminal prenylated Cys by ICMT (isoprenylcysteine carboxyl methyltransferase) [Citation12,Citation13]. N-, H-, and K-Ras4A (the alternative splicing variant of K-Ras) are further modified with the addition of palmitic acids on one or two other Cys residues near the prenylated Cys [Citation11], allowing Ras to interact with and localize to the PM. K-Ras4B (hereafter, K-Ras) is unique in that it has a single farnesyl chain preceded by a polybasic domain of six Lys residues [Citation14]. The strong positive charge of this polybasic domain allows K-Ras to interact with anionic phospholipids in the PM through electrostatic interaction [Citation15,Citation16].
Broadly, four alternative approaches of developing therapies for K-Ras-driven cancers are being pursued: 1) dissociation of K-Ras from the PM; 2) direct allosteric inhibition of K-Ras; 3) inhibition of K-Ras downstream effectors; and 4) dysregulation of cell metabolism. In this review, we will focus on the first two approaches and discuss in detail recent progresses and challenges to inhibiting aberrant K-Ras functions by small molecule inhibitors that dislodge it from the PM or directly bind to K-Ras and allosterically modulate its biochemical activities (). We will then provide an overview of the latter two approaches, and conclude with a brief perspective.
Figure 1. Schematics showing promising approaches to inhibiting oncogenic K-Ras: K-Ras activity can be inhibited directly or by agents that facilitate its dissociation from the plasma member. PDEδ binds to the PM-dissociated K-Ras and unloads it in the perinuclear region, whence K-Ras is translocated to the PtdSer- and SM-enriched recycling endosome (RE) for redelivery to the PM by vesicular transport. Disrupting its interaction with PDEδ or RE reduces the concentration of K-Ras at the PM. Also, perturbation of SM/Cer metabolism and distribution, which regulates PM PtdSer content, depletes PtdSer of the PM, resulting in K-Ras PM dissociation. Dysregulating RE activity by APEH inhibition further results in the mislocalization of PtdSer and K-Ras from the PM. PS – phosphatidylserine, SM – sphingomyelin, Cer – ceramide, ASM – acid sphingomyelinase, APEH – acylpeptide hydrolase, PDEδ – phosphodiesterase δ, G01 – a synthetic small molecule inhibitor of APEH
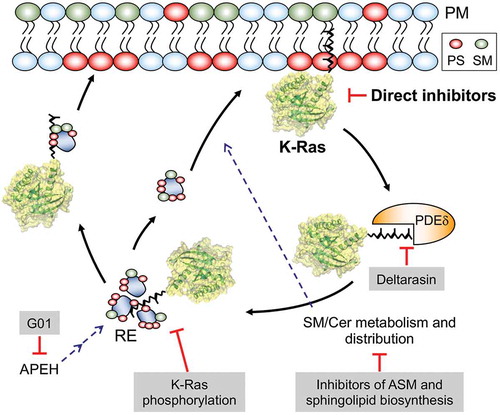
Dissociation of K-Ras from the PM
The first attempt at blocking the interaction of Ras with the PM was via the inhibition of farnesylation by farnesyltransferase inhibitors (FTIs). FTIs were highly effective in cell culture and mouse models of H-Ras tumors, but failed in K-Ras tumors because of geranylgeranylation, an alternative prenylation pathway that effectively subverted the intended therapeutic mechanism [Citation17]. Despite the clinical failure of FTIs as an anti-K-Ras drugs, inhibition of PM interaction remains a valid therapeutic approach to abrogating K-Ras oncogenic activity. Recent studies identified new molecular mechanisms that regulate K-Ras PM interaction and its signaling: depleting the phosphatidylserine (PtdSer) content of the PM, enhancing K-Ras phosphorylation, and disrupting K-Ras interaction with its chaperone protein phosphodiesterase (PDE6δ).
PM PtdSer depletion by perturbing sphingomyelin metabolism
PtdSer is asymmetrically concentrated on the inner leaflet of the PM and is responsible for the significant negative electrostatic potential of the PM [Citation18]. K-Ras interacts with the PM through the combined effects of the polybasic domain and the farnesyl chain of the lipid anchor, which provides high selectivity for PtdSer over other anionic phospholipids [Citation15]. Depletion of PtdSer mislocalizes K-Ras from the PM and blocks K-Ras signaling by disrupting nanoclusters, an essential Ras signaling platform [Citation16,Citation19,Citation20]. Recent studies have shown that sphingomyelin (SM) metabolism modulates K-Ras PM localization by regulating the PtdSer content of the PM, thus opening new opportunities for inhibiting K-Ras.
Two classes of inhibitors that modulate SM metabolism have been reported (). The first is inhibitors of acid sphingomyelinase (ASM), which converts SM to ceramide in the lysosome. It was found that fendiline, a potent inhibitor of ASM redistributes PtdSer and K-Ras (but not H-Ras) from the PM to endomembranes [Citation21,Citation22]. Supplementation with exogenous PtdSer restores K-Ras PM interaction in fendiline-treated cells, suggesting that the K-Ras PM mislocalization is through PtdSer depletion at the inner PM leaflet [Citation21]. Fendiline also reduces cellular ceramide level and induces SM endosomal accumulation. Supplementation with recombinant ASM or exogenous ceramide restores K-Ras and PtdSer back to the PM in fendiline-treated cells, suggesting that ASM-mediated cellular balance of SM/ceramide regulates PtdSer localization at the PM, resulting in K-Ras PM interaction [Citation21]. A wide variety of ASM inhibitors including tricyclic antidepressants are also shown to deplete PM PtdSer and mislocalize K-Ras from the PM [Citation23]. Consistent with this, K-Ras is shown to be mislocalized from the PM in patient-derived Niemann-Pick type A and B cell lines [Citation21]; Niemann-Pick type A and B diseases are lysosomal storage disorders caused by inactivating and partial-loss-of-function mutations, respectively, in the SMPD1 gene that encodes ASM [Citation24]. ASM inhibitors disrupt oncogenic K-Ras signaling and its PM nanoclusters, and block the growth of a range of human cancer cells expressing oncogenic mutant K-Ras but not wild-type K-Ras [Citation22,Citation23,Citation25]. Taken together, these studies identify ASM as an attractive target for the development of anti-K-Ras therapies.
