
ABSTRACT
KRAS genes belong to the most frequently mutated family of oncogenes in cancer. The G12C mutation, found in a third of lung, half of colorectal and pancreatic cancer cases, is believed to be responsible for a substantial number of cancer deaths. For 30 years, KRAS has been the subject of extensive drug-targeting efforts aimed at targeting KRAS protein itself, but also its post-translational modifications, membrane localization, protein–protein interactions and downstream signalling pathways. So far, most KRAS targeting strategies have failed, and there are no KRAS-specific drugs available. However, clinical candidates targeting the KRAS G12C protein have recently been developed. MRTX849 and recently approved Sotorasib are covalent binders targeting the mutated cysteine 12, occupying Switch II pocket.
Herein, we describe two fragment screening drug discovery campaigns that led to the identification of binding pockets on the KRAS G12C surface that have not previously been described. One screen focused on non-covalent binders to KRAS G12C, the other on covalent binders.
Introduction
KRAS belongs to the RAS super-family of small GTPases that play a critical role as regulators of many cellular processes [Citation1,Citation2]. Activating mutations of KRAS are some of the most frequently found in human cancers [Citation3], and they are associated with poor prognosis, such as that seen, for example, in early-stage non-small cell lung cancer [Citation4]. Among these mutations, glycine 12 and glycine 13 mutations are the most frequently observed [Citation5–7], with the G12C mutation one of the three most frequent mutations of this amino acid, along with G12V and G12D [Citation8]. The therapeutic importance of KRAS and its mutants are well known and have been validated experimentally [Citation9], but KRAS was thought to be undruggable for the reasons described below.
KRAS, like other small GTPases (for an extensive review, see [Citation10]), alternates between two different conformations, the ‘OFF’ state where the protein binds the nucleotide guanosine diphosphate (GDP) and the ‘ON’ state where GDP has been replaced by guanosine triphosphate (GTP). The nucleotide is very tightly bound to the protein and the γ-phosphate of GTP stabilizes two loops, switch I and switch II, which are involved in protein–protein interactions (for review on RAS structures see [Citation11]) and are instrumental for KRAS signalling as well as for KRAS cycling. The structure of KRAS, whether in complex with GDP or GTP, is compact, and its surface was thought to be undruggable, as it lacked apparent binding pockets.
In 1998, binders were identified by NMR [Citation12] which seemed to interact preferentially with the GDP form of KRAS. This discovery was followed by new experimental identification of Ras-inhibitor binding interface using a water-soluble Ras ligand in 2009 [Citation13]. In 2012, the assumed undruggable nature of KRAS was further revised with the discovery of small ligands binding to a newly identified pocket created by the rotation of Tyr71 [Citation14,Citation15]. It was later shown that covalent ligands of KRAS G12C could open a previously unknown pocket under switch II in the presence of GDP [Citation16,Citation17]. Furthermore, the presence of the ligand modified switch II conformation and prevented binding of KRAS to its protein partners. These findings revived the interest in the search for small molecule drugs targeting KRAS and led to the discovery of promising new compounds [Citation18–22], which occupy the switch II pocket (for review see [Citation23]). The above-mentioned discovery encouraged the continued search for new pharmacologically relevant mechanism of small molecule binding to KRAS G12C mutation in KRAS. Moreover, these findings represent an opportunity to exploit the solvent exposed side chain of the mutated cysteine in KRAS G12C for covalent targeting, and to discover new binding pockets in this and other mutants, capable of being targeted with reversible synthetic binders.
In order to discover new binding modes and to identify new chemical matter, we performed fragment screening targeting the GDP-loaded form of KRAS G12C. Fragments are molecules of small molecular weight that generally follow the ‘Rule of 3’ [Citation24,Citation25], which, if found binding to the target of interest, are expected to have a better ligand efficiencies [Citation26] than initial hits identified by more traditional approaches.
As with previous strategies used to identify and develop covalent ligands, we firstly determined molecules that reversibly bound to the site of interest, then ‘weaponized’ these molecules with a chemical motif that was expected to form a covalent bond to the proximal cysteine [Citation27,Citation28]. Our first fragment screening campaign was, therefore, focused on the biophysical identification of reversible binders to GDP-loaded KRAS G12C.
We subsequently screened a library of covalent acrylamide-containing fragments. Positives from the two screens were submitted to crystallization trials to identify their binding modes in the GDP-loaded KRAS G12C protein. Whereas the screen of non-covalent fragments did not lead to the discovery of new binding modes, the co-structure of KRAS G12C with several covalent fragments rendered two new pockets that, to our knowledge, have not previously been reported.
Materials and Methods
Recombinant production of human wild type & G12C KRAS proteins
Synthetic genes coding for either the wild type (WT) or the G12C mutant forms of human KRAS (amino acid position 1 to 169, GenBank accession number NM_004985) were ordered from GenArt, with Escherichia coli codon optimization. A polyhistidine tag and a TEV protease site were introduced at 5ʹ of each coding sequence. The expression cassettes were subcloned into a pET-29a vector (Novagen) and recombinant plasmids were used for transforming BL21(DE3) E. coli competent cells (Novagen). After selection and cell banking, recombinant clones were grown into a proprietary chemically defined medium at 37°C in 15 L bioreactors. KRAS gene expression was induced with 0.5 mM IPTG at 20°C, and the cells harvested after 18 h and the resulting pellets were frozen and stored at −20°C.
