
ABSTRACT
While Rho-signalling controlling vascular contraction is a canonical mechanism, with the modern approaches used in research, we are advancing our understanding and details into this pathway are often uncovered. RhoA-mediated Rho-kinase is the major regulator of vascular smooth muscle cells and a key player manoeuvring other functions in these cells. The discovery of new interactions, such as oxidative stress and hydrogen sulphide with Rho signalling are emerging addition not only in the physiology of the smooth muscle, but especially in the pathophysiology of vascular diseases. Likewise, the interplay between ageing and Rho-kinase in the vasculature has been recently considered. Importantly, in smooth muscle contraction, this pathway may also be affected by sex hormones, and consequently, sex-differences. This review provides an overview of Rho signalling mediating vascular contraction and focuses on recent topics discussed in the literature affecting this pathway such as ageing, sex differences and oxidative stress.
Introduction
Smooth muscle cells (SMCs) are essential components of the visceral organs and the walls of blood vessels. These cells not only keep the tonus but also drive active alterations in the volume and size of hollow organs, except for the heart, which has a unique type of striated muscle. SMCs are a highly specialized type of cell with contraction as their main function. To achieve their primary purpose, SMCs have developed mechanisms that allow for mechanical coupling. These mechanisms involve many different proteins and free calcium (Ca2+), a key determinant of many physiological functions. Smooth muscle contraction is triggered, and to a lesser extent sustained, by an increase in the concentration of intracellular free Ca2+. Accordingly, in SMCs, many signalling pathways stimulate the interaction between Ca2+ and calmodulin (Cam), an intermediate calcium-binding messenger protein, which in turn, leads to the phosphorylation of myosin light chain (MLC) by activation of the enzyme MLC kinase (MLCK), ultimately resulting in contraction. Additionally, alterations in Ca2+ sensitivity, a phenomenon leading to a higher force of contraction at an equal concentration of intracellular Ca2+ [Citation1], regulates this mechanical coupling process. It is widely accepted that Ca2+ sensitization arises via one specific monomeric G protein from the Rho family and that this pathway actively enables smooth muscle contraction.
Rho proteins are part of the larger Ras (Rat sarcoma virus) superfamily of guanosine triphosphate hydrolase enzyme (GTPase), which includes more than 100 members, all considered crucial regulators of many physiological mechanisms that affect cell behaviour (for review, see [Citation2]). These GTPases cycles between two modes, an active (GTP) and an inactive (GDP) state. Their activation process is catalysed by guanine-nucleotide exchange factors (GEFs). Among the Rho proteins, the small monomeric GTPase, RhoA (RAS homolog family, member A), is a well-recognized regulator of many aspects related to cellular process, including changes in the actin cytoskeleton, and consequently, smooth muscle tone regulation. Of note, there are many GTPases involved in smooth muscle functions, but this review focus on RhoA-mediated influence on vascular smooth muscle contraction. RhoA in association with its effector, Rho-kinase, is a chief orchestrator of vascular contraction by Ca2+ sensitization. While Rho-kinase has two isoforms known as ROCK1 (ROKβ) and ROCK2 (ROKα), we only refer to the nomenclature ROCK in this review when specifying its subtype. Currently, substantial knowledge about the function of RhoA/Rho-kinase in the regulation of vascular smooth muscle is available. However, it is still insufficient, especially with regards to changes in this pathway that are driven by the process of ageing or by sex-difference. Likewise, the interplay between Rho-signalling and oxidative stress in the smooth muscle contraction is still poorly understood, and it is notably highlighted in serious vascular diseases. Here, we provide an overview of the physiological mechanism by which Rho signalling critically regulates vascular contraction, emphasizing the crosstalk between this pathway and three new important variables currently discussed in the literature, oxidative stress, ageing, and sex.
Vascular smooth muscle contraction
Smooth muscle shares anatomical characteristics with skeletal and cardiac muscles, such as the contractile proteins actin and myosin. However, they differ in many physiological processes leading to contraction. Specifically, SMCs, longitudinally or circularly arranged in layers, constitute the tubes and walls of many organs within the body. The primary function of this type of muscular tissue is force generation. These cells use cross-bridge cycling between actin and myosin, and a change in myosin, the thick filament, initiates this process. The contractile response happens by the sliding of actin over myosin filaments, shortening the SMC. Moreover, SMCs express many other proteins that participate in the contractile process, which are affected not only by hormones but also by local chemical signals.
Overall, contraction in vessels lead to the generation of a Ca2+ -dependent biphasic curve, which is characterized by a fast- phasic- and a slow- tonic- component [Citation3]. The former is mediated by Ca2+ efflux from the sarcoplasmic reticulum, and the latter is sustained by Ca2+ influx from the extracellular milieu [Citation4,Citation5]. Briefly, this ion triggers the underlying signalling pathways foremost smooth muscle contraction. The process is initiated to a more significant extent by agonists that can induce contraction without membrane depolarization. This well-known contractile mechanism includes a G protein-coupled receptor that is activated when bound by hormones or neurotransmitters. Following this process, that is primarily the conversion of phosphatidylinositol 4,5-bisphosphate (PIP2) into inositol trisphosphate (IP3), diacylglycerol (DAG) and IP3 together help promote the release of Ca2+ from the sarcoplasmic reticulum and further elevates intracellular Ca2+ levels. Ca2+ entering through membranes is a key mechanism to many crucial physiological processes. In the SMC, the high influx of Ca2+ into the cytosol binds and activates Cam, a Ca2+ binding protein that plays a central role in this event. When bound to Ca2+, this protein mediates contraction by activating the actin bound myofilament enzyme, MLCK, which must phosphorylate the 20-kDa MLC of myosin within sarcomeres, allowing the interaction between myosin and actin (). Conversely, the phosphorylated MLC can be dephosphorylated by the enzyme MLC phosphatase (MLCP), ultimately resulting in a reversal of contraction [Citation1,Citation6].
Figure 1. Canonical pathways in smooth muscle contraction. GPCR is known to be involved in smooth muscle contraction through two main mechanisms. Once activated, the α subunit associated with the receptor dissociates and activates phospholipase C (PLC), which functions to convert phosphatidylinositol 4,5-bisphosphate (PIP2) to inositol trisphosphate (IP3). IP3 further induces the release of Ca2+ from the sarcoplasmic reticulum and thus results in an increase in intracellular Ca2+ levels. Ca2+ binds to calmodulin (CaM) which activates myosin light chain kinase (MLCK), responsible for phosphorylating myosin light chain (MLC), therefore inducing contraction. Moreover, contraction could also be promoted through Ca2+ induced activation of PKC. This stimulates activation of CPI, an inhibitor of myosin light chain phosphatase (MLCP), consequently preventing the removal of the phosphate group and terminating contraction. The second mechanism entails GPCR activation of a GEF that works to activate Rho through its conversion from Rho-GDP to Rho-GTP. Rho further activates its associated kinases, ROCK1 and ROCK2, which have shown capabilities of inhibiting eNOS, consequently decreasing NO production and relaxation. ROCK1/2 can also promote CPI and directly inhibit MLCP, inducing contraction. Various studies have shown the regulatory mechanisms of RhoE, GEM, and arachidonic acid on RhoA and RhoA-kinase. Other possible regulators are currently still under investigation. Created with BioRender.com
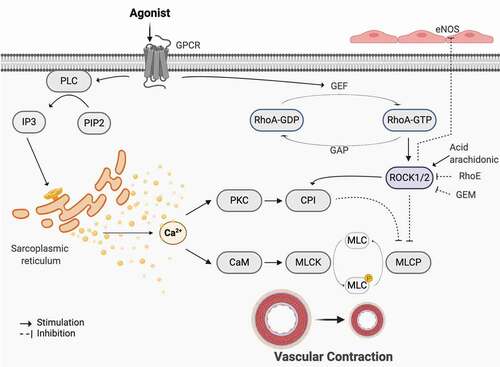
Not only fluctuation in cytosolic calcium affects different pathways, including Rho-signalling, but those pathways can also influence Ca2+ handling. To date, it is undeniable that the cytosolic levels of Ca2+, as well as how the contractile muscle proteins sense transient changes in the levels of this ion, a process known as calcium sensitization [Citation1], regulate smooth muscle contraction. Calcium sensitization maintains force generation in the vessels following dissipation of the initial Ca2+ signal. Rho signalling, a family of small GPTases predominantly located at the plasma membrane [Citation7], are very important in this process.