The second group of inhibitors that modulate SM metabolism target enzymes involved in sphingomyelin biosynthesis. Perturbing the cellular SM/ceramide balance by dysregulating enzymes involved in sphingomyelin biosynthesis is shown to disrupt K-Ras PM interaction and its signal output. In a genetic study using RNA interference against C. elegans genes encoding enzymes in the SM/ceramide biosynthesis pathway, the authors found that knockdown of 14 enzymes suppresses the LET-60 G13D (a K-RasG13D ortholog in C. elegans)-induced multivulva phenotype [Citation23]. Furthermore, pharmacological agents targeting these enzymes in mammalian cells deplete PtdSer and mislocalize K-Ras from the PM [Citation23]. These compounds also disrupt the K-Ras nanoclustering and inhibit the proliferation of pancreatic cancer cell lines expressing oncogenic mutant K-Ras [Citation23]. Although these pharmacological agents either increase or decrease cellular SM levels, they all perturb cellular SM distribution. Based on these observations, the authors proposed that a correct cellular distribution of SM at appropriate concentrations is required for maintaining PtdSer and K-Ras at the PM, and that pharmacological tools targeting the sphingolipid pathways may provide novel therapeutic strategies for the treatment of K-Ras-dependent cancers [Citation23,Citation26].
PM PtdSer depletion by perturbing recycling endosomal activity
A recent study identified acylpeptide hydrolase (APEH) as a new novel protein that regulates K-Ras and PtdSer PM localization. APEH is a ubiquitously expressed cytosolic enzyme that catalyses the removal of N-acylated amino acids from acetylated peptides, and it is involved in the ubiquitin-proteasome protein degradation machinery [Citation27,Citation28]. APEH knockdown or inhibition mislocalizes K-Ras and PtdSer from the PM, which is rescued by ectopic expression of APEH [Citation29]. The study found that APEH-mediated PM mislocalization of K-Ras and PtdSer depends on suppression of recycling endosome (RE) function. PtdSer is the most abundant lipid in REs among intracellular organelles [Citation30], and perturbation in endosomal sorting of PtdSer results in the PM PtdSer depletion and K-Ras PM mislocalization [Citation31]. Also, the RE operation in concert with PDE6δ and Arl-2/3 maintains K-Ras at the PM (discussed in detail later) [Citation32,Citation33]. The authors suggest that the failure to maintain PM PtdSer content in APEH-knockdown cells is at least in part due to aberrant RE function. Furthermore, APEH knockdown or inhibition abrogates Ras/MAPK signaling in cells expressing oncogenic mutant K-Ras and inhibits the growth of K-Ras-positive cancer cells [Citation29,Citation34]. Taken together, this study proposes that perturbation of RE activity could be an attractive approach for inhibiting K-Ras activity, and that APEH is a novel drug target for a potential anti-K-Ras therapeutic.
Enhancing K-Ras phosphorylation
Protein kinase C (PKC) directly phosphorylates K-Ras at Ser181 and to a lesser extent at Ser171 and Thr183, redistributing K-Ras from the PM to the endomembranes and mitochondria, triggering enhanced apoptosis [Citation35]. PKC activators can further suppress the growth of K-Ras tumors in nude mice by stimulating K-Ras phosphorylation [Citation35,Citation36], suggesting that K-Ras phosphorylation is a valid target for blocking oncogenic K-Ras signaling. A recent study identified cyclic GMP-dependent protein kinase 2 (PKG2) as a new K-Ras kinase that phosphorylates K-Ras at Ser181 in response to the AMPK-eNOS-sGC-PKG2 pathway activation [Citation33] (). Using pharmacological agents targeting enzymes in the pathway, it was found that direct or indirect activation of AMP-activated protein kinase (AMPK) stimulates the activity of endothelial nitric oxide synthase (eNOS), one of the AMPK downstream effectors. This in turn elevates cellular NO levels, which promotes soluble guanylyl cyclase (sGC) activity, generating cGMP from GTP. Activated PKG2, but not PKG1, is recruited to the PM and phosphorylates K-Ras at Ser181 by the elevated cGMP. Unlike PKC, which acutely mislocalizes K-Ras from the PM after phosphorylation, K-Ras is mislocalized from the PM at t1/2 = 40min after PKG-mediated phosphorylation. The authors proposed that phosphorylated K-Ras by PKG is not instantly dissociated from the PM, but rather progressively lost via endocytic recycling [Citation33].
