
ABSTRACT
Oncogenic protein kinase C epsilon (PKCε) promotes the formation of membrane ruffles and motility in non-small cell lung cancer (NSCLC) cells. We found that PKCε is down-regulated when NSCLC cells undergo epithelial-to-mesenchymal transition (EMT) in response to TGF-β, thus becoming dispensable for migration and invasion in the mesenchymal state. PKCε silencing or inhibition leads to stress fibre formation, suggesting that this kinase negatively regulates RhoA activity. Ruffle formation induced by PKCε activation in the epithelial state is dependent on PI3K, but does not involve the PI3K-dependent Rac-GEFs Ect2, Trio, Vav2 or Tiam1, suggesting alternative Rac-GEFs as mediators of this response. In the proposed model, PKCε acts as a rheostat for Rho GTPases that differs in the epithelial and mesenchymal states.
Small GTPases, their effectors and regulators are subject to multiple mechanisms of control, including phosphorylation by protein kinases, which are capable of modifying their activity, subcellular localization, and expression. These regulatory events are key mechanisms by which small G-proteins in turn control a number of cellular functions, such as proliferation, gene expression, migration, and adhesion.
The Protein kinase C (PKC) family of kinases control signalling pathways involved in proliferation, migration, and invasion, as well as other processes that contribute to tumorigenesis and metastastic dissemination of cancer cells [Citation1–4]. Human PKCs comprise 10 different serine-threonine kinases classified into three groups: calcium/diacylglycerol (DAG)-dependent ‘classical/conventional’ PKCs (cPKCs α, βI, βII and γ), calcium-independent/DAG-regulated ‘novel’ PKCs (nPKCs δ, ε, η and θ), and calcium/DAG-insensitive ‘atypical’ PKCs (aPKCs ζ and ι). cPKCs and nPKCs represent the main cellular targets for phorbol ester tumour promoters, natural products that mimic the action of endogenous DAG and cause enzyme activation. It is well recognized that changes in expression and/or activity of PKC isozymes determine their roles either as cancer promoters or cancer suppressors in a stringent isozyme- and cell type-specific manner.
The DAG/phorbol ester responsive PKCε has been recognized as an oncogenic kinase and a biomarker for cancer progression [Citation5–7]. Multiple studies established that PKCε is aberrantly up-regulated in solid tumours such as prostate, breast, and lung cancer. Moreover, its overexpression has been linked to disease development and metastatic dissemination of tumour cells [Citation8,Citation9]. Studies in mouse models revealed that prostate-specific overexpression of PKCε, concomitant with the loss of the tumour suppressor Pten, leads to the formation of preneoplastic lesions and invasive adenocarcinomas [Citation10]. Furthermore, PKCε overexpressing/Pten-deficient prostate cancer cells display higher proliferative, migratory, and invasive phenotypes that are dependent on NF-κB-mediated production of the chemokine CXCL13 and COX-2-mediated production of prostaglandin E2 (PGE2) [Citation11,Citation12]. Additionally, PKCε facilitates transendothelial migration of prostate cancer cells and their seeding in the bone metastatic niche [Citation13].
The ability of PKCε to promote cancer cell motility has been tightly linked to the activation of Rho GTPases. Early studies identified RhoA and RhoC as downstream effectors for PKCε-mediated motility and invasion in head and neck squamous cell carcinoma (HNSCC) cells [Citation14]. A similar pro-metastatic role for PKCε via RhoC has been reported in triple-negative breast cancer cells [Citation9]. Previous work from our laboratory demonstrated that activation of PKCε is a crucial step for actin cytoskeletal reorganization via Rac1 in NSCLC cells. Stimulation of A549 lung cancer cells with phorbol 12-myristate 13-acetate (PMA) induces the striking formation of lamellipodia and membrane ruffles. The PMA effect can be impaired by silencing PKCε expression as well as by the specific PKCε inhibitor εV1-2, thus demonstrating the involvement of this kinase as a mediator of actin cytoskeleton reorganization in tumour cells. Moreover, the migratory, invasive, and metastatic capacities of NSCLC cells also rely on PKCε activation. In addition, ectopic expression of the Rac GTPase Activating protein (GAP) β2-chimaerin in NSCLC cells impairs Rac1 activation, ruffle formation, and cell motility [Citation15]. Similar to PMA, the synthetic DAG-lactone AJH-836, a DAG-mimetic compound that preferentially activates PKCε relative to other PKCs, promotes ruffle formation in NSCLC cells in a PKCε-depending manner [Citation16].