RhoA, RhoB, and RhoC encompass the Rho subfamily within the Rho GTPase family and are all expressed in vascular smooth muscle cells. Although they are closely related, RhoA is widely accepted as necessary for smooth muscle contraction, especially in blood vessels, and also it is the most studied in the vasculature. Considering the vasculature, where the smooth muscle is present in circumferential layers, these cells are essential because when contracted, the diameter of the vessel decreases, which plays an important role in blood flow regulation, and therefore, blood pressure. Additionally, the force generated by the smooth muscle mechanical action is essential to support the basal vessel tone. Hence, a delicate balance between contraction and relaxation is crucial for maintaining vascular physiology and body homoeostasis.
RhoA/Rho-kinase signalling in vascular smooth muscle contraction
Rho proteins are molecular switches with two well-defined stage: an inactive state (GDP-bound) and an active state (GTP-bound). Mostly, membrane receptors activate G-protein that is coupled to Rho through guanine nucleotide exchange factors (GEFs). GEFs are multidomain proteins that are regulated by extracellular signals. The negative regulation of Rho can be not only by GTPase activating proteins (GAPs) that stimulates its intrinsic GTPase activity through hydrolysis of GTP, but also by guanidine nucleotide dissociation inhibitors (GDIs), which removes Rho proteins from the membrane [Citation8]. GDI also prevents GTPase activity and inhibits GAPs.
The translocation of RhoA to the membrane and its downstream target are role players in Ca2+ sensitization and contraction [Citation9]. RhoA-GTP activates a downstream target, which phosphorylates MYPT1, the myosin targeting subunit of MLCP, resulting in inhibition of the phosphatase [Citation10]. As pointed out, contraction is initiated by an increase in the levels of intracellular Ca2+ and by the Ca2+ sensitization event. The former activates MLCK, and the latter is achieved, partly, by inhibition of MLCP [Citation11,Citation12]. Consequently, the balance between MLCK and MLCP activation determines relaxation or contraction, as extensively reviewed elsewhere [Citation5,Citation13]. The most detailed Rho downstream target effector molecule in functional research is Rho-kinase, a critical downstream effector of RhoA and a versatile regulator of smooth muscle contraction [Citation14,Citation15]. Rho-kinase has two isoforms, known as ROCK1 (ROKβ) and ROCK2 (ROKα), which are located in chromosome 18 and 12, respectively. Noteworthy, not only RhoA but also other small G proteins such as RhoE, Gem, and Rad can modulate Rho-kinase [Citation16]. While the mechanisms for inhibition of Rho-kinase by Gem and Rad are not well-defined, RhoE inhibits ROCK1, avoiding the attachment of RhoA, by binding to its N-terminal catalytic domain [Citation17,Citation18].
Rho-kinase fits in the group of serine/threonine protein kinases, and its molecular weight is about 160 kDa. It is distributed throughout the body and expressed in many tissues, reflecting, its vast diversity in cellular localization [Citation14]. These two kinases, ROCK1 and ROCK2, have 92% similarity in their kinase domains and 65% homology in their amino acid sequence [Citation19]. Both isoforms are widely expressed in vertebrates vascular SMCs, and endothelial cells. Additionally, they play a role not only in physiological conditions but also in many vascular diseases, such as hypertension, diabetes, atherosclerosis and erectile dysfunction [Citation20–25]. While ROCK2 is greatly expressed in the brain and skeletal muscle [Citation19], SMCs express both ROCK1 and ROCK2 [Citation26], in spite of the fact that ROCK2 is the major isoform regulating vascular contractility [Citation15]. Considering that ROCK1(-/-) KO animals present birth defects and that ROCK2(-/-) KO animals die embryonically due to placental dysfunction [Citation27,Citation28], it is reasonable to assume that according to the type of cells, ROCK1 and ROCK2 might affect independent mechanisms, as suggested by other studies [Citation29]. The tissue prevalence of different isoforms of Rho-kinase in the organs is a branch of investigation about this enzyme that raised in the last decade. While the advance of the technology used in science, through the use of inhibitors, knockdown, and germ-line knockout, enhanced our knowledge about Rho-signalling, we still need to overcome many limitations, such as off-target effects.
The key element in smooth muscle contraction is the balance between MLCK and MLCP. Thus, alterations in this balance favouring phosphorylation of MLC lead to an enhancement in the contractile mechanism of the vascular layer [Citation30]. MLCP embraces three subunits: a catalytic subunit called PP1cδ (major enzyme in MLC20 dephosphorylation), the regulatory subunit MYPT1, and a 20 kDa subunit of unknown function. MYPT1 modulates Ca2+-dependent phosphorylation of MLC by MLCK. Rho-kinase can affect MLC via two mechanisms, as follows: direct or indirect phosphorylation of MLC [Citation31], the latter is achieved by phosphorylating MYPT1 and inactivating it. MLCP inactivation is the product of the phosphorylation of Thr696 and Thr850, two inhibitory sites on MYPT1 [Citation32]. Additionally, another phosphorylation-dependent inhibitory protein of MLCP, CPI-17, which is specifically expressed in smooth muscle [Citation33], can be activated by Rho-kinase, or protein kinase C (PKC) independently of RhoA/Rho-kinase [Citation34,Citation35].
Activation of CPI-17 by Rho-kinase, inhibits MLCP, and consequently, leads to smooth muscle contraction [Citation36]. Therefore, MYPT1 and CPI-17 phosphorylation are key factors in sensitizing smooth muscle to Ca2+ [Citation37]. Interestingly, MYPT1 has many serine and threonine phosphorylation sites, six are involved in smooth muscle contractility, and MYPT1 inhibitory mechanisms can diverge between smooth muscles [for review, see [Citation38]]. Additionally, evidence suggests that Rho-kinase might regulated not only at epigenetic level in smooth muscle cells [Citation39], but also at the transcriptional level, as observed in vascular smooth muscle in response to AngII [Citation40,Citation41]. Likewise, lipids such as the arachidonic acid can also regulate Rho-kinase by increasing its activity through the C-terminal regulatory region [Citation42]. Noteworthy, a great deal of emerging evidence shows that the redox sensitivity of Rho GTPases and ROCK undergo post-translational oxidative modifications [for review see [Citation43]]. In physiological conditions this is reversible, and ROS can also be generated by many stimuli, including GPCR agonists, being recognized as bona fide second messengers. Nonetheless, in many diseases ROS acts as damaging bi-products and can influence pro-contractile pathways in which altered vascular reactivity and augmented contraction are observed [Citation44].
Many molecular details involved in the Rho-signalling and smooth muscle contraction, such as the crosstalk between Rho-kinase and ROS, and new sites for phosphorylation, were revealed in the past couple of years, advancing our understanding of the possibilities of different combinations in the regulatory mechanism according to the location of the smooth muscle in the body. Therefore, new regulatory mechanisms that are dependent upon the phosphorylation of RhoA/Rho-kinase are possible to be identified shortly.