Stimulation of the AMPK-eNOS-sGC-PKG pathway shows anti-cancer response. Chronic treatment with pharmacological agents that activate components in the AMPK-eNOS-sGC-PKG inhibits the growth of non-small cell lung cancer cells expressing oncogenic mutant K-Ras [Citation33]. Also, metformin, an antidiabetic drug that activates AMPK through lowering cellular ATP levels [Citation37], is reported to be associated with a 31% reduction in cancer risk in a meta-analysis of 5 observational studies of all cancer types [Citation38]. It also showed inhibitory activity in tumor growth in preclinical endometrial cancer models, with the greatest response being in cells expressing oncogenic mutant K-Ras [Citation39]. Another activator of the pathway is sildenafil also known as Viagra, which inhibits PDE5 to elevate cellular cGMP levels and thereby activate PKG. Oral administration of sildenafil suppresses colorectal cancer in mice induced by the potent carcinogens azoxymethane/dextran sulfate sodium [Citation40]. Furthermore, PKG2-null mice develop crypt hyperplasia in the colonic epithelium, while ectopic PKG2 expression in colorectal cancer cell lines inhibits proliferation [Citation41]. In contrast, some components of the AMPK-eNOS-PKG pathway may promote tumorigenesis. For example, advanced PDACs with a genetic deficiency of eNOS or treatment of mice with a NOS inhibitor suppresses the development of preinvasive pancreatic lesions and shows a tendency toward an extended lifespan [Citation42]. Also, acute PKG activation in vascular smooth muscle cells enhances MAPK signaling [Citation43]. One explanation for the inconsistency in the anti-cancer response of the pathway is the extent of K-Ras phosphorylation. Using computational modeling, biochemistry and electron microscopy techniques, it has been shown that PKG2-mediated K-Ras phosphorylation acutely increases both phosphatidylinositol 3-kinase/Akt and Raf/MAPK activation by altering K-Ras PM nanoclustering. This is attenuated by a progressive loss of phosphorylated K-Ras from the PM, which subsequently abrogates K-Ras signaling [Citation33]. Based on these data, it is possible that in certain tissues, AMPK-eNOS-PKG signaling is sufficient to phosphorylate K-Ras but not enough to remove it from the PM, resulting in elevated K-Ras signal output. Clearly, more work is required to fully establish the therapeutic potential of K-Ras phosphorylation. In particular, further characterization of cancer types sensitive to the pharmacological activators of the AMPK-eNOS-sGC-PKG pathway will need to be elucidated, and the long-term beneficial effects of the activators need to be analysed in the context of K-Ras-positive cancers.
Inhibiting K-Ras interaction with its chaperone protein
Maintenance of K-Ras at the PM requires the activities of the chaperone protein PDE6δ. The non-catalytic δ subunit of PDE6 binds to endocytosed K-Ras via the farnesyl tail and releases it to the perinuclear membrane in an Arl2-dependent manner. K-Ras then electrostatically interacts with the RE and returns to the PM [Citation32,Citation44,Citation45]. Disrupting the interaction of K-Ras/PDE6δ by PDE6δ knockdown or a PDE6δ inhibitor, deltarasin, mislocalizes K-Ras from the PM [Citation32,Citation46]. Deltarasin blocks the interaction by binding to a hydrophobic pocket of PDE6δ, to which the farnesyl group of K-Ras would bind [Citation46]. PDE6δ inhibition further shows anti-K-Ras activity in human cancer cells. Genetic or pharmacologic inhibition of PDE6δ blocks proliferation and survival of colorectal cancer cells expressing oncogenic mutant K-Ras, whereas the growth of isogenic cell lines in which the oncogenic K-Ras has been removed, or cell lines with oncogenic mutant B-Raf or EGFR overexpression are not affected by PDE6δ inhibition [Citation47]. Deltarasin treatment also inhibits the growth of pancreatic cancer cells expressing oncogenic mutant K-Ras in vitro and in vivo [Citation46]. In addition to inhibiting K-Ras signal output by blocking K-Ras/PDE6δ interaction, deltarasin has K-Ras-independent effects. Deltarasin also elevates autophagy through activating the AMPK-mTOR pathway [Citation48], and autophagy inhibition in deltarasin-treated cancer cells potentiates deltarasin-mediated cell death. This led the authors to propose that deltarasin therapy in combination with an autophagy inhibitor can be a good strategy for treating K-Ras-driven cancers [Citation48].
Despite the reported anti-cancer activities of deltarasin, it is unclear whether the effects are specifically through K-Ras inhibition. PDE6δ binds to many farnesylated Ras superfamily members including H-Ras, Rheb, and Arl2/3 [Citation45,Citation49], which may account for the elevated autophagy in deltarasin-treated cells. Furthermore, K-Ras knockout in mice is embryonic lethal, whereas PDE6δ knockout mice are viable and fertile [Citation50,Citation51], suggesting that K-Ras is functional in the absence of PDE6δ. Taken together, although the effects of PDE6δ inhibition by deltarasin in K-Ras-driven cancers are exciting, translation into the clinic may require further characterization of its K-Ras specificity.
Direct inhibition of K-Ras
As described in section 1, considerable effort has been devoted toward indirectly inhibiting aberrant K-Ras functions, such as by dislodging it from the PM [Citation22,Citation31] or inhibiting its partner proteins [Citation46,Citation52]. This was because direct inhibition has been deemed impossible in part because Ras has high (picomolar) affinity for GDP and GTP that exist at high (~0.5mM) concentration in the cell [Citation7,Citation53]. Another challenge is the conservation of the nucleotide-binding pocket among a diverse group of small GTPases with unrelated functions [Citation1,Citation54]. These issues made competitive inhibition of K-Ras impractical and avoiding off-target effects difficult. In principle, many of these challenges could be overcome by allosteric inhibitors, but first it was necessary to establish that that Ras is an allosteric enzyme. The first clue about the allosteric nature of Ras emerged from molecular dynamics (MD) simulation studies of H-Ras in a simplified model membrane [Citation55]. Additional studies of Ras dynamics in solution and when membrane-bound (reviewed in [Citation3,Citation5,Citation56]), first led to the recognition that Ras has two lobes engaged in long-range coupled motions [Citation6] and then the suggestion that Ras is potentially druggable by allosteric mechanisms [Citation57]. The concept of Ras allostery was initially somewhat controversial given its small size and shallow surface that lacks any obvious ligand binding site outside of the canonical nucleotide binding site. This has changed by the identification of up to four allosteric ligand binding sites first using computational approaches [Citation58–64], and then using NMR or crystallographic studies of ligands bound to these pockets [Citation65–68] (). Many ligands that directly bind to the allosteric sites of K-Ras and modulate its functions have been reported [Citation58,Citation63,Citation65,Citation69,Citation70], including small-molecules [Citation58,Citation70,Citation71], peptidomimetics [Citation72,Citation73], monobodies [Citation74], and even DARPins [Citation75,Citation76].