Searching for PKCε-regulated Rac-GEFs in NSCLC cells
Guanine nucleotide-exchange factors (GEFs) activate Rac1 by promoting the release of GDP from the inactive state resulting in the association of GTP and thus activation of Rac1 signalling. The Rho-GEF family comprises >80 members, and ~40 members of this family have been reported to have activity towards Rac1 [Citation17]. Based on structural considerations, Rho-GEFs can be separated into two classes: the Dbl and the DOCK GEFs. The Dbl class of GEFs has a DH (Dbl-homology) domain responsible for GEF activity, often found in tandem with a PH (pleckstrin-homology) domain acting primarily as a lipid-binding domain. PH domains have been associated with multiple cellular functions such as phosphoinositide-mediated membrane localization and allosteric modulation of GEF activity [Citation18]. Many Rac-GEFs are indeed dependent on the PI3K product PIP3 for their targeting and activation [Citation19]. On the other hand, DOCK GEFs hold two highly conserved regions known as DOCK-homology region 1 and 2 (DHR1 and DHR2), with DHR1 involved in phospholipid-binding and membrane targeting, and DHR2 responsible for nucleotide exchange activity [Citation20].
Taking into consideration the paramount relevance of Rac-GEFs in cancer cell motility, a logical inquiry was whether (and which) Rac-GEFs mediate Rac1 activation downstream of PKCε in NSCLC cells. To achieve this goal, we investigated if actin cytoskeleton reorganization induced by phorbol ester stimulation involves PI3K. These experiments revealed that pharmacological inhibition of PI3K with its specific inhibitor LY294002 abrogates ruffle formation by PMA in A549 cells (). Thus, our prediction is that PI3K-dependent Rac-GEFs potentially mediate this response. Towards this aim, we selected Rac-GEFs already reported to play important roles in NSCLC. Specifically, we focused on Ect2, Trio, Vav2 and Tiam1, which are recognized regulators of actin cytoskeleton reorganization in lung cancer cells [Citation21–24]. Each individual Rac-GEF was transiently silenced in A549 with specific RNAi duplexes, followed by the examination of membrane ruffle formation in response to PMA, which was determined using a densitometric approach [Citation16]. Unexpectedly, none of the selected Rac-GEF knockdowns had any inhibitory effect on ruffle formation (). This suggests that these PI3K-dependent Rac-GEFs do not mediate the PKCε response, and therefore do not contribute towards actin cytoskeleton reorganization in NSCLC cells in the context of the PKCε/PI3K/Rac1 pathway. We speculate that other Dbl Rac-GEFs, or alternatively selected members of the DOCK family, act as PKCε effectors for Rac1 activation and ruffle formation in NSCLC cells. Future studies to address this issue would include a systematic analysis of all PI3K-dependent Rac-GEFs expressed in NSCLC cells. Another likely scenario is that the lack of effect is due to compensatory roles by Rac-GEFs, and that simultaneous silencing of multiple Rac-GEFs would be required to unmask this potential effect. If experiments ultimately reveal that more than one Rac-GEF is required for ruffle formation/motility in this context, this may suggest distinctive involvement in the response due to differential intracellular localization of signalling activation. Finally, it is plausible that alternative PKCε-regulatory mechanisms involving Rac-GTPase Activating Proteins (Rac-GAPs), responsible for accelerating GTP hydrolysis, or Rho GDP-dissociation inhibitors (Rho-GDIs) are controlled by the PKCε/PI3K pathway. These regulatory processes may depend on direct phosphorylation by PKCε or may occur through indirect mechanisms. These multiple scenarios are currently under investigation in our laboratory.
