
ABSTRACT
N-terminal methylation (Nα-methylation) by the methyltransferase NRMT1 is an important post-translational modification that regulates protein–DNA interactions. Accordingly, its loss impairs functions that are reliant on such interactions, including DNA repair and transcriptional regulation. The global loss of Nα-methylation results in severe developmental and premature aging phenotypes, but given over 300 predicted substrates, it is hard to discern which physiological substrates contribute to each phenotype. One of the most striking phenotypes in NRMT1 knockout (Nrmt1−/−) mice is early liver degeneration. To identify the disrupted signaling pathways leading to this phenotype and the NRMT1 substrates involved, we performed RNA-sequencing analysis of control and Nrmt1−/− adult mouse livers. We found both a significant upregulation of transcripts in the cytochrome P450 (CYP) family and downregulation of transcripts in the major urinary protein (MUP) family. Interestingly, transcription of both families is inversely regulated by the transcription factor zinc fingers and homeoboxes 2 (ZHX2). ZHX2 contains a non-canonical NRMT1 consensus sequence, indicating that its function could be directly regulated by Nα-methylation. We confirmed misregulation of CYP and MUP mRNA and protein levels in Nrmt1−/− livers and verified NRMT1 can methylate ZHX2 in vitro. In addition, we used a mutant of ZHX2 that cannot be methylated to directly demonstrate Nα-methylation promotes ZHX2 transcription factor activity and target promoter occupancy. Finally, we show Nrmt1−/− mice also exhibit early postnatal de-repression of ZHX2 targets involved in fetal liver development. Taken together, these data implicate ZHX2 misregulation as a driving force behind the liver phenotype seen in Nrmt1−/− mice.
KEYWORDS:
Introduction
N-terminal methylation (Nα-methylation), occurring on the α-amino group at the N-terminus of proteins, is a post-translational modification (PTM) that can affect protein stability and nucleic acid-binding activity [Citation1,Citation2]. Nα-methylation was first observed as a PTM many decades ago [Citation3], but it was only 12 years ago that the responsible methyltransferase was identified as N-terminal RCC1 methyltransferase 1 (NRMT1) [Citation4]. NRMT1 is the major eukaryotic Nα-trimethylase that sequentially mono-, di-, and trimethylates protein N-termini after removal of the initiating methionine (Met) [Citation4]. It was originally thought that NRMT1 could only act on N-termini with the canonical consensus sequence X-Pro-Lys, where X is Ala, Pro, or Ser [Citation3]. However, comprehensive sequence analysis identified a greatly expanded non-canonical consensus sequence, which allows A/P/S/G/M in the first position, A/P/S/G/M/E/N/Q in the second position, and K/R in the third position after Met cleavage [Citation5]. This expanded consensus sequence predicts over 300 potential NRMT1 targets, with dozens being verified in recent years [Citation5–11].
The most well-studied function of Nα-methylation by NRMT1 is its ability to promote DNA-binding of its substrates [Citation2,Citation4,Citation6,Citation8,Citation10]. The first characterized substrate of NRMT1, regulator of chromatin condensation 1 (RCC1), requires Nα-methylation for association with chromatin and subsequent proper spindle assembly and chromosome segregation during mitosis [Citation2,Citation4]. The use of a methyl-deficient mutant with Glu replacing Lys in the third position of the RCC1 N-terminal consensus sequence (K4Q mutant) revealed that RCC1 does not co-localize with chromatin when unmethylated [Citation2], and knockdown of NRMT1 recapitulates this phenotype [Citation4]. In vitro binding assays confirmed an increased association of methylated RCC1 to DNA and not histone proteins [Citation2]. RCC1 was the first protein shown to require Nα-trimethylation by NRMT1 for proper association with DNA, but several other similarly regulated proteins have now been identified, including the centromere protein B (CENP-B) [Citation6], damaged DNA-binding protein 2 (DDB2) [Citation8], and myosin light chain 9 (MYL9) [Citation10].
CENP-B, which has a GPK canonical consensus sequence, binds to a CENP-B box sequence in centromeric DNA, and a K4Q CENP-B mutant was unable to bind the CENP-B box as strongly as wild-type (WT) [Citation6]. WT DDB2, which has an APK canonical consensus sequence, was recruited to DNA damage foci at a greater rate than the K4Q unmethylatable DDB2 mutant [Citation8], and a K4Q mutant of the transcription factor MYL9 decreased its occupancy at target promoters [Citation10]. The N-terminal Ser-Ser-Lys (SSK) consensus sequence of MYL9 makes it the first non-canonical target identified whose DNA binding is regulated by Nα-methylation [Citation10]. Interestingly, the SSK consensus sequence also allows for Nα-acetylation of MYL9, and while Nα-methylation regulates its nuclear role as a transcription factor, we have also shown that Nα-acetylation regulates its cytoplasmic roles in cell migration [Citation10].
To more comprehensively identify additional targets of NRMT1 regulation, we performed RNA-sequencing (RNA-seq) analysis on the livers of Nrmt1−/− mice. Livers were chosen because one of the most striking phenotypes of the Nrmt1−/− mice is the early degeneration of this tissue [Citation12]. When compared to transcript levels in WT mice livers, hundreds of differentially expressed genes were identified in Nrmt1−/− mice. Most notably, transcripts belonging to the cytochrome P450 (CYP) family were more highly expressed in Nrmt1−/− livers, and transcripts of the major urinary protein (MUP) family showed reduced expression in Nrmt1−/− livers. Interestingly, these families are inversely regulated by a common transcription factor, zinc fingers and homeoboxes 2 (ZHX2). ZHX2 both represses the expression of many CYP genes and activates the expression of many MUP genes [Citation13,Citation14].
ZHX2 is a member of a family of transcriptional factors that also includes ZHX1 and ZHX3, all of which can homo- or heterodimerize [Citation15,Citation16]. The structure of ZHX2, including two zinc finger motifs and five homeodomains, suggests that it interacts with both other proteins and nucleic acids [Citation17,Citation18]. ZHX2 has been implicated in liver development through its postnatal repression of fetal developmental regulators alpha-fetoprotein (AFP), glypican-3 (GPC3), and lncRNA H19 [Citation19,Citation20]. It has also been implicated in numerous diseases, including liver and renal cancers [Citation21–24]. In hepatocellular carcinoma (HCC), ZHX2 acts as a tumor suppressor and its loss from the nucleus leads to increased proliferation through increased expression of its targets cyclin A and cyclin E [Citation22]. However, in clear cell renal cell carcinoma (ccRCC), ZHX2 acts as an oncogene by activating the nuclear factor kappa B (NF-ĸB) pathway, and its loss slows cell growth [Citation24].