Figure 2. Druggable allosteric pockets on the catalytic domain of K-Ras. The sites most frequently targeted by published small molecules are pocket p1 near the core beta-sheet (pink) and pocket p2 between switch 2 and helix 3 (green). Pocket p3 near the C-terminus (blue) and pocket p4 behind switch 1 (yellow) are somewhat shallow and more polar than p1 and p2. See Grant et al. [Citation63], for a detailed structural analysis of the four pockets and Gupta et al. [Citation84], for a comparison of their druggability profile. Image reproduced from Grant et al. [Citation63]
![Figure 2. Druggable allosteric pockets on the catalytic domain of K-Ras. The sites most frequently targeted by published small molecules are pocket p1 near the core beta-sheet (pink) and pocket p2 between switch 2 and helix 3 (green). Pocket p3 near the C-terminus (blue) and pocket p4 behind switch 1 (yellow) are somewhat shallow and more polar than p1 and p2. See Grant et al. [Citation63], for a detailed structural analysis of the four pockets and Gupta et al. [Citation84], for a comparison of their druggability profile. Image reproduced from Grant et al. [Citation63]](/cms/asset/3f0cf1d8-c8d5-4cdb-aae6-b78343e7e2f9/ksgt_a_1655883_f0002_oc.jpg)
To our knowledge, none of the reported non-covalent K-Ras inhibitors have made it to clinical trial. In this regard, covalent inhibitors including GDP analogues [Citation77] or other small-molecules [Citation65] targeting G12C K-Ras may have a better chance of eventual success [Citation78], but their application is likely limited to a few cancer types such as small cell lung cancer [Citation79]. Therefore, non-covalent allosteric inhibition will still be required to target many critical K-Ras mutations including G12D, G12V, G13D and Q61H which together account for >78% of all K-Ras-associated cancers [Citation4,Citation79]. While the examples cited above highlight the vulnerabilities of K-Ras, no approach seems to emerge that is capable of identifying sufficiently potent and selective inhibitors that overcome the persistent problems of weak affinity and pan-Ras activity (see refs [Citation80–83] for recent examples). The solution may lie in ensemble-based approaches, considering the highly dynamic nature of K-Ras. Indeed, a recent report found a ~ 10% success rate of predicted-to-confirmed K-Ras binders using this approach [Citation84]. A similar approach yielded a highly promising pyrazolopyrimidine-based lead compound that appears to be selective toward GTP-bound K-Ras and disrupts effector binding and reduces signal transduction through mutant K-Ras [Citation85]
Recent reports on K-Ras dimerization and clustering [Citation84,Citation86–94] opened up alternative ways of thinking about Ras inhibition, such as preventing dimer formation as exemplified by two recent reports [Citation76,Citation95,Citation96]. Another potentially fertile area of intervention is the concept of membrane reorientation, which was originally proposed based on simulations and then verified experimentally [Citation55,Citation97–103]. Future studies may discover inhibitors that target the inter-switch pocket and stabilize a membrane orientation state that is incapable of effector interaction, as found in two recent reports [Citation104,Citation105].
Other approaches
Arguably the most extensively studied approach of indirectly inhibiting Ras functions is the inhibition of its downstream effectors. These targets contribute to Ras-dependent cancer initiation and/or maintenance. A number of promising inhibitors targeting a variety of Ras effectors have been developed and tested in clinical trials. A key limitation of this approach is that inhibition of one Ras effector pathway can be compensated for by other Ras downstream effectors, and inhibition of multiple Ras signaling pathways are lethal for normal cells, resulting in high toxicity [Citation7]. One example is the B-Raf-specific inhibitors. These inhibitors produce excellent responses in patients with B-Raf mutant melanoma [Citation106], but patients develop resistance because the inhibitors paradoxically activate the MAPK cascade in melanoma cells expressing oncogenic mutant N- or K-Ras via a mechanism that involves C-Raf hyperactivation [Citation20,Citation107,Citation108]. A somewhat related approach is to inhibit proteins that have synthetic lethal interaction with oncogenic K-Ras; i.e., inhibiting proteins whose loss of function is lethal only in the presence of oncogenic K-Ras. This approach has been inspired most strongly by the successful use of PARP inhibitors in the clinic to treat BRCA-defective cancers [Citation109]. Genome-wide RNA interference screenings identified several genes required for the survival of cancer cells harboring oncogenic mutant K-Ras [Citation109]. However, the screening data showed lack of overlap between the results, with the possible exception of proteasome components [Citation109], and pharmacological inhibitors of the identified genes have not been proven as targeting cancer cells harboring oncogenic K-Ras [Citation110].