Figure 1. LY294002 abrogates ruffle formation in NSCLC cells. A549 cells were serum starved for 24 h and then treated with PMA (0.1 μM, 30 min), in the absence or presence of the PI3K inhibitor LY294002 (20 μM, added 1 h before and kept during PMA stimulation). Cells were fixed and stained with rhodamine-phalloidin [Citation16]. Panel A. Representative micrographs of ruffle formation are shown. Panel B. Quantification of ruffle area/cell was done as previously described [Citation33]. Data are expressed as mean ± S.E.M. (n = 3). *, p < 0.05
![Figure 1. LY294002 abrogates ruffle formation in NSCLC cells. A549 cells were serum starved for 24 h and then treated with PMA (0.1 μM, 30 min), in the absence or presence of the PI3K inhibitor LY294002 (20 μM, added 1 h before and kept during PMA stimulation). Cells were fixed and stained with rhodamine-phalloidin [Citation16]. Panel A. Representative micrographs of ruffle formation are shown. Panel B. Quantification of ruffle area/cell was done as previously described [Citation33]. Data are expressed as mean ± S.E.M. (n = 3). *, p < 0.05](/cms/asset/82c65f6f-f9c1-4c40-8944-114f3f717693/ksgt_a_1684785_f0001_b.gif)
Figure 2. Effect of silencing specific Rac-GEFs on PMA-induced ruffle formation in NSCLC cells. A549 cells were transfected with specific RNAi duplexes (Dharmacon) targeting individual Rac-GEFs. After 24 h, cells were serum starved for an additional 24 h, and then treated with PMA (0.1 μM, 30 min). Ruffle formation was evaluated by microscopy after rhodamine-phalloidin staining. Panel A. Representative Q-PCR showing the specific depletion of Ect2, Trio, Vav2, and Tiam1. NTC, non-target control RNAi. ****, p < 0.0001. Panel B. Representative micrographs of ruffle formation are shown. Panel C. Quantification of ruffle area/cell was done as previously described [Citation16]. Results are expressed as percentage relative to the PMA response in parental (mocked-transfected) cells (dotted line). Data are expressed as mean ± S.E.M. (n = 3). **, p < 0.01
![Figure 2. Effect of silencing specific Rac-GEFs on PMA-induced ruffle formation in NSCLC cells. A549 cells were transfected with specific RNAi duplexes (Dharmacon) targeting individual Rac-GEFs. After 24 h, cells were serum starved for an additional 24 h, and then treated with PMA (0.1 μM, 30 min). Ruffle formation was evaluated by microscopy after rhodamine-phalloidin staining. Panel A. Representative Q-PCR showing the specific depletion of Ect2, Trio, Vav2, and Tiam1. NTC, non-target control RNAi. ****, p < 0.0001. Panel B. Representative micrographs of ruffle formation are shown. Panel C. Quantification of ruffle area/cell was done as previously described [Citation16]. Results are expressed as percentage relative to the PMA response in parental (mocked-transfected) cells (dotted line). Data are expressed as mean ± S.E.M. (n = 3). **, p < 0.01](/cms/asset/dfe0934f-403e-4a44-bfb5-745bba726c4a/ksgt_a_1684785_f0002_b.gif)
The PKCε paradox in epithelial-to-mesenchymal transition (EMT)
In order to escape the primary tumour, cancer cells undergo a morphological transformation that abolishes cell-cell contacts and enhances their invasive capacity. This process, known as epithelial-to-mesenchymal transformation (EMT), is driven by transcription factors, including Snail, Slug, Twist1, Zeb1, and Zeb2. Loss of E-cadherin is a hallmark of EMT and contributes to the transformation of the cuboidal epithelial shape into a fusiform and elongated mesenchymal morphology. Prominent alterations in the actin cytoskeleton structure that involve the action of specific Rho GTPases occur during EMT [Citation25,Citation26].