When the same story has two sides: vascular smooth muscle relaxation
Vascular relaxation requires nitric oxide (NO) from endothelial cells, which targets smooth muscle cells and exerts its effects by stimulating the soluble guanylate cyclase (sGC) and the subsequent increase in intracellular cGMP level leading to activation of cGMP-dependent protein kinase (PKG) [Citation45]. The serine residue at position 188 in the RhoA sequence is a PKG target downstream to NO [Citation46]. Thus, NO via cGMP-PKG regulates RhoA expression and inhibits RhoA-induced Ca2+ sensitization in smooth muscle [Citation47]. NO is the most powerful physiological endothelial relaxing factor and it negatively regulates RhoA/Rho-kinase activation in the vasculature.
Endothelial functionality plays an important role in the physiological maintenance of appropriate vascular tone and contraction. Activation of Rho-kinase in endothelial cells has been shown to mediate the destabilization of eNOS mRNA [Citation48] as well as, negatively regulates eNOS phosphorylation via inhibition of protein kinase B/Akt, which in turn, decreases NO availability impairing relaxation [Citation49]. In the last decade, it has been reported and better investigated a functional link between RhoA/Rho-kinase and NO/cGMP pathways in the vasculature, highlighting the possibility of new pharmacological targets within these pathways. Currently, it is well accepted that RhoA/Rho-kinase mediates vascular dysfunction in many pathological conditions [Citation25], and different Rho-kinase inhibitors have been investigated as potential drugs to mitigate the damage. Currently, the isoquinoline sulphonamide, fasudil (HA-1077), a Ca2+ antagonist that causes relaxation in basilar arteries [Citation50], is the most used Rho-kinase inhibitor. Although different types of Rho-kinase inhibitors have been investigated in clinical trials for treating vascular problems, the low specificity of these inhibitors for ROCK1 and ROCK2 is still a significant limitation. Fasudil, for example, fails to distinguish between the two ROCK isoforms [Citation51].
Similar to NO, hydrogen sulphide (H2S) is also implicated in smooth muscle relaxation [for review see [Citation52]]. It is increasingly being considered as an important molecule in the vasculature as the enzymes responsible for generating H2S are widely expressed in arteries and veins [Citation53]. H2S can be generated by endogenous enzymes and monoenzymatic pathways. In the vasculature, endogenous H2S is mainly produced by an enzyme called cystathione gamma-lyase (CSE), which is physiologically activated by Ca-Cam and have been linked to many cardiovascular diseases [Citation54–56]. Two enzymes have been described to generate H2S and all are expressed in vascular cells [Citation57]. H2S inhibits smooth muscle contractions in isolated rabbit aortic strips and affects Rho-kinase activity by impairing RhoA activity through S-sulfhydration [Citation58]. An interaction between Rho-kinase, and H2S has been suggested in cavernosal smooth muscle, a highly specialized vascular structure. In this type of SMCs, a Rho-kinase inhibitor significantly decreased H2S-induced relaxations [Citation59]. Corroborating these data, endogenous H2S inhibits receptor-induced contractions and a Rho-kinase inhibitor (Y-27,632) increased H2S formation in vitro. Furthermore, the expression of pMYPT1, a substrate of Rho-kinase, was decreased in the presence of exogenous H2S compared to control [Citation60]. H2S exhibits antioxidant and vasoactive properties. Yet, downstream effectors of this gasotransmitter in the vessels, the chemical nature of the molecule responsible for the biological activity of H2S, or its implications in diseases, is still a growing field.
Another important aspect to be considered is the fact that RhoA is a key regulator of immunity, and recently, receptors of the innate immune system such as the Toll-like receptors (TLR) have been shown to be involved in chronic diseases that are characterized by a sustained increase in smooth muscle contraction leading to an impaired vasorelaxation [Citation61–63]. Ex vivo pharmacological activation of TLR1/2 reduced the relaxation response to Rho-kinase inhibition in cavernosal smooth muscle, suggesting the involvement of innate immune receptors in smooth muscle relaxation via Rho-kinase [Citation64]. Although there is no direct evidence of TLRs mediating hypercontractility in smooth muscle via activation RhoA/Rho-kinase, this could be a possibility, and therefore deserves further investigation as it could be mediating vascular pathologies. Specifically, cumulative evidence indicates that TLR4, which is the most studied TLR in the vascular system, might be mediating smooth muscle hypercontractility associated with vascular dysfunction in pathological conditions, especially in hypertensive animal models [Citation65–67].
Rho-Kinase and ageing: when ‘old’ means new
Ageing is a natural physiological process associated with changes in many biological mechanisms. In the last few years, pharmacological studies have found that, during ageing, many changes in smooth muscle contraction are associated with Rho-kinase. A study on the age-related elevation of blood pressure described the presence of increased expression of Gαq/11 proteins, which are involved in the control of vascular smooth muscle tone [Citation68]. Fluctuations in the levels of this protein are associated not only with age [Citation69] but also hyperglycaemia in smooth muscle cells [Citation70]. The G proteins involved in this pathway associate with PIP2, IP3, and DAG, leading to Ca2+-dependent activation of RhoA pathways [Citation71–73] and its related proteins, such as ROCK1 and ROCK2 [Citation74]. Rho-kinase functions to phosphorylate CPI-17, and consequently, activates an inhibitor of MLCP (MYPT-1). Similarly, Rho-kinase can directly phosphorylate MYPT-1, rendering it inactive. Considering this, the increase in the G proteins involved in this pathway would, subsequently, increase the activation of Rho-kinase during ageing, leading to prolonged contraction, and potentially, vascular dysfunction. For example, in small mesenteric arteries, ROCK2 is directly involved in the ageing-induced increase in myogenic tone [Citation75], a vital process that regulates blood flow to key organs (brain, heart, and kidney). A recent study showed that murine arteries have a more abundant expression of Gαq/11 proteins within vascular SMCs, as well as endothelial cells during ageing. Specifically, small mesenteric arteries of young and adult mice were analysed, and Rho-kinase expression was found to be significantly elevated in the aged group [Citation68]. Similarly, elevated RhoA/Rho-kinase activity is described in young adult subjects with vascular problems [Citation76].
Noteworthy, ageing might remodel the vascular wall by different mechanisms, including stimulation of RhoA/Rho-kinase signalling. Several factors may play a role in causing age-related dysfunction of smooth muscle, but evidence suggests that the Rho pathway may play either an indirect or a direct role in ageing within vascular SMCs, eventually driving vascular dysfunction. One mechanism by which Rho-kinase mediates vascular remodelling could be by the long-term inhibition of NO [Citation77], and it is widely accepted that many age-related processes are modulated by decreasing NO availability [Citation78,Citation79], such as angiogenesis [Citation80]. Abnormalities in angiogenesis, an essential adaptive response to physiological stress, are associated with age and NO-related pathways mediate this process [Citation81]. Indeed, it has been suggested that RhoA affects the permeability of the endothelium as well as the angiogenic process [Citation82]. A significant reduction in NO availability within the aortic endothelial tissue via decreased NOS expression was observed in older mice compared to younger matches [Citation83]. This is of little surprise because, as we age, there is a reduction in endothelium-dependent relaxation [Citation84]. While underlying mechanisms related to age-induced endothelial dysfunction have not been ultimately revealed, it seems that it involves an increase in membrane permeability, inflammation, and oxidative stress.
ROS production and ageing are entangled physiological events as they are associated with alterations in NO availability [Citation85] and an enhancement in superoxide production [Citation84]. An imbalance in the antioxidant system is a hallmark of ageing, and it has been demonstrated that ROS mediates vascular contraction by activating RhoA/Rho-kinase [Citation86]. Importantly, age has been associated with decline in sex hormones, such as testosterone, which induces vascular smooth muscle cell migration via NADPH oxidase pathway [Citation87]. Interestingly, erectile dysfunction, a highly prevalent vascular condition in the ageing male [Citation88], entailing Rho-kinase upregulation and oxidative stress [Citation89,Citation90], is also associated with low testosterone [Citation91,Citation92] .