Another approach to targeting K-Ras-driven cancers is dysregulation of cell metabolism. Cancer cells alter their metabolism to meet the increased energy requirement for their growth, and oncogenic K-Ras promotes such metabolic rewiring, although the specifics may differ depending on tumor type and genetic context [Citation111,Citation112]. In pancreatic cancer cells that are K-Ras- and autophagy-dependent for their growth, oncogenic K-Ras/MAPK signaling upregulates autophagy in part by impairing other K-Ras- or MAPK-driven metabolic processes [Citation113], and concomitant inhibition of K-Ras/MAPK and autophagy synergistically inhibits tumor growth [Citation113–115]. A clinical trial (NCT03825289) has been initiated recently to test the efficacy of a combined therapy of MEK inhibitors with hydroxychloroquine, an autophagy inhibitor.
Perspectives
K-Ras has been considered as undruggable for many years despite its critical role in many human cancers. However, recent intensive studies identified a number of vulnerabilities that rendered K-Ras druggable. In this review, we have focused on two approaches that we believe are most promising: dislodging K-Ras from the PM and directly targeting its catalytic domain with small molecule allosteric inhibitors. Although K-Ras is localized primarily at the PM for stimulating its downstream effectors, the mechanisms of its trafficking to and maintenance at the PM are not fully elucidated. A better understanding of these processes is beginning to provide valuable insights into novel approaches of developing anti-K-Ras therapeutics. Furthermore, since the dynamics of K-Ras differs when it is in solution and membrane-bound, approaches that account for these differences may yield novel lead compounds with desirable modes of action such as disrupting effector binding. Focus on allosteric instead of competitive inhibition and emphasis on dynamics where Ras isoforms diverge might also allow for the discovery of K-Ras-selective inhibitors.
Combination therapy for cancer treatment is a well-established practice. With only a few exceptions, cytotoxic cancer chemotherapy is most effective when applied as a concurrent treatment of a cocktail of drugs with different mechanisms of action. It would be intriguing to examine the effects of combining K-Ras inhibitors that target different aspects of its activities. Ultimately no single inhibitor, even one that is K-Ras selective, may treat all of the many types of K-Ras-driven cancers, and different mutations drive different cancers. That said, the ability to treat any one of these cancers would itself be a major breakthrough, and will open opportunities for rational re-design and combination therapies until a truly personalized therapy targeting a specific K-Ras mutation becomes a reality.
Acknowledgments
This work was supported by grants from the National Institute of General Medical Sciences (Grant No. R01GM124233 to A.A.G.) and the National Cancer Institute (Grant No. R00CA188593 to K-J.C.). We thank the Texas Advanced Computing Centre (TACC) and the Extreme Science and Engineering Discovery Environment (XSEDE grant No MCB150054 to A.A.G.) for computational resources.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299–1304.
- Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517–531.
- Grant BJ, Gorfe AA, McCammon JA. Large conformational changes in proteins: signaling and other functions. Curr Opin Struct Biol. 2010;20:142–147.
- Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457–2467.
- Prakash P, Gorfe AA. Lessons from computer simulations of Ras proteins in solution and in membrane. Biochim Biophys Acta. 2013;1830:5211–5218.
- Gorfe AA, Grant BJ, McCammon JA. Mapping the nucleotide and isoform-dependent structural and dynamical features of Ras proteins. Structure. 2008;16:885–896.
- Cox AD, Fesik SW, Kimmelman AC, et al. Drugging the undruggable RAS: Mission possible? Nat Rev. 2014;13:828–851.
- Cox AD, Der CJ. Ras history: The saga continues. Small GTPases. 2010;1:2–27.
- Jackson JH, Cochrane CG, Bourne JR, et al. Farnesol modification of Kirsten-ras exon 4B protein is essential for transformation. Proc Natl Acad Sci U S A. 1990;87:3042–3046.
- Willumsen BM, Christensen A, Hubbert NL, et al. The p21 ras C-terminus is required for transformation and membrane association. Nature. 1984;310:583–586.
- Hancock JF, Magee AI, Childs JE, et al. All ras proteins are polyisoprenylated but only some are palmitoylated. Cell. 1989;57:1167–1177.
- Gutierrez L, Magee AI, Marshall CJ, et al. Post-translational processing of p21ras is two-step and involves carboxyl-methylation and carboxy-terminal proteolysis. Embo J. 1989;8:1093–1098.
- Clarke S, Vogel JP, Deschenes RJ, et al. Posttranslational modification of the Ha-ras oncogene protein: evidence for a third class of protein carboxyl methyltransferases. Proc Natl Acad Sci U S A. 1988;85:4643–4647.
- Hancock JF, Paterson H, Marshall CJ. A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell. 1990;63:133–139.
- Zhou Y, Prakash P, Liang H, et al. Lipid-sorting specificity encoded in K-Ras membrane anchor regulates signal output. Cell. 2017;168:239–251 e216.
- Yeung T, Gilbert GE, Shi J, et al. Membrane phosphatidylserine regulates surface charge and protein localization. Science. 2008;319:210–213.
- Hancock JF. Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol. 2003;4:373–384.
- Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys. 2010;39:407–427.
- Tian T, Harding A, Inder K, et al. Plasma membrane nanoswitches generate high-fidelity Ras signal transduction. Nat Cell Biol. 2007;9:905–914.
- Cho KJ, Kasai RS, Park JH, et al. Raf inhibitors target ras spatiotemporal dynamics. Curr Biol. 2012a;22:945–955.
- Cho KJ, van der Hoeven D, Zhou Y, et al. Inhibition of acid sphingomyelinase depletes cellular phosphatidylserine and mislocalizes K-Ras from the plasma membrane. Mol Cell Biol. 2015;36:363–374.