Several studies have described important roles for individual members of the PKC family, including PKCε, in the regulation of EMT [Citation27–30]. In prostate cancer, the microRNA miR-205 impairs PKCε expression, which in turns has a suppressive effect on the EMT process, leading to reduced migration and invasion [Citation31]. Furthermore, overexpression of PKCε in a non-tumorigenic mammary cell line triggers significant changes in the expression of EMT markers consistent with a mesenchymal phenotype, and induces cell migration and resistance to anoikis [Citation32]. In a recent study, we investigated the potential relationship between PKCε and EMT in NSCLC cells [Citation33]. Unlike other cancers, PKCε is dispensable for EMT in NSCLC cells induced by transforming growth factor-β (TGF-β), as revealed by the lack of effect of PKCε silencing on cell morphology and the expression of E-cadherin. Similar results have been observed for other DAG/phorbol ester responsive PKCs expressed in NSCLC cells, namely PKCα and PKCδ. Along the same line, none of these PKCs were shown to be involved in the activation of early steps in the TGF-β signalling cascade, such as phosphorylation of Smad2/3 [Citation34].
An unexpected finding from our studies was the pronounced down-regulation of PKCε expression during mesenchymal transformation. Notably, TGF-β treatment caused ~80% reduction in PKCε protein levels without affecting the expression of other PKCs. Moreover, despite the observed PKCε down-regulation, there was a marked increase in basal Rac1-GTP levels in TGF-β-treated NSCLC cells. Taking into consideration the established PKCε requirement for motility and invasion of NSCLC cells in an ‘epithelial’ state, these results were counterintuitive and argue for paradoxical roles of PKCε in epithelial or mesenchymal NSCLC cells [Citation34].
The elevated activation of Rac1 in mesenchymally transformed NSCLC cells prompted us to examine the expression pattern of Rac-GEFs. Analysis of the expression of 26 Rac-GEFs revealed significant changes after TGF-β treatment, mainly up-regulation of the Dbl Rac-GEFs NGEF, RasGRF2, Tiam2, Trio, and Vav2, as well as the DOCK GEFs DOCK2 and DOCK4. Re-expression of PKCε in mesenchymally-transformed A549 cells either reverse or have no effect on Rac-GEF up-regulation (see examples for DOCK4 and Trio in ), suggesting that these changes could be either dependent or independent of PKCε (). Among these Rac-GEFs, Trio and Tiam2 have been implicated in the migratory activity of mesenchymally transformed NSCLC cells [Citation34].
Figure 3. Differential regulation of selected Rac-GEFs by PKCε in NSCLC cells. Epithelial A549 cells were treated with TGF-β (10 ng/ml, 6 days). TGF-β transformed cells were transfected with either pcDNA-PKCε-FLAG or empty vector, and 30 h later expression levels for DOCK4 and Trio were compared to epithelial A549 cells. Panel A. Protein levels of DOCK4 and Trio were determined by western blot using specific antibodies. A representative experiment is shown. Panel B. Densitometric analysis, normalized to actin. Data are expressed as percentage expression in mesenchymal cells relative to epithelial cells, and represent the mean ± S.E.M. (n = 3). Dotted line, epithelial A549 cells. *, p < 0.05; **, p < 0.01; n.s. non significant
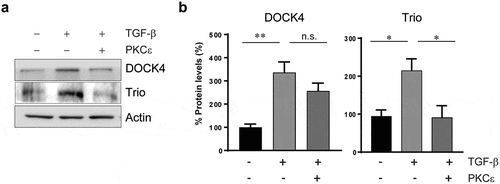
A striking observation is that PMA failed to induce ruffles in TGF-β-treated NSCLC cells, unlike we previously described for cells in the ‘epithelial’ state. This may be a consequence of the down-regulated PKCε expression after TGF-β treatment [Citation34]. Thus, a possible scenario is that the PKCε/PI3K/Rac1 axis observed in ‘epithelial’ NSCLC cells is absent in the mesenchymal state. Rac1 activation may involve distinctive Rac-GEFs in epithelial and mesenchymal NSCLC cells. In this regard, Van Aelst and co-workers reported that DOCK4 up-regulation by TGF-β in lung adenocarcinoma cells is crucially involved in Rac1-dependent invasion and metastasis, a result that unveils a specific Rac1-dependent mechanism in mesenchymally transformed cells [Citation35].