Another potential mechanism affected by ageing is the fact that this process modifies the expression of endothelial markers that are implicated in the regulation of vasomotor tone. The levels of endogenous mediators, such as angiotensin II (Ang II), which promotes the generation of ROS, are increased with ageing [Citation93] and in the majority of CVDs. Indeed, during ageing, there is a reduction in the bioavailability of NO, which can be attributed to the increase in the activity of the NADPH oxidase enzyme [Citation94], which produces ROS. The reaction between ROS and NO leads to the formation of peroxynitrite, which associates with vascular ageing [Citation95] as it reduces eNOS expression via RhoA stimulation, and therefore, it might induce vascular dysfunction in pathological conditions [Citation96]. ROS also affect Rho-GDI. Oxidation of Rho-GDI contributes to cell proliferation via nuclear factor-κB activation [Citation97], which mediates the induction of many pro-inflammatory genes.
In a genetic disease, where ageing is accelerated and it is manifested in childhood, Hitchinson-Gilford progeria syndrome (HGPS), ROS contributes to not only the early onset of the senescent phenotype but also to its progression [Citation98]. The declined antioxidant capacity and, consequently, increased ROS generation, drive the mechanisms leading to Hitchinson’s diseases [Citation99]. Not surprisingly, cardiovascular complications are the primary cause of death in this condition. There is a paucity of information about smooth muscle contraction throughout this pathology. However, in a mouse model of premature ageing (LmnaG609G/G609G) mimicking HGPS, VSMCs were affected, and arterial stiffness and inward remodelling were observed [Citation100]. A recent study identified that VSMCs are the primary cell type generating contractile impairment in this animal model [Citation100]. Furthermore, the pharmacological blockade of Rho-kinase reduced ROS levels in this condition [Citation101], corroborating the connection between ageing and ROS via Rho-kinase.
While some evidence portrays the occurrence and elevation of RhoA and other associated proteins within ageing in vascular tissue, contributing to vascular SMC dysfunctionality, there is not extensive research regarding RhoA/Rho-kinase signalling in ageing. To date, how this pathway may play a role in vascular dysfunction accompanying ageing is still not fully elucidated. Overall, the mechanisms mediating disruption of homoeostasis during ageing and contributing to pathological conditions are far away to be completely understood. Therefore, further studies are warranted to understand the specific process by which RhoA/Rho-kinase plays a role in vascular ageing.
Rho-kinase and sex differences: when we cannot fit both sexes into one package, there is potential for discoveries
Previous studies have shown gender differences in vascular reactivity in response to a variety of agonists [Citation102]. For example, in the aorta, it has been observed that there are sex differences in calcium-mediated vascular reactivity [Citation103], and recently, it has been demonstrated that genes on the sex chromosomes regulate aortic vascular biology [Citation104]. The low incidence of CVDs in women, possibly due to endogenous oestrogen production, suggests the presence of vascular protective effects in this sex [Citation105]. Ubiquitously, agonist-induced endothelium-dependent constriction is significantly higher in vessels derived from male animals compared with those of females. Pressure-induced constriction is higher in arteries derived from males versus females [Citation106–108], suggesting sexual dimorphism in the vascular auto-regulatory control. The first study addressing whether the function or expression of Rho-kinase would be influenced by gender was performed in 2004. The authors showed that in female rat basilar arteries, oestrogen suppresses vascular Rho-kinase [Citation109]. Similarly, serotonin has a more prominent effect in the stimulation of RhoA/Rho-kinase in the aorta of males compared with females, which indicates that regulation of the calcium sensitivity mechanism contributes to sex differences in contraction [Citation110].
In human coronary VSMCs, oestrogen inhibits Rho-kinase expression in a dose-dependent manner [Citation111]. The oestrogen receptor antagonist, ICI 182,720, prevents against the inhibitory effects of oestrogen on VSMCs, suggesting that the effects of oestrogen in this cell population are primarily mediated via transcriptional mechanisms stimulated by oestrogen receptors [Citation112]. Activation of G protein-coupled oestrogen receptor 1 in porcine coronary arteries induces relaxation via inhibition of RhoA/Rho-kinase [Citation113]. Not only oestrogen, but also testosterone levels plays a role in RhoA/Rho-kinase in the vascular tissue. It has been recently described that testosterone and oestrogen acting jointly with Ephb6, a type B of tyrosine kinase receptor, to regulate smooth muscle contractility in small arteries and blood pressure. Increased contractility and RhoA activation were observed in small mesenteric arteries from castrated Ephb6 gene KO males compared with their WT counterparts [Citation114]. Additionally, another study showed that deletion of a cell surface molecule ligand for Eph tyrosine kinase receptor named EFNB3 (Ephrin B3), caused an increased blood pressure and vascular contractility in a sex-dependent manner [Citation115].
Noteworthy, testosterone plays an especial role in male cavernosal tissue. Low testosterone levels are associated to increased expression of RhoA and Rho-kinase signalling is regulated by testosterone levels in vascular diseases such as diabetes [Citation116]. Testosterone deficiency is known to induced endothelial dysfunction, and consequently, vascular problems [Citation117]. Note that androgens do not have the same effects in all type of smooth muscle cells [Citation118,Citation119]. Considerably, vascular responses to Angiotensin II (AngII), a potent endogenous peptide, are influenced by sex differences [Citation104,Citation120], and there is an extensive literature detailing sex difference in the renin-angiotensin-aldosterone system (RAAS), which is particularly important to design tailored therapies for vascular diseases [Citation121,Citation122]. AngII can increases contractility and decreases vasodilatation via AT1 receptors and molecular mechanisms associated to these changes includes upregulation of RhoA/Rho-kinase [Citation123,Citation124]. Yet, the sex difference and hormonal influence in the vasculature affecting RhoA/Rho-kinase signalling is an open field with many overlapping variables, especially ageing, which implies reduced testosterone levels in male and oestrogen in females, driving different outcomes in the vascular smooth muscle contractility.
The crosstalk between NO and Rho-kinase is also associated with sex differences. In the spiral modiolar artery, the myogenic tone, as well as calcium sensitivity, is increased by blocking nitric oxide synthase (NOS) in males, but not in females [Citation125]. This result suggests sex variations in the myogenic tone response based on a sex difference in the regulation of Rho-kinase. In basal conditions, levels of ROS production in smooth muscle cell cultures extracted from the aorta of males were different compared to females [Citation126]. In an experimental model of Type 2 diabetic mice, sex-dependent difference in Rho activation contributes to contractile dysfunction in the aorta. Interestingly, contractile dysfunction in vessels happens in both sexes, but there were no differences in the expression of RhoA, ROCK I and ROCK II between the aortas of males and females. However, activation of RhoA/Rho-kinase was greater in the aorta from diabetic compared to non-diabetic males, and no differences were observed in vascular activation of RhoA/Rho-kinase in females, suggesting contractile vascular dysfunction in males dependent upon activation of RhoA/Rho-kinase in diabetic conditions [Citation127].
Importantly, oestrogen is also associated with NO signalling. Physiological levels of systemic 17β-oestradiol can increase basal NO release in endothelial cells, which affects the diameter of pressurized arteries [Citation106]. Because there is an intricate connection between RhoA/Rho-kinase and NO signalling, it is possible that the effect of oestrogen on NO signalling could be indirectly affecting Rho-kinase in smooth muscle. Still, many questions need to be ruled out. For example, whether the Rho GTPase regulatory proteins or translocation of RhoA to the plasma membrane is different in male and female have not been elucidated. Sex-dependent alterations in Rho-kinase activation seem to contribute to contractile dysfunction during pathological conditions [Citation127], and unveiling these mechanisms could contribute to better tailored medical intervention in pathological conditions.