- van der Hoeven D, Cho KJ, Ma X, et al. Fendiline inhibits K-Ras plasma membrane localization and blocks K-Ras signal transmission. Mol Cell Biol. 2013;33:237–251.
- van der Hoeven D, Cho KJ, Zhou Y, et al. Sphingomyelin metabolism is a regulator of KRAS function. Mol Cell Biol. 2017;38.
- Schuchman EH, Desnick RJ. Types A and B Niemann-Pick disease. Mol Genet Metab. 2017;120:27–33.
- Petersen NH, Olsen OD, Groth-Pedersen L, et al. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell. 2013;24:379–393.
- Maekawa M, Lee M, Wei K, et al. Staurosporines decrease ORMDL proteins and enhance sphingomyelin synthesis resulting in depletion of plasmalemmal phosphatidylserine. Sci Rep. 2016;6:35762.
- Shimizu K, Kiuchi Y, Ando K, et al. Coordination of oxidized protein hydrolase and the proteasome in the clearance of cytotoxic denatured proteins. Biochem Biophys Res Commun. 2004;324:140–146.
- Palumbo R, Gogliettino M, Cocca E, et al. APEH inhibition affects osteosarcoma cell viability via downregulation of the proteasome. Int J Mol Sci. 2016;17.
- Tan L, Cho KJ, Kattan WE, et al. Acylpeptide hydrolase is a novel regulator of KRAS plasma membrane localization and function. J Cell Sci. 2019;132:jcs232132.
- Uchida Y, Hasegawa J, Chinnapen D, et al. Intracellular phosphatidylserine is essential for retrograde membrane traffic through endosomes. Proc Natl Acad Sci U S A. 2011;108:15846–15851.
- Cho KJ, Park JH, Piggott AM, et al. Staurosporines disrupt phosphatidylserine trafficking and mislocalize Ras proteins. J Biol Chem. 2012b;287:43573–43584.
- Schmick M, Vartak N, Papke B, et al. KRas localizes to the plasma membrane by spatial cycles of solubilization, trapping and vesicular transport. Cell. 2014;157:459–471.
- Cho KJ, Casteel DE, Prakash P, et al. AMPK and endothelial nitric oxide synthase signaling regulates K-Ras plasma membrane interactions via cyclic GMP-dependent protein kinase 2. Mol Cell Biol. 2016;36:3086–3099.
- Tan L, Cho KJ, Neupane P, et al. An oxanthroquinone derivative that disrupts RAS plasma membrane localization inhibits cancer cell growth. J Biol Chem. 2018;293:13696–13706.
- Bivona TG, Quatela SE, Bodemann BO, et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell. 2006;21:481–493.
- Wang MT, Holderfield M, Galeas J, et al. K-Ras promotes tumorigenicity through suppression of non-canonical wnt signaling. Cell. 2015;163:1237–1251.
- Hardie DG. Neither LKB1 nor AMPK are the direct targets of metformin. Gastroenterology. 2006;131:973. author reply 974–975.
- Decensi A, Puntoni M, Goodwin P, et al. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila). 2010;3:1451–1461.
- Iglesias DA, Yates MS, van der Hoeven D, et al. Another surprise from metformin: novel mechanism of action via K-Ras influences endometrial cancer response to therapy. Mol Cancer Ther. 2013;12:2847–2856.
- Islam BN, Sharman SK, Hou Y, et al. Sildenafil suppresses inflammation-driven colorectal cancer in mice. Cancer Prev Res (Phila). 2017;10:377–388.
- Wang R, Kwon IK, Thangaraju M, et al. Type 2 cGMP-dependent protein kinase regulates proliferation and differentiation in the colonic mucosa. Am J Physiol Gastrointest Liver Physiol. 2012;303:G209–219.
- Lampson BL, Kendall SD, Ancrile BB, et al. Targeting eNOS in pancreatic cancer. Cancer Res. 2012;72:4472–4482.
- Komalavilas P, Shah PK, Jo H, et al. Activation of mitogen-activated protein kinase pathways by cyclic GMP and cyclic GMP-dependent protein kinase in contractile vascular smooth muscle cells. J Biol Chem. 1999;274:34301–34309.
- Chandra A, Grecco HE, Pisupati V, et al. The GDI-like solubilizing factor PDEdelta sustains the spatial organization and signalling of Ras family proteins. Nat Cell Biol. 2012;14:148–158.
- Hanzal-Bayer M, Renault L, Roversi P, et al. The complex of Arl2-GTP and PDE delta: from structure to function. Embo J. 2002;21:2095–2106.
- Zimmermann G, Papke B, Ismail S, et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638–642.
- Klein CH, Truxius DC, Vogel HA, et al. PDEdelta inhibition impedes the proliferation and survival of human colorectal cancer cell lines harboring oncogenic KRas. Int J Cancer. 2019;144:767–776.
- Leung ELH, Luo LX, Liu ZQ, et al. Inhibition of KRAS-dependent lung cancer cell growth by deltarasin: blockage of autophagy increases its cytotoxicity. Cell Death Dis. 2018;9:216.
- Ismail SA, Chen YX, Rusinova A, et al. Arl2-GTP and Arl3-GTP regulate a GDI-like transport system for farnesylated cargo. Nat Chem Biol. 2011;7:942–949.
- Johnson L, Greenbaum D, Cichowski K, et al. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997;11:2468–2481.
- Zhang H, Li S, Doan T, et al. Deletion of PrBP/delta impedes transport of GRK1 and PDE6 catalytic subunits to photoreceptor outer segments. Proc Natl Acad Sci U S A. 2007;104:8857–8862.
- Hauschild A, Grob JJ, Demidov LV, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–365.