Given our RNA-seq data and the fact that ZHX2 has a non-canonical Ala-Ser-Lys (ASK) NRMT1 consensus sequence, we decided to test whether ZHX2 is a direct target of NRMT1 and if Nα-methylation of ZHX2 regulates its function as a transcription factor. Here, we verify that NRMT1 can methylate ZHX2 in vitro, and through the use of a K4Q unmethylatable ZHX2 mutant, we also find that Nα-methylation of ZHX2 is required for activation of its target genes and increases target gene promoter occupancy. Additionally, we show that while Nrmt1−/− mice postnatally repress Afp expression similar to WT mice, expression of both Gpc3 and H19 is de-repressed by 21 days after birth. Taken together, we find that ZHX2 is a novel target of NRMT1 and loss of Nα-methylation leads to aberrant regulation of ZHX2 function as a transcription factor, resulting in altered liver metabolic and developmental pathways.
Materials and methods
RNA sequencing
RNA was isolated from the livers of three 3-month-old male control C57BL/6J and C57BL/6J-Nrmt1−/− (Nrmt1−/−) mice using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA) as previously described [Citation12]. Two mRNA libraries per sample were made per the standard Illumina protocol and sequenced on the Illumina NextSeq 500 System at the University of Louisville Center for Genetics and Molecular Medicine. Eleven of the 12 libraries produced analyzable data, so all but one of the control samples are represented as duplicates. There were 23,545 genes represented in the 11 samples. Genes with low expression were eliminated, and those represented by at least 1 count per 1 million reads in at least three samples were included in subsequent analysis. Across the 11 samples, 12,612 genes were retained. The trimmed mean of M-values (TMM) method [Citation25] implemented in edgeR (3.4) was used to calculate normalization factors and examine inter-sample reproducibility. The reads were mapped to the Ensembl Mus musculus genome provided by Illumina and STAR (2.5.3a) aligner using default settings. The dispersion parameter for each gene was estimated using the quantile-adjusted conditional maximum likelihood (qCML) method, appropriated for experiments with a single factor. The functions estimateCommonDisp and estimateTagwiseDisp were used to estimate dispersion. Differential expression was tested using an exact test based on qCML methods. The Benjamini–Hochberg correction was used with a false discovery cutoff of 0.1. With these parameters, 102 genes (0.809%) were shown positively regulated in control vs. Nrmt1−/− mice and 119 genes (0.944%) were negatively regulated. The heatmap of the top 50 positively and negatively regulated genes was generated using the heatmap.2 function of gplots package in R [Citation26].
Gene ontology (GO) analysis
Gene ontology (GO) analysis of the top 50 positively regulated and negatively regulated transcripts in the Nrmt1−/− mice and control mice was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID) (ver 6.8). Genes were grouped by both molecular function (MF) and biological process (BP).
Molecular cloning
Full-length wild-type (WT) or methylation-deficient mutant K4Q (ASQ) ZHX2 were amplified from human ZHX2 cDNA in pOTB7 (Horizon Discovery, Waterbeach, United Kingdom) and cloned into the XbaI and BamHI sites of pKGFP2 or pKH3 (generous gifts from Dr. Ian Macara) to put a C-terminal GFP or FLAG tag on ZHX2, respectively. The 5’ primer for the WT constructs was 5’-TCTAGAATGGCTAGCAAACGAAAATC-3’ and the 5’ primer for the ASQ constructs was 5’-TCTAGAATGGCTAGCCAACGAAAATC-3’. The 3’ primer for both GFP constructs was 5’-GGATCCGGCCTGGCCAGCCTCTGCAG-3’. The 3’ primer for both FLAG constructs was 5’-GCGGATCCTCATTTATCATCATCATCTTTATAATCGGCCTGGCCAGCCTCTGCAG-3’. To make an expression vector that simultaneously produces NRMT1 and NRMT2, each was cloned into pETDuet-1 (EMD Millipore, Burlington, MA). First, the human NRMT1 ORF [Citation4] was amplified to introduce a 5’ HindIII restriction site and a 3’ NotI restriction site with a stop codon and subcloned into pETDuet-1 using HindIII and NotI to put an N-terminal His tag on NRMT1. Next, the human NRMT2 ORF [Citation27] was amplified to introduce a 5’ NdeI restriction site and a 3’ BglII restriction site with a stop codon. As NRMT1 naturally contains a BglII restriction site, QuikChange site-directed mutagenesis (Agilent, Santa Clara, CA) was used on the NRMT1-pETDuet-1 vector to introduce a silent mutation at Ile214 (ATC to ATA) and disrupt the BglII site. The amplified NRMT2 ORF was then subcloned into this NRMT1-pETDuet-1 vector using NdeI and BglII, which will result in the production of untagged NRMT2. The following primers were used: 5HindIIINRMT1: 5’-GCAAGCTTACGAGCGAGGTGATAGAAGAC-3’; 3NotIStopNRMT1: 5’-CCGCGGCCGCTCATCTCAGGGCAAAGCTATA-3’; hsNRMT1BglIIQC For: 5’-GGAGAACCTCCCCGATGAGATATACCATGTCTATAGC-3’; hsNRMT1BglIIQC Rev: 5’-GCTATAGACATGGTATATCTCATCGGGGAGGTTCTCC-3’; 5NdeINRMT2: 5’-GGCATATGGCCCACCGGGGAGCCCA-3’; 3BglIINRMT2Stop: 5’-GCAGATCTTCAGGAGTGTCTGTCGCTGTG-3’. All constructs generated were verified by DNA sequencing.