PKCε as a negative RhoA regulator
A detailed analysis of epithelial NSCLC cells revealed that PKCε RNAi silencing caused major changes in the actin cytoskeletal network. Intriguingly, a significant increase in stress fibres was observed upon PKCε depletion, as recapitulated by PKCε inhibition with εV1-2 [Citation15]. Likewise, mesenchymally transformed NSCLC cells, which have down-regulated PKCε expression, display a prominent stress fibre network [Citation34]. Since RhoA plays a major role in stress fibre formation [Citation36–38], we therefore examined the possibility that loss of PKCε expression and/or activity results in elevated RhoA activity. Quite remarkably, treatment of ‘epithelial’ NSCLC cells with εV1-2 increased RhoA-GTP levels [Citation34]. Altogether, these results support the concept that there is an inverse correlation between the activation status of PKCε and RhoA. Although the mechanisms behind this effect are not known, one possibility is that PKCε negatively regulates the activity of GEFs responsible for RhoA activation. As discussed above, mesenchymally transformed cells up-regulate NGEF, Trio and Vav2, GEFs that not only display exchange activity towards Rac1 but also towards RhoA [Citation39–41]. Other Rho-GEFs are also up-regulated in TGF-β-transformed NSCLC cells, specifically ARHGEF1, ARHGEF2, ARHGEF11, ARHGEF12, ARHGEF28 and Net1 (). Functional studies would be required to determine whether these Rho-GEFs are functionally linked to stress fibre formation via RhoA in NSCLC cells. It is also conceivable that the activity of Rho-GAPs responsible for RhoA inactivation is differentially regulated in epithelial and mesenchymal NSCLC cells. Indeed, Rho-GAPs could be tightly regulated via phosphorylation mechanisms, including by PKC isozymes [Citation42–45].
Table 1. Expression of selected Rho-GEFs in ‘epithelial’ vs. ‘mesenchymal’ NSCLC cells. mRNA was obtained from A549 cells before (‘epithelial’) and after TGF-β treatment (10 ng/ml) for 6 days (‘mesenchymal’). cDNA was prepared by reverse transcription, and expression of Rho-GEFs was determined by Q-PCR. Results were normalized to a housekeeping gene (UBC). Results are expressed as fold-change in ‘mesenchymal’ cells relative to ‘epithelial’ cells. Data represents the mean ± S.E.M. of 3 independent experiments
An alternative mechanism that may explain the elevated RhoA activity in PKCε-depleted/inhibited cells is a change in RhoA post-translational modifications. RhoA is subject to phosphorylation, ubiquitylation, SUMOylation, and AMPylation [Citation46–48]. For example, studies have shown that kinases such as PKA or PKG modify the activity and stability of RhoA via phosphorylation [Citation48,Citation49]. Remarkably, an inverse correlation between RhoA and PKCε expression levels has been found during megakaryocyte differentiation, suggesting a potential role for this kinase in promoting RhoA degradation. Lastly, several studies have suggested ubiquitylation as a potential mechanism that controls RhoA expression through proteasomal degradation [Citation50]. Besides, TGF-β-regulated ubiquitylation enzymes, such as SMURF1, USP15, or Nedd4L, may also contribute to control the PKCε-RhoA interplay [Citation51–54].