- Singh H, Longo DL, Chabner BA. Improving Prospects for Targeting RAS. J Clin Oncol. 2015;33:3650–3659.
- Rojas AM, Fuentes G, Rausell A, et al. The Ras protein superfamily: evolutionary tree and role of conserved amino acids. J Cell Biol. 2012;196:189–201.
- Gorfe AA, Hanzal-Bayer M, Abankwa D, et al. Structure and dynamics of the full-length lipid-modified H-Ras protein in a 1,2-dimyristoylglycero-3-phosphocholine bilayer. J Med Chem. 2007;50:674–684.
- Prakash P, Gorfe AA. Overview of simulation studies on the enzymatic activity and conformational dynamics of the GTPase Ras. Mol Simul. 2014;40:839–847.
- Gorfe AA. Mechanisms of allostery and membrane attachment in Ras GTPases: implications for anti-cancer drug discovery. Curr Med Chem. 2010;17:1–9.
- Hocker HJ, Cho KJ, Chen CY, et al. Andrographolide derivatives inhibit guanine nucleotide exchange and abrogate oncogenic Ras function. Proc Natl Acad Sci U S A. 2013;110:10201–10206.
- McCarthy M, Prakash P, Gorfe AA. Computational allosteric ligand binding site identification on Ras proteins. Acta Biochim Biophys Sin (Shanghai). 2016;48:3–10.
- Hocker HJ, Rambahal N, Gorfe AA. LIBSA–a method for the determination of ligand-binding preference to allosteric sites on receptor ensembles. J Chem Inf Model. 2014;54:530–538.
- Prakash P, Sayyed-Ahmad A, Gorfe AA. pMD-membrane: a method for ligand binding site identification in membrane-bound proteins. PLoS Comput Biol. 2015b;11:e1004469.
- Prakash P, Hancock JF, Gorfe AA. Binding hotspots on K-ras: consensus ligand binding sites and other reactive regions from probe-based molecular dynamics analysis. Proteins. 2015a;83:898–909.
- Grant BJ, Lukman S, Hocker HJ, et al. Novel allosteric sites on Ras for lead generation. PLoS One. 2011;6:e25711.
- Sayyed-Ahmad A, Gorfe AA. Mixed-probe simulation and probe-derived surface topography map analysis for ligand binding site identification. J Chem Theory Comput. 2017;13:1851–1861.
- Ostrem JM, Peters U, Sos ML, et al. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551.
- Maurer T, Garrenton LS, Oh A, et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci U S A. 2012;109:5299–5304.
- Rosnizeck IC, Spoerner M, Harsch T, et al. Metal-bis(2-picolyl)amine complexes as state 1(T) inhibitors of activated Ras protein. Angew Chem Int Ed Engl. 2012;51:10647–10651.
- Rosnizeck IC, Graf T, Spoerner M, et al. Stabilizing a weak binding state for effectors in the human ras protein by cyclen complexes. Angew Chem Int Ed Engl. 2010;49:3830–3833.
- Lim SM, Westover KD, Ficarro SB, et al. Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew Chem Int Ed Engl. 2014;53:199–204.
- Shima F, Yoshikawa Y, Ye M, et al. (2013). In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proceedings of the National Academy of Sciences of the United States of America 110(20), 8182–8187.
- Sun Q, Burke JP, Phan J, et al. Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angew Chem Int Ed Engl. 2012;51:6140–6143.
- Upadhyaya P, Qian Z, Habir NA, et al. Direct ras inhibitors identified from a structurally rigidified bicyclic peptide library. Tetrahedron. 2014;70:7714–7720.
- Trinh TB, Upadhyaya P, Qian Z, et al. Discovery of a direct ras inhibitor by screening a combinatorial library of cell-permeable bicyclic peptides. ACS Comb Sci. 2016;18:75–85.
- Spencer-Smith R, Koide A, Zhou Y, et al. Inhibition of RAS function through targeting an allosteric regulatory site. Nat Chem Biol. 2017;13:62–68.
- Pluckthun A. Designed ankyrin repeat proteins (DARPins): binding proteins for research, diagnostics, and therapy. Annu Rev Pharmacol Toxicol. 2015;55:489–511.
- Bery N, Legg S, Debreczeni J, et al. KRAS-specific inhibition using a DARPin binding to a site in the allosteric lobe. Nat Commun. 2019;10:2607.
- Xiong Y, Lu J, Hunter J, et al. Covalent guanosine mimetic inhibitors of G12C KRAS. ACS Med Chem Lett. 2017;8:61–66.
- Rudolph J, Stokoe D. Selective inhibition of mutant Ras protein through covalent binding. Angew Chem Int Ed Engl. 2014;53:3777–3779.
- Stephen AG, Esposito D, Bagni RK, et al. Dragging ras back in the ring. Cancer Cell. 2014;25:272–281.
- Cruz-Migoni A, Canning P, Quevedo CE, et al. Structure-based development of new RAS-effector inhibitors from a combination of active and inactive RAS-binding compounds. Proc Natl Acad Sci U S A. 2019;116:2545–2550.
- Quevedo CE, Cruz-Migoni A, Bery N, et al. Small molecule inhibitors of RAS-effector protein interactions derived using an intracellular antibody fragment. Nat Commun. 2018;9:3169.
- Bery N, Cruz-Migoni A, Bataille CJ, et al. BRET-based RAS biosensors that show a novel small molecule is an inhibitor of RAS-effector protein-protein interactions. Elife. 2018;7:e37122.
- Welsch ME, Kaplan A, Chambers JM, et al. Multivalent small-molecule pan-RAS inhibitors. Cell. 2017;168:878–889 e829.