Perspectives and Challenges
Many advances in understanding the regulation of smooth muscle contraction by Rho signalling were achieved over the past 30 years. The canonical pathway by which RhoA/Rho-kinase regulates smooth muscle has been extensively revised in the literature as it elicits Ca2+ sensitization, and ultimately, contributes to smooth muscle contraction. Noteworthy, contraction is not the sole outcome following the activation of this pathway. In fact, RhoA/Rho-kinase plays a significant role in many other aspects of cell function not explored in this review such as cell differentiation and cell migration, which can also impact smooth muscle contraction. Furthermore, the diversity in Rho-kinase expression is clearly observed in the different smooth muscles distributed throughout the body, performing a specific physiological function in each organ. Importantly, many mechanisms driving the regulation of RhoA activity still need to be researched and uncovered. For example, RhoA activity involves protein kinases, such as cAMP-dependent PKA and type I cGMP-dependent PKG1, but it is still unknown whether other types of PKG would regulate RhoA activity. Likewise, many still unclear physiological compounds may affect Rho-kinase, such as the recently uncovered crosstalk between Rho-kinase and H2S (). Furthermore, new sites of phosphorylation have been unveiled, and various combined mechanisms are possible to regulate the activity of Rho-kinase. Noteworthy, the need for specificity in the blockade of ROCK 1 and ROCK 2 isoforms is critical to advance therapeutic strategies focusing on diseases-associated Rho-kinase.
Lastly, but not least, how age and gender differences influence and affect the regulation of smooth muscle contraction by RhoA/Rho-kinase are crucial topics that need to be investigated and deciphered (). At this point, it seems that increased ROS levels are the link between ageing and RhoA/Rho-kinase mediating hypercontractility. However, many other factors may be involved in smooth muscle regulation during the process of ageing, especially because an age-related decline in function is a natural physiological phenomenon in all organ and systems. Additionally, ageing per se implies low-grade inflammation, increased vascular permeability, and other processes that indirectly might affect smooth muscle contraction regulation by Rho-kinase. Regarding sex-differences driving smooth muscle contraction, link between RhoA/Rho-kinase and oestrogen is noticeable. A body of evidence suggests that greater vascular protection in female versus male is associated with oestrogen levels. This hormone exerts an inhibitory effect on Rho-kinase in vascular SMCs. Despite that, sex differences in smooth muscle contraction may be related to the effect of sex hormones on vascular calcium not only via Rho-kinase but also via PKC activity as well as other protein kinases. Undoubtedly, sex hormones have differential effects in both males and females, and it is reasonable to consider that the vascular effects, and consequently, smooth muscle contraction, of sex hormones is different in the two sexes. However, it is imperative to perform more studies to elucidate vascular protection- associated sex hormones as well as how these hormones affect Rho signalling in smooth muscle contraction. Notably, an important point to be considered is that dysregulation in NO pathways and oxidative stress in both physiological variables, ageing and sex-differences, might indirectly affect the regulation of smooth muscle contraction by RhoA/Rho-kinase signalling.
Figure 2. Current details into the mechanism by which Rho signalling regulates smooth muscle contraction. Not only the well-described gas NO might inhibit Rho-kinase, but also H2S, which the physiological functions and targets are currently under extensive investigation (first box). Gender differences and ageing also are highlighted in the literature as they drive differences in smooth muscle contraction by affecting Rho-kinase. Oestrogen inhibits Rho-kinase via specific G-protein coupling receptor (GPCER1) in young females (second box), and crosstalk between Rho-kinase and ROS is observed during ageing followed by decreasing NO availability (third box). Created with BioRender.com
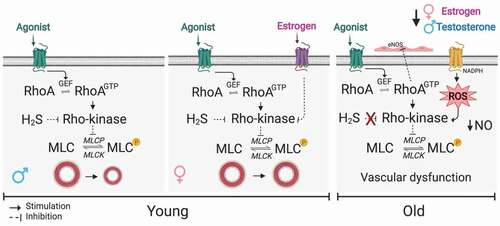
In summary, while many details were uncovered over the past decades regarding the mechanism by which Rho signalling regulates smooth muscle contraction, there are, as yet, many unanswered questions, and this area of research should be more thoroughly examined.
Acknowledgments
R.C.W is supported by the National Institute of Health (NIH- HL-134604) and DiaComp Pilot Feasibility Program, NIDDK.
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
References
- Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236.
- Colicelli J. Human RAS superfamily proteins and related GTPases. Sci STKE. 2004;250:re13.
- Fransen P, Van Hove CE, Leloup AJA, et al. Dissecting out the complex Ca2+-mediated phenylephrine-induced contractions of mouse aortic segments. PLoS One. 2015;10(3):e0121634.
- Kim HR, Appel S, Vetterkind S, et al. Smooth muscle signalling pathways in health and disease. J Cell Mol Med. 2008;12(6A):2165–2180.
- Brozovich FV, Nicholson CJ, Degen CV, et al. Mechanisms of vascular smooth muscle contraction and the basis for pharmacologic treatment of smooth muscle disorders. Pharmacol Rev. 2016;68(2):476–532.
- Khalil RA Regulation of Vascular Smooth Muscle Function. In: Colloquium Series on Integrated Systems Physiology: From Molecule to Function. San Rafael, CA: Morgan & Claypool Life Sciences; 2010. p. 1–62.
- Madaule P, Axel R. A novel ras-related gene family. Cell. 1985;41(1):31–40.
- Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol Rev. 2013;93(1):269–309.
- Gong MC, Fujihara H, Somlyo AV, et al. Translocation of rhoA associated with Ca2+ sensitization of smooth muscle. J Biol Chem. 1997;272(16):10704–10709.
- Feng J, Ito M, Ichikawa K, et al. Inhibitory phosphorylation site for Rho-associated kinase on smooth muscle myosin phosphatase. J Biol Chem. 1999;274(52):37385–37390.
- Kimura K, Ito M, Amano M, et al. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho- kinase). Science. 1996;273(5272):245–248.
- Somlyo AP, Somlyo AV. Signal transduction by G-proteins, Rho-kinase and protein phosphatase to smooth muscle and non-muscle myosin II. J Physiol. 2000;522(Pt 2):177–185.
- Webb RC. Smooth muscle contraction and relaxation. Am J Physiol - Adv Physiol Educ. 2003;27(4):201–206.
- Matsui T, Amano M, Yamamoto T, et al. Rho-associated kinase, a novel serine/threonine kinase, as a putative target for small GTP binding protein Rho. Embo J. 1996;15(9):2208–2216.
- Wang Y, Zheng XR, Riddick N, et al. ROCk isoform regulation of myosin phosphatase and contractility in vascular smooth muscle cells. Circ Res. 2009;104(4):531–540.
- Amin E, Dubey BN, Zhang SC, et al. Rho-kinase: regulation, (dys)function, and inhibition. Biol Chem. 2013;394(11):1399–1410.
- Riento K, Guasch RM, Garg R, et al. RhoE Binds to ROCK I and Inhibits Downstream Signaling. Mol Cell Biol. 2003;23(12):4219–4229.
- Riento K, Totty N, Villalonga P, et al. RhoE function is regulated by ROCK I-mediated phosphorylation. Embo J. 2005;24(6):1170–1180.
- Nakagawa O, Fujisawa K, Ishizaki T, et al. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 1996;392(2):189–193.