Cell culture and lipofectamine transfection
Human renal carcinoma 786-O cells (ATCC, Manassas, VA) were cultured in RPMI 1640 media (Corning, Corning, NY) supplemented with 10% fetal bovine serum (FBS; Atlanta Biologicals, Atlanta, GA) and 1% penicillin–streptomycin (P/S; Thermo Fisher Scientific). WT human colorectal carcinoma HCT116 cells (generous gift from Dr. Ian Macara) and HCT116 cells with CRISPR/Cas9-mediated NRMT1 knockout [Citation28] were cultured in McCoy’s media (Thermo Fisher Scientific), supplemented with 10% FBS and 1% P/S. All the cells were grown at 37°C and 5% CO2 on tissue culture treated plastic (Corning). For quantitative real-time PCR analysis (qRT-PCR) analysis, 786-O or HCT116 cells were transfected with 1.5 μg WT-ZHX2-GFP or K4Q-ZHX2-GFP using Lipofectamine 2000 reagent (Thermo Fisher Scientific) per manufacturer’s instructions. After 24 hours, GFP expression was confirmed with an EVOS fluorescent microscope (Thermo Fisher Scientific) and cells were harvested for mRNA.
Quantitative real time PCR analysis
Mouse livers or 786-O pellets were homogenized in TRIzol (Invitrogen) and mixed with chloroform to extract RNA. RNA was pelleted in isopropanol, washed with ethanol, and resuspended in 40–100 μl sterile water. cDNA was synthesized from 1 μg RNA using the SuperScript First-Strand Synthesis System (Invitrogen) and diluted 1:10 in sterile water. For quantitative RT-PCR, 2 μl of each sample was used with 2X SYBR green Master Mix and the CFX96 Touch Real-Time PCR Detection System (Bio-Rad). Samples were run in triplicate. Transcript expression levels were determined using the ΔΔCT quantification method. Primer sequences (Invitrogen) were as follows: mouse β-tubulin forward 5’- TGTCCTGGACAGGATTCGC-3’ and reverse 5’-CTCCATCAGCAGGGAGGTG-3’; mouse Cyp17a1 forward 5’- CAATGACCGGACTCACCTCC-3’ and reverse 5’- GATCACTGTGTGTCCTTCG-3’; mouse Cyp2a4 forward 5’-CGTTGGGAACTTCCTTCAGC-3’ and reverse 5’-CTTCCTTGACTGTCTCCTGT-3’; mouse Mup1 forward 5’-CACACTACAGATCGCTGCCT-3’ and reverse 5’-CAGGATGGAATGCAGATCAC-3’; mouse Mup7 forward 5’-TACCAAAATGAAGATGCTGC-3’ and reverse 5’-ATAGTATGCCATTCCCCA-3’; human GAPDH forward 5’-ACAGCCTCAAGATCATCAGCAA-3’ and reverse 5’-CCATCACGCCACAGTTTCC-3’; human BCL2 forward 5’-ATCGCCCTGTGGATGACTGAGT-3’ and reverse 5’-GCCAGGAGAAATCAAACAGAGGC-3’; human ICAM1 forward 5’-CTCAGTTTCCCAGCGACAGG-3’ and reverse 5’-GGAAGCTGCGTGATCCCTAC-3’; human IL8 forward 5’-TCTGACATAATGAAAAGATGAGGGT-3’ and reverse 5’-CCTTCCGGTGGTTTCTTCCT-3’ [Citation24]; mouse Afp forward 5’-ATCAGTGTCTGCTGGCACGCA-3’ and reverse 5’-GGCTGGGGCATACATGAAGGGG-3’ [Citation13]; mouse H19 forward 5’- CAGTGATCGGTGTCTCGAAG-3’ and reverse 5’-CTGTCACATTGACCACACCT-3’; and mouse Gpc3 forward 5’-GTCACCAGGTCCGTTCTTTC-3’ and reverse 5’-GTAGTTCTTGGCATGGCGAA-3’ [Citation29].
Chromatin immunoprecipitation
Human 786-O cells were plated at a density of 200,000 cells on 60 mm plates (Corning). The next day, cells were either left untransfected (control) or transfected with 1.5 μg of WT-ZHX2-FLAG or K4Q-ZHX2-FLAG using Lipofectamine 2000 (Thermo Fisher Scientific) per manufacturer’s instructions. Cells were harvested after 24 h with trypsin, cross-linked with 1% formaldehyde for 10 min at RT, quenched with 1X glycine for 5 min at RT, resuspended in 600 µl ChIP lysis buffer (150 mM NaCl, 25 mM Tris–HCl, pH 7.5, 5 mM EDTA, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate (NaDoc)), and sonicated 10 × 10 s at 30% amplitude to fragment DNA. The sonicated DNA was saved as input, and 8 μg of each sample was used for immunoprecipitation. ZHX2-FLAG was immunoprecipitated from lysates for 1 h at 4°C using 10 μl Pierce Anti-DYKDDDDK (FLAG) magnetic agarose beads (Thermo Fisher Scientific) after lysates had been pre-cleared for 1 h at 4°C using 5 μl ChIP-grade protein G agarose beads (Cell Signaling Technologies, Danvers, MA). After immunoprecipitation, beads were washed one time in high salt ChIP wash buffer (500 mM NaCl, 20 mM Tris–HCl, pH 8.0, 0.1% SDS, 1% NP-40, 2 mM EDTA), one time in LiCl ChIP wash buffer (0.25 M LiCl, 10 mM Tris–HCl, pH 8.0, 1% NaDoc, 1% NP-40, 1 mM EDTA) and two times in Tris–EDTA (TE). Bound material was eluted 2X in 100 μl elution buffer (1% SDS, 0.1 M NaHCO3) for 15 min at RT. De-cross-linking was performed by adding 8 μl 5 M NaCl to each sample and incubating them overnight at 65°C. Eluates were then treated with 1 μl of RNase A (24 mg/ml stock, Thermo Scientific), 4 μl 0.5 M EDTA, 8 μl 1 M Tris-HCl, and 1 μl of proteinase K (10 mg/ml stock, Millipore Sigma) and incubated at 45°C for 1 h. The remaining immunoprecipitated DNA was isolated using a PCR purification kit (Qiagen, Hilden, Germany) and resuspended in 30 μl of TE buffer. The input samples were processed alongside ChIP samples after elution. Equal amounts of purified DNA were amplified by qRT-PCR with 2X SYBR green Master Mix and the CFX-96 Touch Real-Time PCR Detection System (Bio-Rad, Hercules, CA). The analysis was performed by normalizing each ChIP sample to 100 ng of input, then comparing the WT or K4Q samples to the untransfected control using the ΔΔCT quantification method. Samples were run in triplicate. The experiment was performed three times. The primer sequences (Invitrogen, Waltham, MA) for the ICAM1 promoter were human ICAM1 forward 5’-CTCAGTTTCCCAGCGACAGG-3’ and reverse 5’-GGAAGCTGCGTGATCCCTAC-3’ [Citation24].