- Gupta AK, Wang X, Pagba CV, et al. Multi-target, ensemble-based virtual screening yields novel allosteric KRAS inhibitors at high success rate. Chem Biol Drug Des. 2019.
- McCarthy MJ, Pagba CV, Prakash P, et al. Discovery of high-affinity noncovalent allosteric KRAS inhibitors that disrupt effector binding. ACS Omega. 2019;4:2921–2930.
- Prior IA, Harding A, Yan J, et al. GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat Cell Biol. 2001;3:368–375.
- Li Z, Janosi L, Gorfe AA. Formation and domain partitioning of H-ras peptide nanoclusters: effects of peptide concentration and lipid composition. J Am Chem Soc. 2012;134:17278–17285.
- Janosi L, Li Z, Hancock JF, et al. Organization, dynamics, and segregation of Ras nanoclusters in membrane domains. Proc Natl Acad Sci U S A. 2012;109:8097–8102.
- Guldenhaupt J, Rudack T, Bachler P, et al. N-Ras forms dimers at POPC membranes. Biophys J. 2012;103:1585–1593.
- Muratcioglu S, Chavan TS, Freed BC, et al. GTP-Dependent K-Ras Dimerization. Structure. 2015;23:1325–1335.
- Li H, Gorfe AA. Aggregation of lipid-anchored full-length H-Ras in lipid bilayers: simulations with the MARTINI force field. PLoS One. 2013;8:e71018.
- Prakash P, Sayyed-Ahmad A, Cho KJ, et al. Computational and biochemical characterization of two partially overlapping interfaces and multiple weak-affinity K-Ras dimers. Sci Rep. 2017;7:40109.
- Sarkar-Banerjee S, Sayyed-Ahmad A, Prakash P, et al. Spatiotemporal analysis of K-Ras plasma membrane interactions reveals multiple high order homo-oligomeric complexes. J Am Chem Soc. 2017;139:13466–13475.
- Sayyed-Ahmad A, Cho KJ, Hancock JF, et al. Computational equilibrium thermodynamic and kinetic analysis of K-Ras dimerization through an effector binding surface suggests limited functional role. J Phys Chem B. 2016;120:8547–8556.
- Ambrogio C, Kohler J, Zhou ZW, et al. KRAS dimerization impacts MEK inhibitor sensitivity and oncogenic activity of mutant KRAS. Cell. 2018;172:857–868 e815.
- Spencer-Smith R, O’Bryan JP. Direct inhibition of RAS: Quest for the Holy Grail? Semin Cancer Biol. 2019;54:138–148.
- Kapoor S, Triola G, Vetter IR, et al. Revealing conformational substates of lipidated N-Ras protein by pressure modulation. Proc Natl Acad Sci U S A. 2012;109:460–465.
- Prakash P, Litwin D, Liang H, et al. Dynamics of membrane-bound G12V-KRAS from simulations and single-molecule FRET in native nanodiscs. Biophys J. 2019;116:179–183.
- Prakash P, Zhou Y, Liang H, et al. Oncogenic K-Ras binds to an anionic membrane in two distinct orientations: a molecular dynamics analysis. Biophys J. 2016;110:1125–1138.
- Abankwa D, Gorfe AA, Inder K, et al. Ras membrane orientation and nanodomain localization generate isoform diversity. Proc Natl Acad Sci U S A. 2010;107:1130–1135.
- Mazhab-Jafari MT, Marshall CB, Smith MJ, et al. Oncogenic and RASopathy-associated K-RAS mutations relieve membrane-dependent occlusion of the effector-binding site. Proc Natl Acad Sci U S A. 2015;112:6625–6630.
- Abankwa D, Hanzal-Bayer M, Ariotti N, et al. A novel switch region regulates H-ras membrane orientation and signal output. Embo J. 2008;27:727–735.
- Weise K, Kapoor S, Denter C, et al. Membrane-mediated induction and sorting of K-Ras microdomain signaling platforms. J Am Chem Soc. 2011;133:880–887.
- Fang Z, Marshall CB, Nishikawa T, et al. Inhibition of K-RAS4B by a unique mechanism of action: stabilizing membrane-dependent occlusion of the effector-binding site. Cell Chem Biol. 2018;25:1327–1336 e1324.
- Jansen JM, Wartchow C, Jahnke W, et al. Inhibition of prenylated KRAS in a lipid environment. PLoS One. 2017;12:e0174706.
- Flaherty KT, Puzanov I, Kim KB, et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N Engl J Med. 2010;363:809–819.
- Wagle N, Emery C, Berger MF, et al. Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol. 2011;29:3085–3096.
- Heidorn SJ, Milagre C, Whittaker S, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–221.
- Downward J. RAS synthetic lethal screens revisited: still seeking the elusive prize? Clin Cancer Res. 2015;21:1802–1809.
- Luo T, Masson K, Jaffe JD, et al. STK33 kinase inhibitor BRD-8899 has no effect on KRAS-dependent cancer cell viability. Proc Natl Acad Sci U S A. 2012;109:2860–2865.
- Ying H, Kimmelman AC, Lyssiotis CA, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670.
- Son J, Lyssiotis CA, Ying H, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–105.
- Bryant KL, Stalnecker CA, Zeitouni D, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2019;25:628–640.
- Lee CS, Lee LC, Yuan TL, et al. MAP kinase and autophagy pathways cooperate to maintain RAS mutant cancer cell survival. Proc Natl Acad Sci U S A. 2019;116(10):4508–4517.
- Kinsey CG, Camolotto SA, Boespflug AM, et al. Protective autophagy elicited by RAF–>MEK–>ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med. 2019;25:620–627.