Protein purification and characterization
A frozen cell pellet was resuspended in 10 volumes of lysis buffer (20 mM Tris, 500 mM NaCl, 2 mM TCEP, pH 8.0), homogenized for 10 min using an Ultra-Turrax disperser (IKA) and the resuspended cell solution was lysed by 3 passages at 350, 700 & 1000 bars respectively on an EmulsiFlex C3 system (Avestin). Lysate was cleared by 120,000 g centrifugation at 4°C for 1 h and the clarified solution was loaded on a Talon SuperFlow column (GE Healthcare) equilibrated with lysis buffer. After a wash of 5 column volumes with lysis buffer, the elution was performed with a linear gradient of 0–300 mM Imidazole in 12 column volumes. Fractions containing the recombinant His-tagged protein were pooled, dialysed overnight at 4°C in a buffer (20 mM Tris, 300 mM NaCl, 1 mM TCEP, 5 mM Imidazole, pH 8.0) containing 2% (w/w) of His-tagged TEV protease. Dialysed solution was then applied on a Ni-Sepharose FF column (GE Healthcare) to remove the His-tag and the His-tagged TEV protease. The His-tagged free KRAS protein was recovered into the flow-through fraction, diluted 10 folds in dilution buffer (20 mM Tris, 1 mM TCEP, pH 8.0) and applied on Q Sepharose FF (GE Healthcare). After a wash of 10 column volumes, recombinant protein was eluted with a gradient (10–100%) of dilution buffer plus 500 mM NaCl in 10 column volumes. Fractions with KRAS protein were pooled, concentrated using Centriprep centrifugal filters (10 kDa MWCO, Merck-Millipore) and applied on a Superdex 200 PG column (GE Healthcare) equilibrated with SEC buffer (20 mM HEPES, 150 mM NaCl, 1 mM MgCl2, pH 7.5). After protein concentration (ca. 8 mg/mL), recombinant KRAS forms were aliquoted and stored at −80°C.
KRAS WT and KRAS G12C proteins were characterized by UV titration with a molar extinction coefficient taking into account the presence of one GDP molecule per protein, electrospray ionization mass spectrometry, capillary gel electrophoreses and analytical size exclusion chromatography.
Ligand-Observed NMR
The KRAS G12C GDP solution was exchanged with the NMR buffer containing 50 mM [D11]-Tris, 150 mM NaCl, 1 mM [D16]-TCEP, 1 mM MgCl2, 100 µM DSS (used as internal reference) in 100% D2O, pH 7.4 by ultrafiltration using 10 kDa cut-off membrane concentration devices (Amicon Ultrafree, Millipore).
The NMR samples were prepared in 200 µL volume using a Gilson robot. Each cocktail of 5 compounds at a final concentration of 1 mM per compound (5% D6-DMSO), was incubated with 10 µM of protein for 1 hour.
All NMR experiments were carried out at 298 K on a Bruker ADVANCE II 600 NMR spectrometer, equipped with a BACS-60 sample changer and a 5 mm TXI Cryoprobe, and processed with TOPSPIN 2.1 software from Bruker. The [Citation1]H with excitation sculpting [Citation29] and saturation transfer difference (STD) with 3 9 19 pulses [Citation30] experiments were recorded using 3 mm tubes. The data were multiplied with an exponential window function of 1 Hz [Citation1H] or 3 Hz (STD) line broadening prior to Fourier transformation.
The STD experiments were recorded with selective frequencies of pre-saturation on the protein of 60 Hz and – 8000 Hz with a saturation time of 2 s, a relaxation delay of 2 s and 256 scans.
To rank the fragments, the STD scaling amplification factor f = ISTD/I 1H x [frag]/[KRAS] [Citation31,Citation32] was used and a cut-off of 2.5 was applied to select moderate (2.5 < f < 5) and strong binders (f > 5) before individual confirmation. This criterion was also used when confirming individual binders.
Thermal shift assay (TSA) by Differential Scanning Fluorimetry (DSF)
The TSA experiments were performed on an CFX384 (Bio-Rad) using a heating range of 20–90°C and a heating rate of 0.5°C/min. 384-well plates were used with 10 μl sample volume including 26 μM KRAS G12C, 150 mM NaCl, 1 mM MgCl2, 20 mM Tris pH 7.5, SyproOrange 20X and the compound. Fragments were tested at 1 mM for screening and prepared in series of 100 µM to 5 mM for dose-response experiments. Melting temperatures were determined by CFX Manager software (v3.1, Bio-Rad) by the first derivative method and dose-response curves were traced in Excel (Microsoft Office). Fragments with ΔTm>0.4°C at 1 mM were selected for dose-response follow-up. Fragments exhibiting a ΔTm saturation signal plateau in dose-response were considered hits.
Surface Plasmon Resonance (SPR)
SPR biosensing experiments were performed on a Biacore4000 equipped with Sensorchip CM5 at 25°C (GE Healthcare). For immobilization, KRAS G12C was prepared at 0.5 μM in 10 mM Na Acetate pH 4.5 (GE Healthcare). HBS-P (10 mM Hepes, 150 mM NaCl, 0.05% surfactant P20, pH 7.4, GE Healthcare) supplemented with 1 mM MgCl2, was used as the running buffer. Immobilization levels used were around 3500 RU.
For the interaction study, HBS-P supplemented with 1 mM MgCl2 and 5% DMSO was used as running buffer. Fragments were tested at 200 μM and 1000 μM for screening and prepared in series of 20 µM up to 1 mM or up to solubility limit for KD determination. Compounds were injected at a flow rate of 30 µL/min for 60 s, and the dissociation was thereafter followed for 120 s. Data processing and analysis were performed by BiaEvaluation software ver. 1.0 (GE Healthcare). Sensorgrams were double-referenced prior to global fitting the concentration series to 1:1 binding with the mass-transport model. Responders with signal ratio below five between 1000 μM and 200 μM were selected for affinity determination. Fragments with KD<2 mM were declared hits.
Screening the library of covalent fragments by Mass spectrometry (MS-modification)
969 fragments bearing an acrylamide moiety were incubated at 500 µM with 10 µM KRAS G12C for 24 h at room temperature (RT), in 20 mM HEPES pH 7.5, 150 mM NaCl, 5% DMSO 10 mM EDTA, in 384-well microtiter plates. Reaction was stopped by adding 0.2% final concentration formic acid. Samples were analysed in a Waters Acquity UPLC I-Class system. Proteins were analysed on a Waters MassPREP desalting column (diphenyl) 5×2.1 mm at 80°C with a flow rate of 0.3 mL/min. For these separations, mobile phase A was aqueous 0.1% (v/v) formic acid and mobile phase B was 0.1% (v/v) formic acid in acetonitrile. Proteins were eluted from the column with a linear gradient ranging from 5 to 90% B over 1.5 min.