Future perspectives
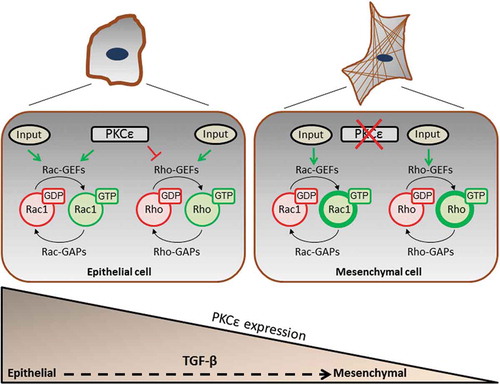
The distinctive involvement of PKCε in the control of actin cytoskeleton reorganization and Rho GTPase function in epithelial and mesenchymal NSCLC cells represents a conceptually novel notion in cell biology. A scheme depicting the key events involved in this paradigm is shown in . The inverse correlation between PKCε and RhoA activation may have significant impact on cell morphology and polarity, and regulation of cancer cells metastatic dissemination. A major challenge would be to elucidate the mechanisms by which PKCε controls Rac1 activation in epithelial NSCLC cells. We predict that PKCε activates specific Rac-GEFs, either by direct phosphorylation or indirectly by controlling their association with specific partners and subcellular localization. To address this issue, it would be important to identify which Dbl-GEFs and/or DOCK-GEFs are expressed in NSCLC cells, followed by a thorough functional analysis to determine their involvement in motility signalling. Since Rac1 controls a number of functions in addition to actin reorganization, such as gene expression and cell metabolism, a conceivable scenario is that individual Rac-GEFs control diverse subcellular pools of Rac1 involved in specific cellular functions [Citation55]. Indeed, Rac-GEFs display a characteristic pattern of subcellular distribution, such as cytoplasmic, nuclear, and perinuclear membrane [Citation20]. Different stimuli involving PKCε activation or other signalling mediators may result in unique patterns of Rac-GEF re-localization depending on particular lipid- and protein-protein associations. In a broader context, the wide complexity of NSCLC genetic driver mutations (e.g. KRas, EGFR, BRaf), translocations (e.g. ELM4-Alk) and genomic amplifications (c-Met) may result in the utilization of different Rac-GEF-mediated pathways and intracellular Rac1 pools. Deciphering this intricate network represents a major challenge towards the identification and characterization of novel cancer-type specific invasion biomarkers in NSCLC.
Figure 4. Model for PKCε regulation of small GTPases in NSCLC cells. In the epithelial state, PKCε promotes Rac1 activation and inhibits RhoA. PKCε is down-regulated in the ‘mesenchymal’ state, becoming a permissive signal for Rho activation and stress fibre formation
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer. 2007;7(4):281–294.
- Isakov N. Protein kinase C (PKC) isoforms in cancer, tumor promotion and tumor suppression. Semin Cancer Biol. 2018;48:36–52.
- Wu-Zhang AX, Newton AC. Protein kinase C pharmacology: refining the toolbox. Biochem J. 2013;452(2):195–209.
- Garg R, Benedetti LG, Abera MB, et al. Protein kinase C and cancer: what we know and what we do not. Oncogene. 2014;33(45):5225–5237.
- Toton E, Ignatowicz E, Skrzeczkowska K, et al. Protein kinase cepsilon as a cancer marker and target for anticancer therapy. Pharmacol Rep. 2011;63(1):19–29.
- Mischak H, Goodnight JA, Kolch W, et al. Overexpression of protein kinase C-delta and -epsilon in NIH 3T3 cells induces opposite effects on growth, morphology, anchorage dependence, and tumorigenicity. J Biol Chem. 1993;268(9):6090–6096.
- Perletti GP, Concari P, Brusaferri S, et al. Protein kinase cepsilon is oncogenic in colon epithelial cells by interaction with the ras signal transduction pathway. Oncogene. 1998;16(25):3345–3348.
- Bae KM, Wang H, Jiang G, et al. Protein kinase C epsilon is overexpressed in primary human non-small cell lung cancers and functionally required for proliferation of non-small cell lung cancer cells in a p21/Cip1-dependent manner. Cancer Res. 2007;67(13):6053–6063.
- Pan Q, Bao LW, Kleer CG, et al. Protein kinase C epsilon is a predictive biomarker of aggressive breast cancer and a validated target for RNA interference anticancer therapy. Cancer Res. 2005;65(18):8366–8371.
- Garg R, Blando JM, Perez CJ, et al. Protein kinase C epsilon cooperates with PTEN loss for prostate tumorigenesis through the CXCL13-CXCR5 pathway. Cell Rep. 2017;19(2):375–388.
- Garg R, Blando J, Perez CJ, et al. Activation of nuclear factor kappaB (NF-kappaB) in prostate cancer is mediated by protein kinase C epsilon (PKCepsilon). J Biol Chem. 2012;287(44):37570–37582.
- Garg R, Blando JM, Perez CJ, et al. COX-2 mediates pro-tumorigenic effects of PKCepsilon in prostate cancer. Oncogene. 2018;37:4735–4749.