- Uehata M, Ishizaki T, Satoh H, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389(6654):990–994.
- Chrissobolis S, Sobey CG. Evidence that Rho-kinase activity contributes to cerebral vascular tone in vivo and is enhanced during chronic hypertension: comparison with protein kinase C. Circ Res. 2001;88(8):774–779.
- Yao L, Chandra S, Toque HA, et al. Prevention of diabetes-induced arginase activation and vascular dysfunction by Rho kinase (ROCK) knockout. Cardiovasc Res. 2013;97(3):509–519.
- Nohria A, Prsic A, Liu PY, et al. Statins inhibit Rho kinase activity in patients with atherosclerosis. Atherosclerosis. 2009;205(2):517–521.
- Toque HA, Nunes KP, Yao L, et al. Activated rho kinase mediates diabetes-induced elevation of vascular arginase activation and contributes to impaired corpora cavernosa relaxation: possible involvement of p38 MAPK activation. J Sex Med. 2013;10(6):1502–1515.
- Nunes KP, Rigsby CS, Webb RC. RhoA/Rho-kinase and vascular diseases: what is the link? Cell Mol Life Sci. 2010;67(22):3823–3836.
- Wei L, Roberts W, Wang L, et al. Rho kinases play an obligatory role in vertebrate embryonic organogenesis. Development. 2001;128(15):2953–2962.
- Shimizu Y, Thumkeo D, Keel J, et al. ROCK-I regulates closure of the eyelids and ventral body wall by inducing assembly of actomyosin bundles. J Cell Biol. 2005;168(6):941–953.
- Thumkeo D, Keel J, Ishizaki T, et al. Targeted Disruption of the Mouse Rho-Associated Kinase 2 Gene Results in Intrauterine Growth Retardation and Fetal Death. Mol Cell Biol. 2003;23(14):5043–5055.
- Zhang Y, Bo J, Taffet GE, et al. Targeted deletion of ROCK1 protects the heart against pressure overload by inhibiting reactive fibrosis. Faseb J. 2006;20(1):916–925.
- Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83(4):1325–1358.
- Amano M, Chihara K, Kimura K, et al. Formation of actin stress fibers and focal adhesions enhanced by Rho- kinase. Science. 1997;275(5304):1308–1311.
- Murányi A, Derkach D, Erdodi F, et al. Phosphorylation of Thr695 and Thr850 on the myosin phosphatase target subunit: inhibitory effects and occurrence in A7r5 cells. FEBS Lett. 2005;579(29):6611–6615.
- Eto M, Ohmori T, Suzuki M, et al. A novel protein phosphatase-1 inhibitory protein potentiated by protein kinase C. isolation from porcine aorta media and characterization. J Biochem. 1995;118(6):1104–1107.
- Koyama M, Ito M, Feng J, et al. Phosphorylation of CPI-17, an inhibitory phosphoprotein of smooth muscle myosin phosphatase, by Rho-kinase. FEBS Lett. 2000;475(3):197–200.
- Eto M, Kitazawa T, Matsuzawa F, et al. Phosphorylation-Induced Conformational Switching of CPI-17 Produces a Potent Myosin Phosphatase Inhibitor. Structure. 2007;15(12):1591–1602.
- Sakai H, Chiba Y, Misawa M. Role of Rho kinase in endothelin-1-induced phosphorylation of CPI-17 in rat bronchial smooth muscle. Pulm Pharmacol Ther. 2007;20(6):734–739.
- Mori D, Hori M, Murata T, et al. Synchronous phosphorylation of CPI-17 and MYPT1 is essential for inducing Ca 2+ sensitization in intestinal smooth muscle. Neurogastroenterol Motil. 2011;23(12):1111–1122.
- Butler T, Paul J, Europe-Finner N, et al. Role of serine-threonine phosphoprotein phosphatases in smooth muscle contractility. Am J Physiol Cell Physiol. 2013;304(6):C485–504.
- Chiba Y, Tanabe M, Goto K, et al. Down-regulation of miR-133a contributes to up-regulation of RhoA in bronchial smooth muscle cells. Am J Respir Crit Care Med. 2009;180(8):713–719.
- Hiroki J, Shimokawa H, Higashi M, et al. Inflammatory stimuli upregulate Rho-kinase in human coronary vascular smooth muscle cells. J Mol Cell Cardiol. 2004;37(2):537–546.
- Jin L, Ying Z, Hilgers RHP, et al. Increased RhoA/Rho-kinase signaling mediates spontaneous tone in aorta from angiotensin II-induced hypertensive rats. J Pharmacol Exp Ther. 2006;318(1):288–295.
- Araki S, Ito M, Kureishi Y, et al. Arachidonic acid-induced Ca2+ sensitization of smooth muscle contraction through activation of Rho-kinase. Pflugers Arch Eur J Physiol. 2001;441(5):596–603.
- Aaron Hobbs G, Zhou B, Cox AD, et al. oxidation, and cell redox control. Small GTPases. 2014;5(2):e28579.
- Higashi M, Shimokawa H, Hattori T, et al. Long-Term Inhibition of Rho-Kinase Suppresses Angiotensin II-Induced Cardiovascular Hypertrophy in Rats In Vivo: effect on Endothelial NAD(P)H Oxidase System. Circ Res. 2003;93(8):767–775.
- Lee MR, Li L, Cyclic KT. GMP causes Ca2+ desensitization in vascular smooth muscle by activating the myosin light chain phosphatase. J Biol Chem. 1997;272(8):5063–5068.
- Sauzeau V, Rolli-Derkinderen M, Marionneau C, et al. RhoA expression is controlled by nitric oxide through cGMP-dependent protein kinase activation. J Biol Chem. 2003;278(11):9472–9480.
- Sauzeau V, Le Jeune H, Cario-Toumaniantz C, et al. Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA- induced ca2+ sensitization of contraction in vascular smooth muscle. J Biol Chem. 2000;275(28):21722–21729.
- Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273(37):24266–24271.
- Ming X-F, Viswambharan H, Barandier C, et al. Rho GTPase/Rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase B/Akt in human endothelial cells. Mol Cell Biol. [Internet]. 2002Dec [cited 2019 Sep 21];22(24):8467–8477. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12446767
- Takayasu M, Suzuki Y, Shibuya M, et al. The effects of HA compound calcium antagonists on delayed cerebral vasospasm in dogs. J Neurosurg. 1986;65(1):80–85.
- Breitenlechner C, Gaßel M, Hidaka H, et al. Protein Kinase a in Complex with Rho-Kinase Inhibitors Y-27632, Fasudil, and H-1152P: structural Basis of Selectivity. Structure. 2003;11(12):1595–1607.
- Dunn WR, Alexander SPH, Ralevic V, et al. Effects of hydrogen sulphide in smooth muscle. Pharmacol Ther. 2016;158:101–113.
- Xu S, Liu Z, Liu P. Targeting hydrogen sulfide as a promising therapeutic strategy for atherosclerosis. Int J Cardiol. 2014;172(2):313–317.
- Yang G, Wu L, Jiang B, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ-lyase. Science. 2008;322(5901):587–590.
- Polhemus DJ, Calvert JW, Butler J, et al. The Cardioprotective Actions of Hydrogen Sulfide in Acute Myocardial Infarction and Heart Failure. Scientifica (Cairo). 2014;2014:768607.
- Pan LL, Liu XH, Gong QH, et al. Role of cystathionine γ-Lyase/hydrogen sulfide pathway in cardiovascular disease: A novel therapeutic strategy? Antioxid Redox Signal. 2012;17(1):106–118.
- Kimura H. Production and physiological effects of hydrogen sulfide. Antioxid Redox Signal. 2014;20(5):783–793. Production and physiological effects of hydrogen s.