The UPLC system was coupled to a Waters SYNAPT G2 QTOF system used in MS mode for acquisition. MS data were automatically processed (peak detection and intact protein spectrum deconvolution with MaxEnt™ algorithm) using Waters OpenLynx software, a qualitative application of Masslynx™, to deliver the relative peak height of each molecular weight detected species.
A subset of fragments was tested on the wild type protein under the same conditions.
EC50 determination by Mass spectrometry
The mutant protein was incubated at 2 µM for 24 h at RT, with 6 concentrations of covalent fragment, ranging from 0.7 to 162 µM, in 20 mM HEPES pH 7.5, 150 mM NaCl, 1 mM MgCl2 and 5% DMSO. The reaction was stopped by adding 0.2% v/v formic acid.
Samples were analysed in MS mode on a QTRAP-5500 (Sciex). The spectra were deconvoluted to obtain the mass and the intensities (areas) of the protein species, with the BioPharmaView™ Sciex software. Using the intensities of the unmodified and modified species a percentage of modification was determined for each concentration of covalent fragment. The results were fitted to a 4-parameter dose-response model to generate EC50.
Peptide mapping
Peptide mapping was performed to determine which cysteines of the recombinant KRAS G12C were modified by the fragments. Samples were prepared using the same condition as for the library screen, then reduced with DTT, alkylated with iodoacetamide, digested with trypsin (2 h, 37°C) and analysed on a Dionex U3000 HPLC system coupled to a Thermo Orbitrap XL mass spectrometer. The peptides were separated on an Acclaim C18 RSLC pepmap column (75 µm*15 cm) with a gradient starting with 96% of mobile phase A (0.1% v/v Formic acid in water) and 4% mobile phase B (0.1% v/v formic acid in acetonitrile). Peptides were eluted in 15 min; at 40% mobile phase B, with a flow rate of 300 nL/min.
The MS/MS analyses were performed for peptide identification in a data-dependent mode in which one cycle of experiments consisted of one full MS scan of 350 to 2000 m/z followed by five sequential MS/MS events. The raw data were queried with Mascot Software [Citation33] (MatrixScience) against KRAS G12C sequence to cover the whole sequence with two types of modifications on cysteines: a carbamidomethylation or attachment of the fragment of interest.
Guanine nucleotide exchange (GEF) assay
0.5 µM of KRASG12C loaded with GDP was pre-incubated with 500 µM of the acrylamide fragment, in 20 mM HEPES-KOH pH7.5, 10 mM MgCl2, 0.05% CHAPS, 5% DMSO. The reaction was performed at RT in non-binding black plates. For the positive control, KRAS G12C was preincubated with DMSO only. After 24 h pre-incubation, the nucleotide exchange reaction was initiated by the addition of a 2ʹ/3ʹ-O-N-methyl-anthraniloyl (mant)-GDP (Jena Biosciences) and hSOS1 catalytic domain mixture (1.25/1.25 µM final) or mant-GDP and GDP mixture (1.25 µM/200 µM) for the negative control. After excitation at 370 nm, the fluorescence emission at 450 nm was monitored for 90 min on a Safire spectrofluorometer (Tecan). The IC50 for each compound was calculated from the initial velocity of the reaction normalized to the positive control and was fitted to a 4-parameter dose-response curve using Xlfit software from IDBS (https://www.idbs.com/excelcurvefitting/).
Nephelometry
Nephelometry detects the presence of particles in a solution by measuring the light scattering of a 635 nm incident laser light at a 80° angle. 100 µl three-fold serial dilutions of the compounds, from 162 to 2 µM, were prepared in MS buffer containing 5% DMSO final in 384 acoustic-dispensing 781,201–906PP (Greiner) microtiter plates. After 1 h incubation at RT, the plates were read with a NEPHELOstar Plus (BMG LABTECH). The 635 nm diffracted photons of each serial dilution were compared to control wells (n = 30 to 60) containing MS buffer with 5% DMSO. The solubility threshold was defined as the mean signal of the control + 3 standard deviations. Compounds are considered soluble at a given concentration if the signal remains below this threshold.
Preparation of the covalent complexes for crystallization
The KRAS G12C protein and the covalent fragment were incubated prior complex crystallization. Four different incubation conditions were initially tested for two different compounds: 1 mM compound during 4 or 23 h at 4 or 20°C, with 1 mM protein. The extent of the covalent modification was determined by mass spectrometry. More than 95% covalent modification was observed after 23 h incubation at 20°C, over 80% at 4°C. These conditions were further refined to allow 2–2.5 ligand excess (depending on ligand solubility) for 24 h at 4°C to increase the covalent modification efficiency. Covalent binding was systematically verified by mass spectrometry prior to crystallization screening.
Crystallization
Non-covalent fragments: Four crystallization conditions have been identified for GDP-bound KRAS G12C, leading to four different crystal forms, in P3, P212121, R3 and C2. Soaking experiments were carried out for all fragments in all four crystal forms, using up to 25 mM fragment (depending on fragment solubility in the crystallization solution) for one and six days. The structure of compound 1 was obtained by soaking a P212121 crystal in 25 mM fragment for one day. The protein was concentrated to 44 mg/ml in 25 mM TRIS pH 8, 100 mM NaCl, 5 mM MgCl2, 1 mM TCEP. Crystals were obtained by vapour diffusion experiments in hanging drop geometry under 23% PEG4000, 100 mM Na Acetate, 100 mM TRIS pH 8.5.
Covalent fragments: The protein was concentrated to 22.8 mg/ml in 20 mM TRIS pH 8, 200 mM NaCl, 2.5 mM MgCl2. All covalent complexes were prepared by incubating the protein in the presence of 2.5 mM ligand and 2.5 mM MgCl2 for 24 h at 4°C. Extensive crystallization screening was then carried out at 20°C.