Western blots
Liver lysates were generated by homogenizing livers into a buffer containing 15 mM Tris (pH 7.6), 0.25 M sucrose, 2 mM EDTA, 1 mM EGTA, 10 mM Na3VO4, 25 mM NaF, 10 mM sodium pyrophosphate, 1 mM PMSF, 1 μg/mL leupeptin, and 1 μg/mL aprotinin. Protein samples were quantified using a Bradford assay, and an equal concentration was loaded for each sample in 5 μl of Laemmli Sample Buffer (60 mM Tris (pH 6.8), 2% SDS, 10% glycerol, 5% β-mercaptoethanol, 0.01% bromophenol blue). Samples were separated on 10% SDS-PAGE gels and transferred to nitrocellulose membranes (Bio-Rad) using a Trans-Blot Turbo Transfer System (Bio-Rad). Membranes were incubated for 1 h at room temperature in a blocking buffer (5% w/v nonfat dry milk in TBS + 0.1% Tween 20 (TBS-T)). Primary and secondary antibodies were incubated in 5% w/v nonfat dry milk in TBS-T according to manufacturer’s instructions. Dilutions used for primary antibodies were rabbit anti-GAPDH (1:1000; 2275-PC-100, Trevigen, Gaithersburg, MD), rabbit anti-β-tubulin (9F3) (1:1000; Cell Signaling Technology), rabbit anti-NRMT1 (D9D6P) (1:1000; Cell Signaling Technology), mouse anti-ZHX2 (D-2) (1:1000; sc-393399, Santa Cruz Antibodies, Dallas, TX), rabbit anti-CYP26A1 (1:1000; PA5-28165, Invitrogen), and rabbit anti-MUP (1:1000; PA5-112879, Invitrogen). Secondary antibodies used were donkey anti-mouse or donkey anti-rabbit (1:5000; Jackson ImmunoResearch, West Grove, PA). Blots were developed on a ChemiDoc Touch imaging system (Bio-Rad) using Clarity Western ECL Substrate (Bio-Rad) or SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific). Quantification of blots was performed using ImageJ 1.53e (NIH, Bethesda, MD).
In vitro methylation assays
Recombinant His6-tagged NRMT1 or His6-tagged NRMT1 + NRMT2 proteins were purified as previously described [Citation4,Citation30]. Briefly, pET15b-NRMT1 [Citation5] or pETDuet-1-NRMT1/NRMT2 was expressed in BL21 Star (DE3) E. coli (Thermo Fisher Scientific) and purified on Ni2+-NTA beads (Qiagen). NRMT1 and NRMT2 complex together [Citation1], so the His-tagged NRMT1 expressed by the pET15b will also pull out the untagged NRMT2. The methyltransferase assays were conducted using the MTase-Glo Methyltransferase Assay (Promega, Madison, WI) following the protocol from the manufacturers. Briefly, 0.2 μM NRMT1 or 0.2 μM NRMT1 + NRMT2 was incubated in wells on a 96-well plate with 40 μM of synthetic peptide substrate corresponding to the first 15 N-terminal amino acids of WT ZHX2 or K4Q ZHX2 (minus the initiating methionine) (AnaSpec, Fremont, CA) in the presence of 40 μM S-adenosyl methionine (SAM). Reactions were incubated at room temperature and stopped after 20 minutes with the addition of 0.5% trifluoroacetic acid. The MTase-Glo detection reagents were added according to the manufacturer’s protocols, and luminescence was measured using the Cytation5 Imaging System (BioTek, Winooski, VT). Background signal was measured for each condition through the inclusion of no substrate control reactions.
Statistical analyses
All statistical analysis was performed using Prism 6 software (GraphPad, San Diego, CA). Statistical test used is denoted in the figure legends. Results shown are mean ± standard error of the mean (SEM).
Results
RNA sequencing of Nrmt1−/− livers
Our initial characterization of the NRMT1 knockout (Nrmt1−/−) mouse was striking, given the widespread aging phenotypes we observed, including extensive liver degeneration [Citation12]. To begin to understand what disrupted signaling pathways lead to this degeneration and what NRMT1 substrates are involved, we performed RNA sequencing (RNA-seq) analysis on livers from three-month-old Nrmt1−/− and control wild type (WT) C57BL/6J male mice. The RNA-seq revealed over 200 transcripts with differential expression between the two groups (102 genes positively regulated in WT, 119 genes negatively regulated).
Aanalysis of the top 50 up- and downregulated genes showed the expected misregulation of genes involved in liver metabolism, small molecule and pheromone processing, and transport (, Supplemental Figures 1–3). Nrmt1 was one of the top ten transcripts with higher expression in WT livers, further validating the sequencing results ()). Interestingly, several genes belonging to the major urinary protein (MUP) family were additionally upregulated in WT/downregulated in the Nrmt1−/− livers, including Mup8, Mup14, Mup7, Mup15, Mup1, Mup12, and Mup21 ()). MUP proteins are secreted by the liver, bind pheromones and other lipophilic molecules, regulate their transportation, and are ultimately secreted in the urine [Citation31]. Gene ontology (GO) analysis of the top 50 genes downregulated in Nrmt1−/− livers confirms enrichment of MUP transcripts in the dataset, showing the highest functional enrichment in genes involved in transporter activity and small molecule and pheromone binding ()). Enriched biological processes include ion and transmembrane transport, as well as negative regulation of lipid biosynthetic processes ()).