- Nalli AD, Wang H, Bhattacharya S, et al. Inhibition of RhoA/Rho kinase pathway and smooth muscle contraction by hydrogen sulfide. Pharmacol Res Perspect. 2017;5(5):e00343.
- Aydinoglu F, Ogulener N. The role of arachidonic acid/cyclooxygenase cascade, phosphodiesterase IV and Rho-kinase in H2S-induced relaxation in the mouse corpus cavernosum. Pharmacol Rep. 2017;69(4):610–615.
- Aydinoglu F, EÖ A, Yılmaz-Oral D, et al. Involvement of RhoA/Rho-kinase in L-cysteine/H 2 S pathway-induced inhibition of agonist-mediated corpus cavernosal smooth muscle contraction. Nitric Oxide - Biol Chem. 2019;85(1):54–60.
- Nunes KP, de Oliveira AA, Mowry FE, et al. Targeting toll‐like receptor 4 signalling pathways: can therapeutics pay the toll for hypertension? Br J Pharmacol. 2018;176(12):1864–1879.
- de Oliveira AA, Davis D, Nunes KP. Pattern recognition receptors as potential therapeutic targets in metabolic syndrome: from bench to bedside. Diabetes Metab Syndr Clin Res Rev. 2019 Mar 1;13(2):1117–1122.
- de Oliveira AA, Webb RC, Nunes KP. Toll-Like Receptor 4 and Heat-Shock Protein 70: is it a New Target Pathway for Diabetic Vasculopathies? Curr Drug Targets. 2018 Nov 27;20(1):51–59.
- Stallmann-Jorgensen I, Ogbi S, Szasz T, et al. A Toll-Like Receptor 1/2 Agonist Augments Contractility in Rat Corpus Cavernosum. J Sex Med. 2015;12:1722–1731.
- Bomfim GF, Dos Santos RA, Oliveira MA, et al. Toll-like receptor 4 contributes to blood pressure regulation and vascular contraction in spontaneously hypertensive rats. Clin Sci. 2012;122(11):535–543.
- Nunes KP, Bomfim GF, Toque HA, et al. Toll-like receptor 4 (TLR4) impairs nitric oxide contributing to Angiotensin II-induced cavernosal dysfunction. Life Sci. 2017;15:219–226.
- Nunes KP, de Oliveira AA, Szasz T, et al., Blockade of Toll-Like Receptor 4 Attenuates Erectile Dysfunction in Diabetic Rats. J Sex Med. [Internet]. 2018;15:1235–1245. Available from:. ;(9):. https://www.ncbi.nlm.nih.gov/pubmed/30145096
- Björling K, Joseph PD, Egebjerg K, et al. Role of age, Rho-kinase 2 expression, and G protein-mediated signaling in the myogenic response in mouse small mesenteric arteries. Physiol Rep. 2018;6(17):e13863.
- Benetos A, Huguet F, Albaladejo P, et al. Role of adrenergic tone in mechanical and functional properties of carotid artery during aging. Am J Physiol - Hear Circ Physiol. 1993;265(4 Pt 2):H1132–8.
- Descorbeth M, Anand-Srivastava MB. High glucose increases the expression of Gq/11α and PLC-β proteins and associated signaling in vascular smooth muscle cells. Am J Physiol - Hear Circ Physiol. 2008;295(5):H2135–42.
- Lutz S, Freichel-Blomquist A, Yang Y, et al. The guanine nucleotide exchange factor p63RhoGEF, a specific link between Gq/11-coupled receptor signaling and RhoA. J Biol Chem. 2005;280(12):11134–11139.
- Vogt S, Grosse R, Schultz G, et al. Receptor-dependent RhoA activation in G12/G13-deficient cells. Genetic evidence for an involvement of Gq/G11. J Biol Chem. 2003;278(31):28743–28749.
- Mederos Y Schnitzler M, Storch U, Gudermann T. Mechanosensitive Gq/11 Protein–Coupled Receptors Mediate Myogenic Vasoconstriction. Microcirculation. 2016;23(8):621–625.
- Wirth A, Wang S, Takefuji M, et al. Age-dependent blood pressure elevation is due to increased vascular smooth muscle tone mediated by G-protein signalling. Cardiovasc Res. 2016;109(1):131–140.
- Mikkelsen MF, Björling K, Jensen LJ. Age-dependent impact of CaV3.2 T-type calcium channel deletion on myogenic tone and flow-mediated vasodilatation in small arteries. J Physiol. 2016;594(20):5881–5898.
- Leguina-Ruzzi A, Pereira J, Pereira-Flores K, et al. Increased RhoA/Rho-Kinase Activity and Markers of Endothelial Dysfunction in Young Adult Subjects with Metabolic Syndrome. Metab Syndr Relat Disord. 2015;13(9):373–380.
- Ikegaki I, Hattori T, Yamaguchi T, et al. Involvement of Rho-kinase in vascular remodeling caused by long-term inhibition of nitric oxide synthesis in rats. Eur J Pharmacol. 2001;427(1):69–75.
- Gao Y. The multiple actions of NO. Pflugers Arch Eur J Physiol. 2010;459(6):829–839.
- Sverdlov AL, Ngo DTM, Chan WPA, et al. Aging of the nitric oxide system: are we as old as our NO? J Am Heart Assoc. 2014;3(4):e000973.
- Rivard A, Fabre JE, Silver M, et al. Age-dependent impairment of angiogenesis. Circulation. 1999;99(1):111–120.
- Lähteenvuo J, Rosenzweig A. Effects of aging on angiogenesis. Circ Res. 2012;110(9):1252–1264.
- Gavard J, Gutkind JS. Protein kinase C-related kinase and ROCK are required for thrombin-induced endothelial cell permeability downstream from Gα12/13 and Gα11/q. J Biol Chem. 2008;283(44):29888–29896.
- Novella S, Dantas AP, Segarra G, et al. Aging-related endothelial dysfunction in the aorta from female senescence-accelerated mice is associated with decreased nitric oxide synthase expression. Exp Gerontol. 2013;48(11):1329–1337.
- Tschudi MR, Barton M, Bersinger NA, et al. Effect of age on kinetics of nitric oxide release in rat aorta and pulmonary artery. J Clin Invest. 1996;98(4):899–905.
- Cernadas MR, De Miguel LS, García-Durán M, et al. Expression of constitutive and inducible nitric oxide synthases in the vascular wall of young and aging rats. Circ Res. 1998;83(3):279–286.
- Jin L, Ying Z, Webb RC. Activation of Rho/Rho kinase signaling pathway by reactive oxygen species in rat aorta. American Journal of Physiology-Heart and Circulatory Physiology. 2004;287:H1495–500.
- Chignalia AZ, Schuldt EZ, Camargo LL, et al. Testosterone induces vascular smooth muscle cell migration by NADPH oxidase and c-Src-dependent pathways. Hypertension. 2012;59(6):1263–1271.
- Rajfer J. Decreased Testosterone in the Aging Male. Rev Urol. 2003;5(Suppl 1):S1.cited 2020 Aug 12. [Internet]. [];():. Available from:. : https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1502317/
- Priviero FBM, Toque HAF, Nunes KP, et al. Impaired Corpus Cavernosum Relaxation Is Accompanied by Increased Oxidative Stress and Up-Regulation of the Rho-Kinase Pathway in Diabetic (Db/Db) Mice. PLoS One. 2016;11(5):e0156030. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4882003/pdf/pone.0156030.pdf
- Nunes KP, Webb RC Mechanisms in Erectile Function and Dysfunction: an Overview. In: Erectile Dysfunction-Disease-Associated Mechanisms and Novel Insights into Therapy. InTech; 2012. p. 3–22.