In presence of compound 2, the protein crystallizes in 100 mM HEPES pH 7.5, 2.25 M Ammonium Sulphate (cryoprotectant 25% glycerol) (14.29 mg/ml protein in 20 mM HEPES 150 mM NaCl 0.2 mM TCEP)
In presence of compound 3, the protein crystallizes in 20% PEG3350, 200 mM Na2HPO4 pH7.0 (cryoprotectant 25% glycerol)
In presence of compound 4, the protein crystallizes in 1.1 M sodium citrate pH 6.0 (cryoprotectant 25% glycerol)
Data processing and refinement
X-Ray diffraction data sets were collected at ESRF or SOLEIL synchrotrons and processed using the Global Phasing Suite [Citation34] and CCP4 [Citation35] software. The structures were determined by molecular replacement using Phaser [Citation36] with a GDP-bound KRAS G12C monomer without Switch II as search model and refinement was carried out by successive rounds of Buster [Citation37] and COOT [Citation38] rebuilding. Processing and refinement data are summarized in .
Results
Non-covalent fragment screening
A non-covalent fragment screening was initiated on KRAS G12C, using three independent biophysical methods as primary screening techniques: DSF (Differential Scanning Fluorimetry), LO-NMR (Ligand Observed Nuclear Magnetic Resonance) and SPR (Surface Plasmon Resonance) (). These three techniques measure interactions based on distinct physical phenomena and all three were used in parallel. The resulting binders were cross validated and fragments that bound in at least two different assays were prioritized for further studies. An internal library of over 4488 fragments was screened by SPR at 200 μM, resulting in the selection of 543 binders (selection criteria described in Material & Methods). These 543 SPR binders were then tested at 2 concentrations (200 μM and 1000 μM) to remove suprastoichiometric binders. The remaining 126 fragments were submitted to a dose-response experiment up to 1000 μM. A total of 19 fragments were finally selected with KD estimates ranging from 0.2 to 6 mM. The screening by DSF of the same library identified 49 fragments having a significant effect on thermal stabilization (DeltaTm>0.4°C at 1000 μM). These positives submitted to a dose-response DSF experiment up to 5000 μM. Based on the saturation plateaus obtained from these experiments, 12 fragments were identified as positive.
Figure 1. Non-covalent fragment screening flowchart.
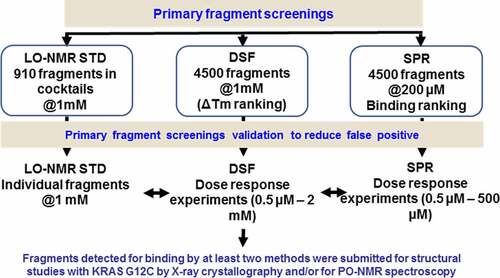
A subset of 910 molecules from the fragment library was also tested in STD-NMR [Citation30] in cocktails of five fragments at 1 mM. On the basis of the results from these cocktail experiments, 121 compounds were further evaluated individually at the same concentration. 42 of these tested fragments were to shown to bind moderately or strongly to KRAS G12C (STD amplification factor above 2.5).
The binders identified by one technique were cross validated using the other two, with 24 fragments showing binding in at least two biophysical methods. These fragments were selected for crystallization trials in complex with GDP-bound KRAS G12C. Soaking experiments at 10 and 25 mM were carried out in four different crystal forms and this led to three structures containing a bound ligand.
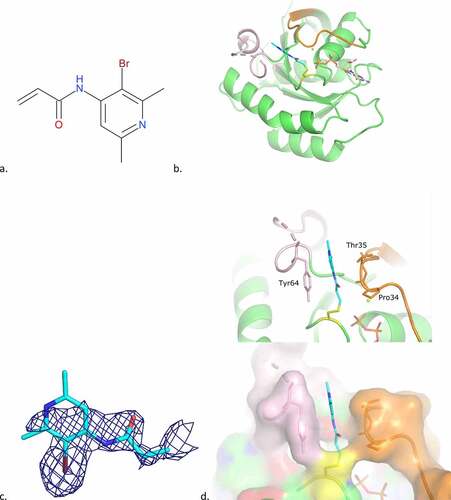
The first two structures surprisingly showed covalently bound fragments to Cys118, and to a lesser extend Cys12. The third structure showed an indole-containing fragment (cpd1) bound to a small hydrophobic pocket in between the α2-helix of switch II and the core β-sheet. This pocket was created as a result of the rotation of Tyr71. This pocket is a known, previously described binding site for indole ligands [Citation14,Citation39]: the switch I/II pocket. The identified fragment adopts the same binding mode as indole molecules already described (4EPV, 4EPY in [Citation14], see ).
Figure 2. Cpd1 binding mode to KRAS G12C GDP.
a) 2D structure of cpd1b Overall binding mode of cpd1. b) The ligand is in cyan. The protein overall structure is in green with switch I (as defined by [Citation40]) in orange, switch II in pink; the GDP is also depicted (in light orange) to the right of Cys12 (coloured yellow),c) Final 2FoFc-sigmaa weighted map around the ligand, contoured at 1σ,d) Superimposition of the 4EPY ligand (in magenta). .