Figure 1. Transcripts downregulated in Nrmt1−/− mice. (a) The top ten transcripts with lower expression in Nrmt1−/− mice include Nrmt1 and Mup8. Extending past the top ten into the top 50 reveals downregulation of several other major urinary protein (Mup) transcripts. Gene ontology analysis provided enriched (b) molecular functions and (c) biological processes of the top 50 genes with downregulated expression.
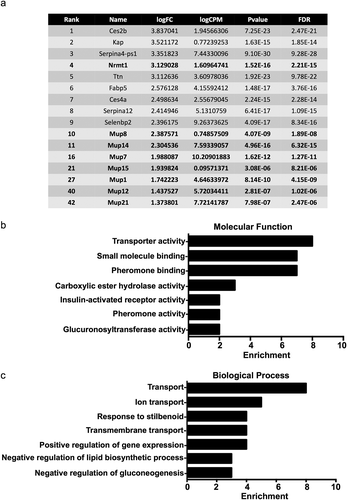
Figure 2. Transcripts upregulated in Nrmt1−/− mice. (a) The top ten transcripts with higher expression in Nrmt1−/− mice include Cyp2a4 and Cyp4a14. Extending past the top ten into the top 50 reveals upregulation of several other cytochrome P450 (Cyp) transcripts. Gene ontology analysis provided enriched (b) molecular functions and (c) biological processes of the top 50 genes with upregulated expression.
Conversely, several genes belonging to the cytochrome P450 (CYP) family showed downregulated expression in WT livers/upregulation in Nrmt1−/− livers, including Cyp2a4, Cyp4a14, Cyp26a1, Cyp17a1, Cyp2b10, Cyp4a10, Cyp4a31, and Cyp2b9 ()). The CYP family is comprised of hemeproteins that regulate steroid hormone synthesis and the metabolism of drugs and other xenobiotics, primarily through their monooxygenase hydroxylation activity [Citation32]. GO analysis of the top 50 genes upregulated in Nrmt1−/− livers confirms enrichment of CYP genes in the dataset, showing the highest functional enrichment in genes involved in iron ion binding, heme binding, and oxidoreductase and monooxygenase activity ()). Also, enriched are epoxygenase, aromatase, and steroid hydroxylase activity, all functions of CYP enzymes [Citation33–35]. Enriched biological processes include oxidation-reduction processes, epoxygenase P450 pathway, steroid metabolic processes, and xenobiotic metabolic processes ()).
Verification of RNA sequencing
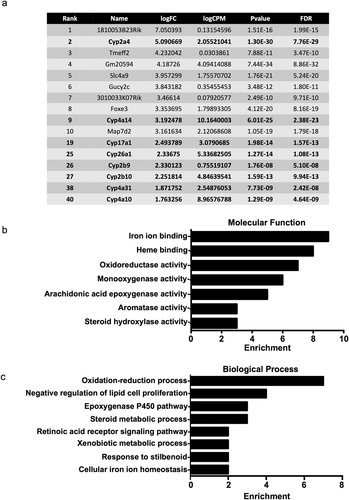
To verify the RNA-seq data at the mRNA level, quantitative real-time PCR (qRT-PCR) was performed. First, as a control, Nrmt1 mRNA levels were compared between WT and Nrmt1−/− livers and found to be significantly higher in WT samples ()). qRT-PCR analysis of two representative Mup genes, Mup1 and Mup7, also showed significantly higher levels in WT samples ()). Mup1 and Mup7 transcripts were chosen for analysis due to their high differential expression between WT and Nrmt1−/− livers and regions of sequence disparity between other Mup genes that allowed for specific primer design. qRT-PCR analysis of two representative Cyp genes from the RNA-seq data, Cyp17a1 and Cyp2a4, also verified the RNA-seq data, showing significantly lower expression in WT livers ()). These Cyp genes were also chosen for their high differential expression in the RNA-seq data and ease of primer design.
Figure 3. qRT-PCR verification of RNA sequencing. (a-c) qRT-PCR analysis of wild-type (WT) and Nrmt1 knockout (Nrmt1−/−) mice shows (a) decreased Nrmt1 expression, (b) decreased Mup1 and Mup7 expression, and (c) increased Cyp17a1 and Cyp2a4 expression in Nrmt1−/− mice. * denotes p < 0.05 and ** denotes p < 0.01 as determined by unpaired t-test. n = 3. Error bars represent ± standard error of the mean (SEM).
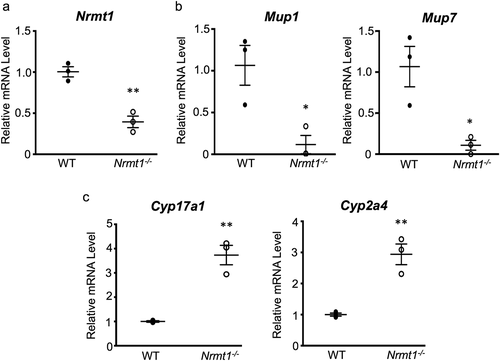
To verify the RNA-seq data at the protein level, protein expression changes between WT and Nrmt1−/− livers were measured by western blot. Again verifying the RNA-seq data, MUP protein expression was significantly higher in the WT liver samples ()). Total MUP protein was analyzed by western blot, as opposed to individual MUP proteins, as the MUP proteins are highly similar and specific antibodies are not readily available. Since at least seven Mup genes are downregulated in Nrmt1−/−livers, we reasoned correctly that this should result in a significant reduction in total MUP protein. We also confirmed that Cyp26a1 protein levels were significantly lower in WT livers as compared to Nrmt1−/− livers ()). Cyp26a1 was chosen for its high differential expression and antibody quality.
Figure 4. Western blot verification of RNA sequencing. (a) Western blot analysis of wild-type (WT) and Nrmt1 knockout (Nrmt1−/−) mouse livers verifies (b) significant downregulation of MUP protein levels in Nrmt1−/− mice and (c) significant upregulation of CYP26A1 protein levels in Nrmt1−/− mice. GAPDH is used as a loading control. ** denotes p < 0.01 and *** denotes p < 0.001 as determined by unpaired t-test. n = 3. Error bars represent ± standard error of the mean (SEM).