- Gutierrez-Uzquiza A, Lopez-Haber C, Jernigan DL, et al. PKCepsilon is an essential mediator of prostate cancer bone metastasis. Mol Cancer Res. 2015;13(9):1336–1346.
- Pan Q, Bao LW, Teknos TN, et al. Targeted disruption of protein kinase C epsilon reduces cell invasion and motility through inactivation of RhoA and RhoC GTPases in head and neck squamous cell carcinoma. Cancer Res. 2006;66(19):9379–9384.
- Caino MC, Lopez-Haber C, Kissil JL, et al. Non-small cell lung carcinoma cell motility, rac activation and metastatic dissemination are mediated by protein kinase C epsilon. PloS One. 2012;7(2):e31714.
- Cooke M, Zhou X, Casado-Medrano V, et al. Characterization of AJH-836, a DAG-lactone with selectivity for novel PKC isozymes. J Biol Chem. 2018;293:8330–8341.
- Marei H, Malliri A. GEFs: dual regulation of Rac1 signaling. Small GTPases. 2017;8(2):90–99.
- Reckel S, Gehin C, Tardivon D, et al. Structural and functional dissection of the DH and PH domains of oncogenic Bcr-Abl tyrosine kinase. Nat Commun. 2017;8(1):2101.
- Campa CC, Ciraolo E, Ghigo A, et al. Crossroads of PI3K and Rac pathways. Small GTPases. 2015;6(2):71–80.
- Lawson CD, Ridley AJ. Rho GTPase signaling complexes in cell migration and invasion. J Cell Biol. 2018;217(2):447–457.
- Zhu G, Zhang Y, Wang Q, et al. The prognostic value of Tiam1 correlates with its roles in epithelial-mesenchymal transition progression and angiogenesis in lung adenocarcinoma. Cancer Manag Res. 2019;11:1741–1752.
- Baker MJ, Cooke M, Kazanietz MG. Nuclear PKCiota-ECT2-Rac1 and ribosome biogenesis: a novel axis in lung tumorigenesis. Cancer Cell. 2017;31(2):167–169.
- Justilien V, Ali SA, Jamieson L, et al. Ect2-dependent rRNA synthesis is required for KRAS-TRP53-driven lung adenocarcinoma. Cancer Cell. 2017;31(2):256–269.
- Havel LS, Kline ER, Salgueiro AM, et al. Vimentin regulates lung cancer cell adhesion through a VAV2-Rac1 pathway to control focal adhesion kinase activity. Oncogene. 2015;34(15):1979–1990.
- Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119(6):1420–1428.
- Stemmler MP, Eccles RL, Brabletz S, et al. Non-redundant functions of EMT transcription factors. Nat Cell Biol. 2019;21(1):102–112.
- Pham TND, Perez White BE, Zhao H, et al. Protein kinase C alpha enhances migration of breast cancer cells through FOXC2-mediated repression of p120-catenin. BMC Cancer. 2017;17(1):832.
- Abera MB, Kazanietz MG. Protein kinase Calpha mediates erlotinib resistance in lung cancer cells. Mol Pharmacol. 2015;87(5):832–841.
- Ouelaa-Benslama R, De Wever O, Hendrix A, et al. Identification of a GalphaGbetagamma, AKT and PKCalpha signalome associated with invasive growth in two genetic models of human breast cancer cell epithelial-to-mesenchymal transition. Int J Oncol. 2012;41(1):189–200.
- Ghoul A, Serova M, Astorgues-Xerri L, et al. Epithelial-to-mesenchymal transition and resistance to ingenol 3-angelate, a novel protein kinase C modulator, in colon cancer cells. Cancer Res. 2009;69(10):4260–4269.
- Gandellini P, Folini M, Longoni N, et al. miR-205 Exerts tumor-suppressive functions in human prostate through down-regulation of protein kinase Cepsilon. Cancer Res. 2009;69(6):2287–2295.
- Jain K, Basu A. Protein kinase C-epsilon promotes EMT in breast cancer. Breast Cancer. 2014;8:61–67.
- Casado-Medrano V, Barrio-Real L, Wang A, et al. Distinctive requirement of PKCepsilon in the control of Rho GTPases in epithelial and mesenchymally transformed lung cancer cells. Oncogene. 2019;38:5396–5412.