![Figure 2. Cpd1 binding mode to KRAS G12C GDP.a) 2D structure of cpd1b Overall binding mode of cpd1. b) The ligand is in cyan. The protein overall structure is in green with switch I (as defined by [Citation40]) in orange, switch II in pink; the GDP is also depicted (in light orange) to the right of Cys12 (coloured yellow),c) Final 2FoFc-sigmaa weighted map around the ligand, contoured at 1σ,d) Superimposition of the 4EPY ligand (in magenta). .](/cms/asset/9142f3f7-07d3-46cd-9ad0-65bfb8ae7668/ksgt_a_1979360_f0002_oc.jpg)
Overall, X-Ray data analysis on 147 soaking experiments for 24 fragments resulted in only 3 interpretable electron density maps that described fragment binding, this is in spite of the high concentrations (up to 50 mM) used in the soaking experiments. These initial results highlight the difficulties in obtaining non-covalent ligand-bound structures in KRAS G12C, even when binding is detected by biophysical methods. These limitations meant this screening approach did not yield new chemical matter or lead to the identification of new binding sites. Contrary to what has been observed in kinases [Citation27], trying to first identify non-covalent binders to KRAS prior to weaponization may not be the best ligand finding approach, as the creation of induced allosteric sites around the switches might require stronger binding affinity than that of non-covalent fragments. As a consequence of our findings, it was decided not to pursue these efforts and to proceed with a covalent fragment screening approach instead, using a newly designed in-house covalent fragment library.
Covalent fragment screening
An internal library of 969 acrylamide-containing fragments incubated at 500 µM for 24 h with KRAS G12C was screened by mass spectrometry in intact mass mode (). The solubility of the fragments in the screening buffer had been previously verified in a nephelometry assay. The incubation was conducted in the presence of 10 mM EDTA to accelerate the rate of nucleotide exchange, as described previously [Citation16]. The results identified 77 fragments as modifying KRAS G12C by more than 50%. These fragments were defined as hits and evaluated further. It was noticed that 37 hits modified the protein at more than one site. In a few rare cases, up to four fragment adducts were present, corresponding to the total number of cysteine residues in the sequence of the recombinant protein. These fragments lacked selectivity presumably leading to a partial or transient unfolding of the mutant protein in the assay conditions. This hypothesis was confirmed by the partial modification of non-solvent exposed cysteines under covalent screening, residues, which under physiological conditions, are unmodified.
Figure 3. Covalent fragment screening flowchart.
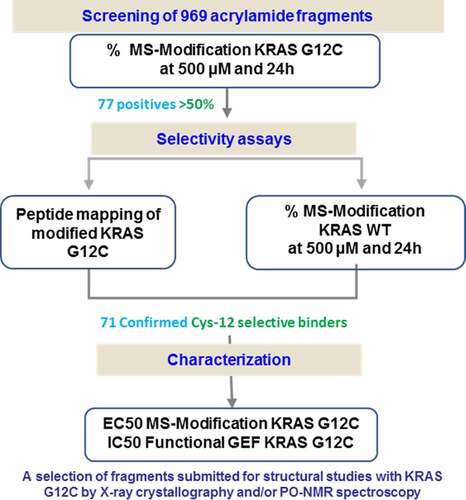
The hits were then tested on the wild-type protein, under the same assay conditions. They were generally selective for the mutant over the wild type protein and the stoichiometry of the wild type protein modification was never superior than 1:1, even for those hits forming more than one covalent adduct on the G12C mutant.
In order to determine which amino acid was covalently bound to the fragment, the mutant protein was also submitted for peptide mapping after incubation with the hits. For 71 hits, the peptide mapping revealed Cysteine 12 to be the modified residue. Cysteine 118 was the only other site identified as being modified by the fragments.
The hits were further characterized in secondary assays. The 71 fragments were tested in a dose response mass spectrometry assay, in buffer supplemented with 1 mM MgCl2. The calculated EC50 for 90% of the hits tested was below 500 µM, confirming the result of the initial screen performed at 500 µM in the presence of EDTA. The 12 most potent hits had an EC50 below 20 µM. Multiple modification was less frequent and less severe in the MgCl2 buffer. The stoichiometric modification of protein was greater than one for 16 fragments at concentrations of 500 µM or below. The conditions in which multiple fragment binding occurred were recorded to keep track of the characteristics of each hit.
In parallel, 28 hits, selected for their chemical diversity, were tested in a dose dependent nucleotide exchange reaction using a GEF (Guanine nucleotide Exchange Factor) assay to study the impact of the covalent modification on the functional activity of KRAS G12C. Of those tested, 5 fragments had an IC50 below 50 µM. The most potent fragments in this test were also potent in the mass spectrometry assay. Furthermore, the hits with low EC50 in the MS assay were not systematically active in the functional assay, suggesting that covalent attachment to Cysteine 12 was not sufficient to impair the functional activity of KRAS G12C.
In total 30 fragments were selected for follow-up based on their results in the secondary assays, as well as the ease with with which they could be chemically modified and grown into lead-like molecules. Structural studies were initiated by both X-Ray crystallography and PO-NMR (Protein Observed NMR). After extensive co-crystallization trials with the 30 fragments, diffracting crystals were obtained for 11 compounds, but only 3 unambiguously bound to the protein. For 8 compounds, the electron density map only showed density for KRAS G12C and GDP. This finding could be attributed to a significant percentage of unbound protein in our assay conditions. In some instances, protein dissolved-from-crystal showed 100% covalent fragment binding, in spite of the ligand not being visible in the final crystal structure. This finding is likely due to fragments not forming significant interactions with the protein and were thus too mobile to contribute to the electron density. Two structures where the fragments were unambiguously detected suggested bindind between symmetrical proteins in the crystal and were subsequently discarded. Cpd2, from the covalent acrylamide-containing fragments library gave rise to a structure of interest.
Switch I/II cleft binding region
In the presence of cpd2, a new crystal form was obtained after two weeks. While switch I was constrained by packing interactions, this was not the case for switch II, which was destabilized and adopted a new conformation, opening a cleft between the two switches (). There was additional density near Cys12, which facilitated the placement of the bromide-containing fragment. This fragment was only partially visible, however, the electron density of the bromide atom confirmed its presence. The fragment stacks along Tyr64 of switch II on one side and Thr35 of switch I on the other, and the bromide moiety points in the direction of the previously identified switch I/II pocket.
Figure 4. Cpd2 binding mode to KRAS G12C GDP.
This allosteric rearrangement, resulting in the creation of a cleft between the switches, was also observed with a 2-amino-pyrimidine ligand, cpd3. To the best of our knowledge, such a binding mode had not been observed in KRAS prior to these two structures.