ZHX2 function is regulated by Nα-methylation
The discovery of both downregulation of Mup genes and upregulation of Cyp genes in Nrmt1−/− livers was interesting, as both are regulated inversely by the transcription factor zinc fingers and homeoboxes 2 (ZHX2) [Citation13,Citation14]. ZHX2 has been shown to both promote Mup expression [Citation13] and repress Cyp expression [Citation14]. Given that the RNA-seq results show lowered Mup expression and increased Cyp expression in Nrmt1−/− livers, they indicate ZHX2 is unable to function optimally as a transcription factor in the absence of NRMT1.
Nα-methylation by NRMT1 has been shown to regulate protein/DNA interactions, and its loss has been shown to disrupt processes that rely on these interactions [Citation2,Citation6,Citation8,Citation10]. Analysis of the N-terminal sequence of ZHX2 showed that it has a non-canonical NRMT1 consensus sequence, Ala-Ser-Lys (ASK – after initiating methionine cleavage), indicating ZHX2 could be a direct target of NRMT1. To verify this, in vitro methylation assays were performed with an N-terminal ZHX2 peptide and recombinant human NRMT1 alone or in combination with recombinant human NRMT2. We have previously shown that non-canonical substrates can be methylated by NRMT1, but this activity is significantly increased by complex formation between NRMT1 and NRMT2 [Citation1]. NRMT1 alone was able to methylate the ZHX2 peptide, and addition of NRMT2 significantly increased this methylation, confirming that ZHX2 is a non-canonical target of NRMT1 ()). NRMT1 alone or in combination with NRMT2 was unable to methylate a ZHX2 peptide with a K4Q mutation, verifying the specificity of NRMT1 for the ASK consensus sequence and confirming that this mutation disrupts methylation of ZHX2 like previous substrates ()).
Figure 5. ZHX2 is methylated by NRMT1 and Nα-methylation regulates ZHX2 function. In vitro methyltransferase assays show (a) ZHX2 peptide is methylated by recombinant NRMT1 but (b) K4Q ZHX2 peptide is not. Addition of NRMT2 also significantly increases the methylation activity of NRMT1 for WT ZHX2 but has no effect on K4Q ZHX2 methylation. (c – left) qRT-PCR analysis of the ZHX2 target transcripts ICAM1, BCL2, and interleukin 8 in untransfected, K4Q-ZHX2-GFP overexpressing, or WT-ZHX2-GFP overexpressing 786-O cells shows a significant increase in expression of all three genes when comparing WT overexpression to untransfected. K4Q overexpression does not significantly alter transcript expression as compared to untransfected. (c – right) Western blot confirms equal expression of WT and K4Q-ZHX2-GFP. Tubulin is used as a loading control. (d) Chromatin immunoprecipitation showing the K4Q mutation reduces ZHX2 occupancy of the ICAM1 promoter to that seen in the untransfected control. * denotes p < 0.05, ** denotes p < 0.01, and **** denotes p < 0.0001 as determined by unpaired t-test. n = 3. Error bars represent ± standard error of the mean (SEM).
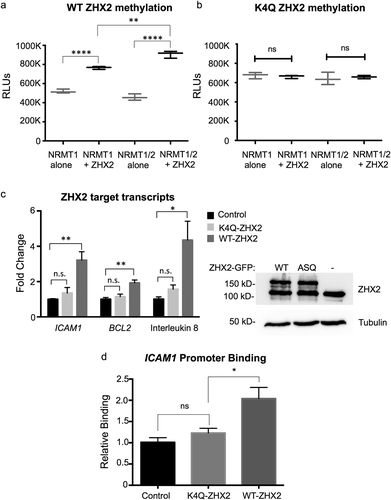
We have previously shown that the loss of Nα-methylation of another non-canonical target, MYL9, impairs its activity as a transcriptional activator [Citation10]. A K4Q mutation of MYL9 inhibits its binding to target promoter regions and decreases their expression levels [Citation10]. Besides the regulation of Mup and Cyp genes in the liver, ZHX2 also regulates the expression of NF-ĸB target genes during clear cell renal cell carcinoma (ccRCC) progression [Citation24]. To test if Nα-methylation of ZHX2 also affects its ability to function as a transcription factor, WT ZHX2-GFP or methyl-deficient K4Q-ZHX2-GFP were overexpressed in 786-O ccRCC cells and expression of the NF-ĸB targets BCL2, IL8, and ICAM1 were quantified using qRT-PCR. Overexpression of WT ZHX2-GFP resulted in increased expression of all three transcripts as compared to untransfected controls ()). However, the overexpression of K4Q-ZHX2-GFP resulted in a significantly lower expression of all three transcripts as compared to WT ZHX2-GFP expression, which was also not significantly different from untransfected controls ()). Western blotting confirmed that altered ZHX2 target expression was not due to unequal expression of the WT and K4Q-ZHX2 proteins ()), and chromatin immunoprecipitation verified the K4Q mutation reduces ZHX2 occupancy of the ICAM1 promoter ()). Finally, expression of WT ZHX2-GFP in cells depleted of NRMT1 was not able to promote expression of BCL2, IL6, or ICAM1 (Supplemental Figure 4). These data support the RNA-seq data and indicate Nα-methylation regulates the DNA-binding and transcription factor activities of ZHX2.
ZHX2 function and liver degeneration
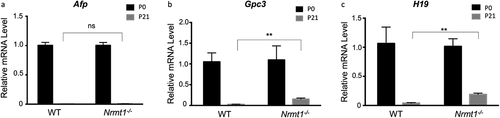
While downregulation of Mup expression and upregulation of Cyp expression have both been shown to disrupt liver metabolic function [Citation36–38] and are likely contributing to the observed liver degenerative phenotypes seen in Nrmt1−/− mice, ZHX2 also plays a role in liver development through its postnatal repression of the liver fetal development regulators AFP, GPC3, and H19 [Citation19,Citation20]. All three are expressed at high levels in the fetal liver but are subsequently silenced during postnatal development by ZHX2 [Citation19,Citation20]. AFP and GPC3 also play a postnatal role in liver stem cell (oval cell) regulation, and abnormal postnatal H19 expression promotes liver fibrosis [Citation39–42]. To determine if early Afp, Gpc3, or H19 repression was altered in Nrmt1−/− mice, we performed qRT-PCR analysis on WT and Nrmt1−/− livers at birth (P0) and 21 days after birth (P21). While we see no significant change in Afp silencing between P0 and P21 ()), Gpc3 and H19 expression is significantly de-repressed in Nrmt1−/− mice by 21 days ()). These data indicate that the transcription factor activity of ZHX2 is altered in Nrmt1−/− mice in a substrate-specific manner and suggest that, in addition to misregulation of liver metabolic pathways, loss of ZHX2 activity contributes to the liver phenotype seen in Nrmt1−/− mice by altering developmental pathways as well.