- Rajfer J. Relationship between testosterone and erectile dysfunction. Rev Urol. 2000;2(2):122–128.
- Sopko NA, Hannan JL, Bivalacqua TJ. Understanding and targeting the Rho kinase pathway in erectile dysfunction. Nat Rev Urol. 2014;11(11):622–628.
- Michel JB, Nadaud S, Philippe M. Circulating and cellular markers of endothelial dysfunction with aging in rats. Am J Physiol - Hear Circ Physiol. 1997;273(4):H1941–8.
- Hamilton CA, Brosnan MJ, McIntyre M, et al. Superoxide excess in hypertension and aging a common cause of endothelial dysfunction. Hypertension. 2001;37(2):529–534.
- Van Der Loo B, Labugger R, Skepper JN, et al. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. 2000;192(12):1731–1744.
- El-Remessy AB, Tawfik HE, Matragoon S, et al. Peroxynitrite mediates diabetes-induced endothelial dysfunction: possible role of Rho kinase activation. Experimental Diabetes Research. 2010;2010:247861.
- Kim JG, Kwon HJ, Wu G, et al. RhoA GTPase oxidation stimulates cell proliferation via nuclear factor-κB activation. Free Radic Biol Med. 2017;103:57–68.
- Green DR, Galluzzi L, Kroemer G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science. 2011;333(6046):1109–1112.
- Kubben N, Zhang W, Wang L, et al. Repression of the antioxidant NRF2 pathway in premature aging. Cell. 2016;165(6):1361–1374.
- Del Campo L, Sánchez-López A, Salaices M, et al. Vascular smooth muscle cell-specific progerin expression in a mouse model of Hutchinson–Gilford progeria syndrome promotes arterial stiffness: therapeutic effect of dietary nitrite. Aging Cell. 2019;18(3):e12936.
- Kang HT, Park JT, Choi K, et al. Chemical screening identifies ROCK as a target for recovering mitochondrial function in Hutchinson-Gilford progeria syndrome. Aging Cell. 2017;16(3):541–550.
- Karanian JW, Ramwell PW. Effect of gender and sex steroids on the contractile response of canine coronary and renal blood vessels. J Cardiovasc Pharmacol. 1996;27(3):312–319.
- Eatman D, Stallone JN, Rutecki GW, et al. Sex differences in extracellular and intracellular calcium-mediated vascular reactivity to vasopressin in rat aorta. Eur J Pharmacol. 1998;361(2–3):207–216.
- Alsiraj Y, Thatcher SE, Charnigo R, et al. Female mice with an XY sex chromosome complement develop severe Angiotensin II-induced abdominal aortic aneurysms. Circulation. 2017;135(4):379–391.
- Barrett-Connor E. Sex differences in coronary heart disease: why are women so superior? The 1995 Ancel Keys Lecture. Circulation. 1997;95(1):252–264.
- Wellman GC, Bonev AD, Nelson MT, et al. Gender Differences in Coronary Artery Diameter Involve Estrogen, Nitric Oxide, and Ca 2+Dependent K+ Channels. Circulation Research. 1996;79(5):1024–1030.
- Gros R, Van Wert R, You X, et al. Effects of age, gender, and blood pressure on myogenic responses of mesenteric arteries from C57BL/6 mice. Am J Physiol - Hear Circ Physiol. 2002;282(1):H380–8.
- Chan MV, Bubb KJ, Noyce A, et al. Distinct endothelial pathways underlie sexual dimorphism in vascular auto-regulation. Br J Pharmacol. 2012;167(4):805–817.
- Chrissobolis S, Budzyn K, Marley PD, et al. Evidence that estrogen suppresses Rho-kinase function in the cerebral circulation in vivo. Stroke. 2004;35(9):2200–2205.
- Nuno DW, Korovkina VP, England SK, et al. RhoA activation contributes to sex differences in vascular contractions. Arterioscler Thromb Vasc Biol. 2007;27(9):1934–1940.
- Hiroki J, Shimokawa H, Mukai Y, et al. Divergent effects of estrogen and nicotine on Rho-kinase expression in human coronary vascular smooth muscle cells. Biochem Biophys Res Commun. 2004;326(1):154–159.
- Kolodgie FD, Jacob A, Wilson PS, et al. Estradiol attenuates directed migration of vascular smooth muscle cells in vitro. Am J Pathol. 1996;148(3):969–976.
- Yu X, Zhang Q, Zhao Y, et al. Activation of G protein-coupled estrogen receptor 1 induces coronary artery relaxation via Epac/Rap1-mediated inhibition of RhoA/Rho kinase pathway in parallel with PKA. PLoS One. 2017;12(3):e0173085.
- Luo H, Wu Z, Tremblay J, et al. Receptor tyrosine kinase Ephb6 regulates vascular smooth muscle contractility and modulates blood pressure in concert with sex hormones. J Biol Chem. 2012;287(9):6819–6829.
- Wang Y, Wu Z, Thorin E, et al. Estrogen and testosterone in concert with EFNB3 regulate vascular smooth muscle cell contractility and blood pressure. Am J Physiol - Hear Circ Physiol. 2016;310(7):H861–H872.
- Vignozzi L, Morelli A, Filippi S, et al. Testosterone regulates RhoA/Rho-kinase signaling in two distinct animal models of chemical diabetes. J Sex Med. 2007;4(3):620–632.
- Fahed AC, Gholmieh JM, Azar ST. Connecting the lines between hypogonadism and atherosclerosis. Int J Endocrinol. 2012;2012:793953.
- Makieva S, Hutchinson LJ, Rajagopal SP, et al. Androgen-induced relaxation of uterine myocytes is mediated by blockade of both Ca2+ flux and MLC phosphorylation. J Clin Endocrinol Metab. 2016;101(3):1055–1065.
- Flores-Soto E, Reyes-García J, Carbajal-García A, et al. Sex steroids effects on guinea pig airway smooth muscle tone and intracellular Ca2+ basal levels. Mol Cell Endocrinol. 2017;439:444–456.
- Sartori-Valinotti JC, Iliescu R, Yanes LL, et al. Sex differences in the pressor response to angiotensin II when the endogenous renin-angiotensin system is blocked. Hypertension. 2008;51(4):1170–1176.
- Henriques TA, Huang J, D’Souza SS, et al. Orchidectomy, but not ovariectomy, regulates angiotensin II-induced vascular diseases in apolipoprotein E-deficient mice. Endocrinology. 2004;145(8):3866–3872.
- Xue B, Johnson AK, Hay M. Sex differences in angiotensin II-induced hypertension. Braz J Med Biol Res. 2007;40(5):727–734.
- Touyz RM. The role of angiotensin II in regulating vascular structural and functional changes in hypertension. Curr Hypertens Rep. 2003;5(2):155–164.
- Savoia C, Tabet F, Yao G, et al. Negative regulation of RhoA/Rho kinase by angiotensin II type 2 receptor in vascular smooth muscle cells: role in angiotensin II-induced vasodilation in stroke-prone spontaneously hypertensive rats. J Hypertens. 2005;23(5):1037–1045.
- Reimann K, Krishnamoorthy G, Wangemann P. NOS Inhibition Enhances Myogenic Tone by Increasing Rho-Kinase Mediated Ca2+ Sensitivity in the Male but Not the Female Gerbil Spiral Modiolar Artery. PLoS One. 2013;8(1):e53655.
- Malorni W, Straface E, Matarrese P, et al. Redox state and gender differences in vascular smooth muscle cells. FEBS Lett. 2008;582(5):635–642.
- Nuno DW, Harrod JS, Lamping KG. Sex-dependent differences in Rho activation contribute to contractile dysfunction in type 2 diabetic mice. Am J Physiol - Hear Circ Physiol. 2009;297(4):H1469–77.