Analysis of the complex between cpd3 and GDP-bound KRAS G12C by mass spectrometry, prior crystallization, showed that the protein was mainly modified on two cysteines, identified as Cys12 and Cys118 by peptide mapping. This finding was confirmed when solving the crystal structure and was more informative than the cpd2 structural data. The structure shows the ligand bound to both Cys12 and Cys118, but with different conformations of the piperidinyl-N-benzyl. The Cys118-bound ligand points towards a symetrically-related protein in the crystal and its benzyl moiety occupies the switch I/II pocket of this protein, thus participating to the crystal packing.
The Cys12-bound ligand lies in the cleft created by the rearrangement of switch II and its pyrimidine ring is sandwiched between Thr35 of Switch I and Glu62 of Switch II, while the nitrogen of the piperidine ring makes a H-bond with the main-chain carbonyl of Glu62. The benzyl moiety of the molecule points out of the protein towards solvent and is stabilized by crystal packing interactions ().
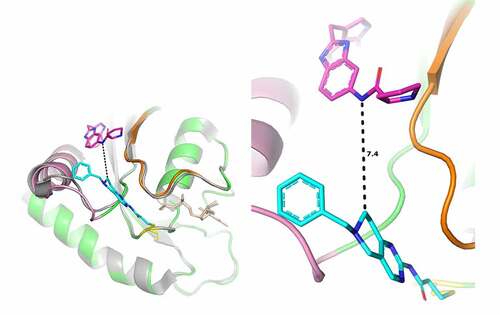
Figure 5. Cpd3 binding mode to KRAS G12C GDP.
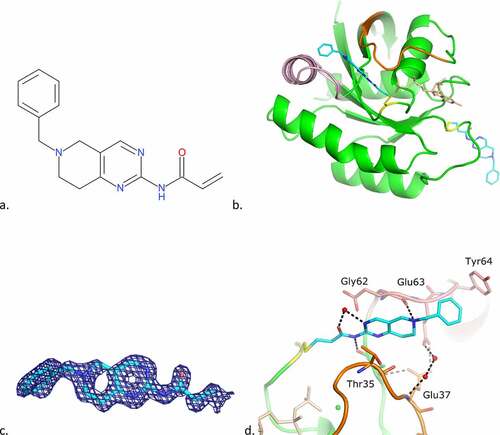
By binding in a cleft between the two switches, cpd3 opens new possibilities of allosteric inhibition. This finding was confirmed in the KRAS G12 functional GEF assay, where the fragment had an IC50 of 27 µM (). The crystal structure of the fragment suggests this ligand could be modified to introduce additional interactions with both switches: one of the nitrogen atoms of the pyrimidine ring faces Ala59 and could be replaced by a carbon to improve van der Waals contacts with the protein. Substituents on the tetrahydropyridine ring could interact with or replace the water network bridging Glu37 of switch I and Glu63 of switch II. Furthermore, the ligand is oriented such that the ‘Tyr71 pocket’ is less than 8 Å away from the tetrahydropyridine ring, suggesting the possibility of ‘growing’ the molecule towards this region (). These chemical modifications would also have to take into account the need to decrease the reactivity of the molecule, to prevent it from binding to Cys118.
Table 1. Activity of the ligands
Table 2. Crystallographic data
Figure 6. Superimposition of the structure of KRAS G12C GDP in complex with cpd3 and with 4EPY ligand.
Switch I binding region
A small chemical programme was carried out around cpd2, in order to explore binding to the cleft region. During this study the crystal structure of cpd4, a fragment from the same series as cpd2, containing a bromo-pyridine ring scaffold, in complex with KRAS G12C GDP was determined. The structure showed another unexpected mode of binding, where cpd4 underwent dimerization through a chemical cross-link between two cpd4 molecules and, while one part of the ligand was covalently bound to Cys12, the other was localized in a new pocket created by a large move of Switch I, and to a lesser extend of Switch II (). The presence of the dimeric form of the ligand both in solution and bound to KrasG12C was confirmed by PO-NMR studies.
Figure 7. Cpd4 binding mode to KRAS G12C GDP.
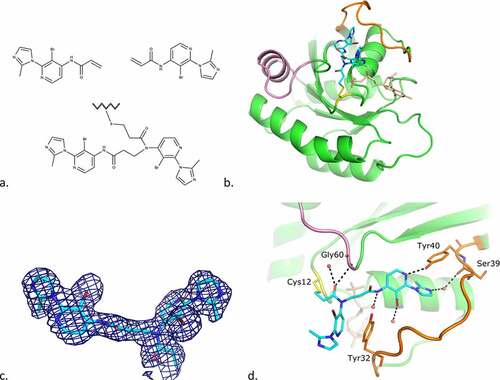
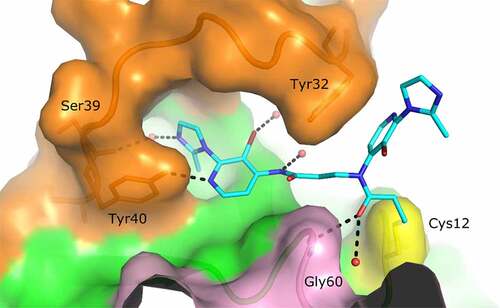
The proximal moiety of the dimer (originally the monomer covalently bound to Cys12) points towards the solvent-exposed region of the protein. The acrylamide carbonyl oxygen interacts with switch II Gly60 main-chain amide and a water molecule. Pi-stacking interactions occur between switch I Tyr32 and the pyridine ring of the inhibitor. The bromide atom of the pyridine ring interacts with Glu63 main-chain carbonyl of a crystal symmetry protein molecule (not shown in ).