Figure 6. Postnatal Gpc3 and H19 expression is de-repressed in Nrmt1−/− livers. qRT-PCR analysis of wild-type (WT) and Nrmt1 knockout (Nrmt1−/−) mouse livers reveals (a) Afp expression is not de-repressed in Nrmt1−/− livers between 0 and 21 days after birth. However, (b) Gpc3 and (c) H19 expression is significantly de-repressed by 21 days. ** denotes p < 0.01 as determined by unpaired t-test. n = 3. Error bars represent ± standard error of the mean (SEM).
Discussion
The numerous phenotypes exhibited by Nrmt1−/− mice, including premature hair loss, cystic ovaries, and liver degeneration [Citation12], speak to the wide variety of targets of Nα-methylation that are misregulated when NRMT1 is lost. Here, we characterize ZHX2 as a novel substrate of NRMT1 and demonstrate how the loss of Nα-methylation disrupts the ability of ZHX2 to regulate its targets in the mouse liver, including Mups, Cyps, Gpc3, and H19, and in renal cancer cells, including NF-ĸB targets.
Though we found many transcripts misregulated in the Nrmt1−/− livers, we predict that those regulated by ZHX2 are a driving force in the observed liver phenotypes, as it has previously been shown that a hypomorphic ZHX2 mutation in BALB/cJ mice contributes to their high susceptibility to liver damage [Citation43]. Exactly which downstream ZHX2 targets are involved in promoting liver degeneration remains to be elucidated, but our data suggest that both metabolic and developmental pathways play a role. Increased Cyp expression has been shown to promote oxidative stress in the liver, and decreased Mup1 expression alters both glucose and lipid metabolism [Citation36–38]. Postnatal Gpc3 expression is normally isolated to hepatic progenitor/oval cells [Citation39], so its de-repression could abnormally expand the postnatal oval cell population. Abnormal postnatal H19 expression promotes liver fibrosis and epithelial–mesenchymal transition (EMT) [Citation40,Citation41], so its de-repression could lead to abnormal morphological changes. While our data suggest that many ZHX2 downstream targets contribute to the observed phenotypes, we saw no difference in Afp postnatal repression at P21, indicating Nα-methylation does not control ZHX2 transcription factor activity at all sites. We predict that the effect of Nα-methylation may be DNA sequence dependent or reliant on whether ZHX2 homodimerizes, heterodimerizes with ZHX1/ZHX3, or is bound to alternate complex members including NF-YA [Citation15,Citation16].
The liver is likely not the only organ in Nrmt1−/− mice where ZHX2 activity is affecting phenotypic outcomes. We have recently shown that the two neural stem cell (NSC) niches in the brain, the subventricular zone (SVZ) of the lateral ventricles and the subgranular zone (SGZ) of the dentate gyrus, exhibit postnatal misregulation of the cell cycle [Citation44]. The NSCs from these niches show precocious proliferation followed by premature depletion of the quiescent stem cell pool [Citation44]. This results in both neurodegenerative phenotypes and cognitive impairments in Nrmt1−/− mice [Citation44]. Through its interactions with ephrin-B1, ZHX2 has been shown to regulate neural progenitor cell maintenance and prevent premature neuronal proliferation [Citation45], indicating it may also be an important downstream target of NRMT1 in the brain. Similar RNA-sequencing analysis of cells in the SVZ and SGZ of Nrmt1−/− mice will help further identify ZHX2 targets regulated by Nα-methylation in the brain. It will also help elucidate tissue-specific signaling pathways regulated by ZHX2.
Besides Nα-methylation, the N-terminal sequence of ZHX2 (ASK) allows it to be targeted for Nα-acetylation [Citation5]. Our lab previously characterized MYL9 as the first protein regulated by both Nα-methylation and Nα-acetylation [Citation10]. While Nα-methylation regulated the nuclear roles of MYL9 as a transcription factor, Nα-acetylation regulated the cytoplasmic roles of MYL9 in cell migration [Citation10]. We have shown here that the nuclear role of ZHX2 as a transcription factor is also regulated by Nα-methylation, but ZHX2 is also found in the cytoplasm. Though there are currently no known cytoplasmic roles of ZHX2, its cytoplasmic localization does increase in disease states, including during hepatocellular carcinoma progression [Citation22]. Future studies will be aimed at identifying novel cytoplasmic functions of ZHX2 and determining if they are regulated by Nα-acetylation.
The 13 amino-terminal acids of ZHX2 are incredibly highly conserved, implying that there is a functional relevance for the N-terminus [Citation46]. Thus, regulation of ZHX2 function through Nα-methylation, which is more rapid than upregulating production of more ZHX2 protein, is a sensitive method for modulating the expression of ZHX2 targets. Both in the mouse liver and in renal cancer cells, the loss of Nα-methylation is sufficient to disrupt the activity of ZHX2 as a transcription factor, and Nα-acetylation may be providing a separate regulatory role as well. Continued characterization of how Nα-PTMs affect ZHX2 function will give us a better understanding of how ZHX2 regulates both developmental and oncogenic processes.
Supplemental Material
Download Zip (2.4 MB)Acknowledgments
We thank Megan Heaney and Lindsay Bonsignore for their contributions to the manuscript and Elizabeth Hudson at the University of Louisville Center for Genetics and Molecular Medicine (CGeMM) for performing the RNA sequencing. This work was supported by a Next-Gen Pilot grant from CGeMM and a research grant from the National Institutes of Health to C.E.S.T [GM144111].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/21541264.2022.2079184
Additional information
Funding
References
- Faughn JD, Dean WL, Schaner Tooley CE. The N-terminal methyltransferase homologs NRMT1 and NRMT2 exhibit novel regulation of activity through heterotrimer formation. Protein Sci. 2018;27(9):1585–1599.