- Brown KA, Aakre ME, Gorska AE, et al. Induction by transforming growth factor-beta1 of epithelial to mesenchymal transition is a rare event in vitro. BCR. 2004;6(3):R215–31.
- Yu JR, Tai Y, Jin Y, et al. TGF-beta/smad signaling through DOCK4 facilitates lung adenocarcinoma metastasis. Genes Dev. 2015;29(3):250–261.
- Burridge K, Guilluy C. Focal adhesions, stress fibers and mechanical tension. Exp Cell Res. 2016;343(1):14–20.
- Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol. 1996;133(6):1403–1415.
- Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70(3):389–399.
- Shamah SM, Lin MZ, Goldberg JL, et al. EphA receptors regulate growth cone dynamics through the novel guanine nucleotide exchange factor ephexin. Cell. 2001;105(2):233–244.
- Blangy A, Vignal E, Schmidt S, et al. TrioGEF1 controls Rac- and Cdc42-dependent cell structures through the direct activation of rhoG. J Cell Sci. 2000;113(Pt 4):729–739.
- Abe K, Rossman KL, Liu B, et al. Vav2 is an activator of Cdc42, Rac1, and RhoA. J Biol Chem. 2000;275(14):10141–10149.
- Holinstat M, Mehta D, Kozasa T, et al. Protein kinase Calpha-induced p115RhoGEF phosphorylation signals endothelial cytoskeletal rearrangement. J Biol Chem. 2003;278(31):28793–28798.
- Jiang W, Betson M, Mulloy R, et al. p190A RhoGAP is a glycogen synthase kinase-3-beta substrate required for polarized cell migration. J Biol Chem. 2008;283(30):20978–20988.
- Levay M, Settleman J, Ligeti E. Regulation of the substrate preference of p190RhoGAP by protein kinase C-mediated phosphorylation of a phospholipid binding site. Biochemistry. 2009;48(36):8615–8623.
- Tripathi BK, Grant T, Qian X, et al. Receptor tyrosine kinase activation of RhoA is mediated by AKT phosphorylation of DLC1. J Cell Biol. 2017;216(12):4255–4270.
- Olson MF. Rho GTPases, their post-translational modifications, disease-associated mutations and pharmacological inhibitors. Small GTPases. 2018;9(3):203–215.
- Lang P, Gesbert F, Delespine-Carmagnat M, et al. Protein kinase A phosphorylation of RhoA mediates the morphological and functional effects of cyclic AMP in cytotoxic lymphocytes. EMBO J. 1996;15(3):510–519.
- Su T, Straight S, Bao L, et al. PKC epsilon phosphorylates and mediates the cell membrane localization of RhoA. ISRN Oncol. 2013;2013:329063.
- Kato K, Yazawa T, Taki K, et al. The inositol 5-phosphatase SHIP2 is an effector of RhoA and is involved in cell polarity and migration. Mol Biol Cell. 2012;23(13):2593–2604.
- Asanuma K, Yanagida-Asanuma E, Faul C, et al. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol. 2006;8(5):485–491.
- Jacob T, Van den Broeke C, Van Waesberghe C, et al. Pseudorabies virus US3 triggers RhoA phosphorylation to reorganize the actin cytoskeleton. J Gen Virol. 2015;96(8):2328–2335.
- Zhu H, Kavsak P, Abdollah S, et al. A SMAD ubiquitin ligase targets the BMP pathway and affects embryonic pattern formation. Nature. 1999;400(6745):687–693.
- Liu WT, Huang KY, Lu MC, et al. TGF-beta upregulates the translation of USP15 via the PI3K/AKT pathway to promote p53 stability. Oncogene. 2017;36(19):2715–2723.
- Qu MH, Han C, Srivastava AK, et al. miR-93 promotes TGF-beta-induced epithelial-to-mesenchymal transition through downregulation of NEDD4L in lung cancer cells. Tumour Biol. 2016;37(4):5645–5651.
- Payapilly A, Malliri A. Compartmentalisation of RAC1 signalling. Curr Opin Cell Biol. 2018;54:50–56.