The distal moiety of the dimer is deeply embedded into the groove formed by structural movements of switches I and II. The carbonyl group of this moiety is H-bonded to the Ala66 main-chain amide of the crystal symmetry molecule. This residue also creates a stacking interaction with the pyridine moiety. The nitrogen atom of the pyridine ring is H-bonded to Tyr40 side-chain hydroxyl and the bromide atom is bonded to a water molecule. The nitrogen atom of the imidazole ring interacts with switch I Ser39 main-chain carbonyl via a water molecule (). As indicated in Material and Methods, the protein buffer contained 2.5 mM MgCl2 and the incubation with the ligand was carried out in the presence of this salt. Nevertheless, the magnesium ion, usually found coordinating phosphates from GDP, was not found in the structure, probably due to the loss of coordination when Thr35 moved with switch I loop.
Cpd4 is active in the functional GEF assay with an IC50 of 49 µM () and its binding mode suggested ways in which activity could be improved. While the solvent exposed proximal part of the dimeric cpd4 is of little interest for affinity optimization, the distal moiety provides potential avenues for improvements (). For instance, the imidazole ring indirectly interacts with KRAS through a water molecule. Modifications on this moiety could lead to direct interactions with Ser39 or polar residues above. The bromide atom points towards a large cavity: a bulkier moiety here might also bring new interactions. Moreover, modifications on the pyridine ring could also bring new interactions.
Figure 8. Surface representation of KRAS G12C GDP in complex with cpd4.
Discussion
The described KRAS G12C fragment campaigns demonstrated the value of changing our approach when looking for covalent ligands that to bind proteins at unknown binding site. Our findings suggest that instead of weaponizing non-covalent binders with a warhead to bind to a proximal cysteine, it was more efficient to first identify ligands that covalently react to the cysteine of interest, to then verify whether they interact productively with the protein, and then optimize the binding interactions.
The covalent fragment screening campaign enabled us to validate in house the Switch II allosteric pocket previously identified by various authors [Citation23] and exemplified by the binding mode of sotorasib (). Screening also led to the discovery of two new allosteric binding regions ().
Figure 9. Overall KRAS G12C structure and surface representation of the four binding pockets identified on KRAS. The orientation of the protein is the same in all pictures.
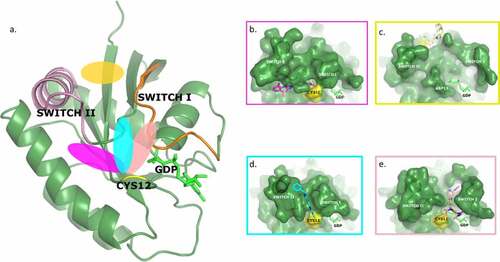
Two different ligands were observed by X-ray and/or NMR in an induced cleft between switch I and switch II (). They make direct and indirect interactions with the two switches, which, with judicious chemical modifications, could be reinforced. Furthermore, this binding mode directs the ligand towards another earlier described allosteric binding pocket [Citation14,Citation15], the switch I/II pocket (), which could be targeted by growing the molecule. Both ligands showed functional activity in a GEF assay, reinforcing the interest of this cleft binding mode. While exploring these ligand interactions, another new binding region that induced a binding pocket under switch I was serendipitously identified (). Again, the bound ligand showed functional activity and a pathway for medicinal chemistry optimization.
For the covalently binding ligands complexes described here, the in silico molecular dynamics simulations generated reproducible results for both ligands (compound 3 and 4), showing the instability of the experimentally derived x-ray structure conformation despite their covalent binding to Cys12. The non-covalent interactions were probably not strong enough to keep the ligand into its initial position as assessed by in silico methods. Optimizing the described crystallographic binding mode by introducing new interactions for the covalently bound ligands should lead to improved ligand binding occupancy and potency for both newly discovered SI/SII cleft and switch I pocket.
During the course of this study, KRAS crystallized in a large number of different space groups. Four different crystal forms were observed for the GDP-form alone, and each covalently-bound protein crystallized in a different crystal form, with different switches conformations. These findings highlight the structural plasticity of the protein.
The current and recently published structural studies of KRAS protein demonstrate a surprisingly high level of protein plasticity, which has led to the identification of allosteric ligandable binding pockets in a protein that was thought for decades to be non-ligandable. In the present study, we have identified two de novo binding modes capturing two new allosteric pockets on the KRAS G12C surface, which could be exploited in the search for new inhibitors of KRAS activity.
Author Contributions
M. Mathieu: x-ray crystallography, management, paper writing
V. Steier: protein biochemistry and crystallization
F. Fassy: mass spectrometry and biochemistry experiments, paper writing
C. Delorme: protein biochemistry
D. Papin: in-silico modelling
B. Genet: mass spectrometry experiments
F. Duffieux: recombinant protein design and production
T.Bertrand: x-ray crystallography, paper writing
L. Delarbre: SPR experiments design and analysis, paper writing
H. Le Borgne: Mass spectrometry experiments
A. Parent: NMR experiments
P. Didier: protein biochemistry
J.P. Marquette: DSF experiments
M. Lowinski: SPR experiments.
J. Houtmann: DSF experiments
A. Lamberton: SPR experiments
L. Debussche: designed and coordinated research
A.Rak: project strategy, data analysis and interpretation, management, paper writing and submission
Competing Interest Statement
All authors: No competing interests besides Sanofi employee (or retired from Sanofi employment) and Sanofi shareholder
Acknowledgments
We thank Dr Kwame Amaning for critical reading of the manuscript. We thank Dr Andreas Karlsson for preparing analysis of the non-covalent fragment library. We thank Dr Eiso AB and Dr Gregg Seagal from ZOBIO for providing incredible PO-NMR support in the project and great collaboration spirit. ESRF and Soleil synchrotrons staffs are acknowledged for assisting in the x-ray data collection.
References
- Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517–531.
- Rajalingam K, Schreck R, Rapp UR, et al. Ras oncogenes and their downstream targets. Biochim Biophys Acta. 2007;1773:1177–1195.
- Cox AD, Fesik SW, Kimmelman AC, et al. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. 2014;13:828–851.
- Scheffler M, Ihle MA, Hein R, et al. K-ras Mutation Subtypes in NSCLC and Associated Co-occuring Mutations in Other Oncogenic Pathways. J Thorac Oncol. 2019;14:606–616.
- Biankin AV, Waddell N, Kassahn, KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405.
- Cancer Genome Atlas Research, N. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550.
- Neumann J, Zeindl-Eberhart E, Kirchner T, et al. Frequency and type of KRAS mutations in routine diagnostic analysis of metastatic colorectal cancer. Pathol Res Pract. 2009;205:858–862.
- Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci. 2016;129:1287–1292.
- Moore AR, Rosenberg SC, McCormick F, et al. RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discov. 2020;19:533–552.
- Mott HR, Owen D. Structures of Ras superfamily effector complexes: what have we learnt in two decades? Crit Rev Biochem Mol Biol. 2015;50:85–133.
- Gasper R, Wittinghofer F. The Ras switch in structural and historical perspective. Biol Chem. 2019;401:143–163.
- Ganguly AK, Wang Y-S, Pramanik BN, et al. Interaction of a novel GDP exchange inhibitor with the Ras protein. Biochemistry. 1998;37:15631–15637.
- Palmioli A, Sacco E, Abraham S, et al. First experimental identification of Ras-inhibitor binding interface using a water-soluble Ras ligand. Bioorg Med Chem Lett. 2009;19:4217–4222.
- Sun Q, Burke JP, Phan J, et al. Discovery of small molecules that bind to K-Ras and inhibit Sos-mediated activation. Angew Chem Int Ed Engl. 2012;51:6140–6143.
- Maurer T, Garrenton LS, Oh A, et al. Small-molecule ligands bind to a distinct pocket in Ras and inhibit SOS-mediated nucleotide exchange activity. Proc Natl Acad Sci U S A. 2012;109:5299–5304.
- Ostrem JM, Peters U, Sos ML, et al. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551.
- Ostrem JM, Shokat KM. Direct small-molecule inhibitors of KRAS: from structural insights to mechanism-based design. Nat Rev Drug Discov. 2016;15:771–785.
- Hallin J, Engstrom LD, Hargis L, et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients, Cancer Discov, 2020; 10(1):54-71.
- Shin Y, Jeong JW, Wurz RP, et al. Discovery of N-(1-Acryloylazetidin-3-yl)-2-(1H-indol-1-yl)acetamides as Covalent Inhibitors of KRAS(G12C). ACS Med Chem Lett. 2019;10:1302–1308.
- Canon J, Rex K, Saiki AY, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217–223.
- Kettle JG, Bagal SK, Bickerton S, et al. Structure-Based Design and Pharmacokinetic Optimization of Covalent Allosteric Inhibitors of the Mutant GTPase KRAS(G12C), J Med Chem, 2020;63(9):4468-4483.
- Fell JB, Fisher JP, Baer BR, et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRAS(G12C) Inhibitor for the Treatment of Cancer. J Med Chem. 2020;63(13):6679–6693.
- Ni D, Li X, He X, et al. Drugging K-Ras(G12C) through covalent inhibitors: mission possible? Pharmacol Ther. 2019;202:1–17.
- Congreve M, Carr R, Murray C, et al. A ‘rule of three’ for fragment-based lead discovery? Drug Discov Today. 2003;8:876–877.
- Jhoti H, Williams G, Rees DC, et al. The ‘rule of three’ for fragment-based drug discovery: where are we now? Nat Rev Drug Discov. 2013;12:644.
- Schultes SDG, C.; Haaksma, E.E.J; de Esch I.J.P.; Leurs, R.; Krämer, O. Ligand Efficiency as a guide in fragment hit selection and optimization. Drug Discov Today. 2010;7:e157-e162.
- Liu Q, Sabnis Y, Zhao Z, et al. Developing irreversible inhibitors of the protein kinase cysteinome. Chem Biol. 2013;20:146–159.
- Weisner J, Gontla R, van der Westhuizen L, et al. Covalent-Allosteric Kinase Inhibitors. Angew Chem Int Ed Engl. 2015;54:10313–10316.
- Hwang T-L, Shaka AJ. Water suppression that works. Excitation sculpting using arbitrary waveforms and pulsed field gradients. Journal of Magnetic Resonance Series A. 1995;112(2):275–279. https://doi.org/10.1006/jmra.1995.1047
- Mayer M, Meyer B. Characterization of Ligand Binding by Saturation Transfer Difference NMR Spectroscopy. Angew Chem Int Ed Engl. 1999;38:1784–1788.
- Bretonnet AS, Jochum A, Walker O, et al. NMR screening applied to the fragment-based generation of inhibitors of creatine kinase exploiting a new interaction proximate to the ATP binding site. J Med Chem. 2007;50:1865–1875.
- Meinecke R, Meyer B. Determination of the binding specificity of an integral membrane protein by saturation transfer difference NMR: RGD peptide ligands binding to integrin. J Med Chem. 2001;44:3059–3065.
- Perkins DN, Pappin DJ, Creasy DM, et al. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(3551–67):3551–3567.
- Vonrhein C, Flensburg C, Keller P, et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D Biol Crystallogr. 2011;67:293–302.
- Winn MD, Ballard CC, Cowtan KD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242.
- McCoy AJ, Grosse-Kuntsleve RW, Adams PD, et al. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674.
- Blanc E, Roversi P, Vonrhein C, et al. Refinement of severely incomplete structures with maximum likelihood in BUSTER–TNT. Acta Crystallogr D Biol Crystallogr. 2004;60:2210–2221.
- Emsley P, Lohkamp B, Scott WG, et al. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501.
- Kessler D, Bergner A, Böttcher J, et al. Drugging all RAS isoforms with one pocket. Future Med Chem. 2020;12(21):1911–1923.
- Hall BE, Bar-Sagi D, Nassar N. The structural basis for the transition from Ras-GTP to Ras-GDP. Proc Natl Acad Sci U S A. 2002;99:12138–12142.