- Chen T, Muratore TL, Schaner-Tooley CE, et al. N-terminal alpha-methylation of RCC1 is necessary for stable chromatin association and normal mitosis. Nat Cell Biol. 2007;9(5):596–603.
- Stock A, Clarke S, Clarke C, et al. N-terminal methylation of proteins: structure, function and specificity. FEBS Lett. 1987;220(1):8–14.
- Tooley CE, Petkowski JJ, Muratore-Schroeder TL, et al. NRMT is an alpha-N-methyltransferase that methylates RCC1 and retinoblastoma protein. Nature. 2010;466(7310):1125–1128.
- Petkowski JJ, Schaner Tooley CE, Anderson LC, et al. Substrate specificity of mammalian N-terminal alpha-amino methyltransferase NRMT. Biochemistry. 2012;51(30):5942–5950.
- Dai X, Otake K, You C, et al. Identification of novel alpha-n-methylation of CENP-B that regulates its binding to the centromeric DNA. J Proteome Res. 2013;12(9):4167–4175.
- Dai X, Rulten SL, You C, et al. Identification and functional characterizations of N-terminal alpha-N-methylation and phosphorylation of serine 461 in human poly(ADP-ribose) polymerase 3. J Proteome Res. 2015;14(6):2575–2582.
- Cai Q, Fu L, Wang Z, et al. alpha-N-methylation of damaged DNA-binding protein 2 (DDB2) and its function in nucleotide excision repair. J Biol Chem. 2014;289(23):16046–16056.
- Sathyan KM, Fachinetti D, Foltz DR. alpha-amino trimethylation of CENP-A by NRMT is required for full recruitment of the centromere. Nat Commun. 2017;8:14678.
- Nevitt C, Tooley JG, Schaner Tooley CE. N-terminal acetylation and methylation differentially affect the function of MYL9. Biochem J. 2018;475(20):3201–3219.
- Jia K, Huang G, Wu W, et al. In vivo methylation of OLA1 revealed by activity-based target profiling of NTMT1. Chem Sci. 2019;10(35):8094–8099.
- Bonsignore LA, Tooley JG, Van Hoose PM, et al. NRMT1 knockout mice exhibit phenotypes associated with impaired DNA repair and premature aging. Mech Ageing Dev. 2015;146–148:42–52.
- Jiang J, Creasy KT, Purnell J, et al. Zhx2 (zinc fingers and homeoboxes 2) regulates major urinary protein gene expression in the mouse liver. J Biol Chem. 2017;292(16):6765–6774.
- Creasy KT, Jiang J, Ren H, et al. Zinc fingers and homeoboxes 2 (Zhx2) regulates sexually dimorphic cyp gene expression in the adult mouse liver. Gene Expr. 2016;17(1):7–17.
- Kawata H, Yamada K, Shou Z, et al. The mouse zinc-fingers and homeoboxes (ZHX) family; ZHX2 forms a heterodimer with ZHX3. Gene. 2003;323:133–140.
- Kawata H, Yamada K, Shou Z, et al. Zinc-fingers and homeoboxes (ZHX) 2, a novel member of the ZHX family, functions as a transcriptional repressor. Biochem J. 2003;373(Pt 3):747–757.
- Bird LE, Ren J, Nettleship JE, et al. Novel structural features in two ZHX homeodomains derived from a systematic study of single and multiple domains. BMC Struct Biol. 2010;10:13.
- Wienk H, Lammers I, Hotze A, et al. The tandem zinc-finger region of human ZHX adopts a novel C2H2 zinc finger structure with a C-terminal extension. Biochemistry. 2009;48(21):4431–4439.
- Perincheri S, Dingle RW, Peterson ML, et al. Hereditary persistence of alpha-fetoprotein and H19 expression in liver of BALB/cJ mice is due to a retrovirus insertion in the Zhx2 gene. PNAS. 2005;102( 2):396–401.
- Morford LA, Davis C, Jin L, et al. The oncofetal gene glypican 3 is regulated in the postnatal liver by zinc fingers and homeoboxes 2 and in the regenerating liver by alpha-fetoprotein regulator 2. Hepatology. 2007;46(5):1541–1547.
- Spear BT, Jin L, Ramasamy S, et al. Transcriptional control in the mammalian liver: liver development, perinatal repression, and zonal gene regulation. Cell Mol Life Sci. 2006;63(24):2922–2938.
- Yue X, Zhang Z, Liang X, et al. Zinc fingers and homeoboxes 2 inhibits hepatocellular carcinoma cell proliferation and represses expression of cyclins A and E. Gastroenterology. 2012;142(7):1559–70.e2.
- Liu Y, Ma D, Ji C. Zinc fingers and homeoboxes family in human diseases. Cancer Gene Ther. 2015;22(5):223–226.
- Zhang J, Wu T, Simon J, et al. VHL substrate transcription factor ZHX2 as an oncogenic driver in clear cell renal cell carcinoma. Science (New York, NY). 2018;361(6399):290–295.
- Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11(3):R25.
- McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40(10):4288–4297.
- Petkowski Janusz J, Bonsignore Lindsay A, Tooley John G, et al. NRMT2 is an N-terminal monomethylase that primes for its homologue NRMT1. Biochem J. 2013;456(3):453–462.
- Shields KM, Tooley JG, Petkowski JJ, et al. Select human cancer mutants of NRMT1 alter its catalytic activity and decrease N-terminal trimethylation. Protein Sci. 2017;26(8):1639–1652.
- Chen X, Yamamoto M, Fujii K, et al. Differential reactivation of fetal/neonatal genes in mouse liver tumors induced in cirrhotic and non-cirrhotic conditions. Cancer Sci. 2015;106(8):972–981.
- Chen T, Brownawell AM, Macara IG. Nucleocytoplasmic shuttling of JAZ, a new cargo protein for exportin-5. Mol Cell Biol. 2004;24(15):6608–6619.
- Zhou Y, Rui L. Major urinary protein regulation of chemical communication and nutrient metabolism. Vitam Horm. 2010;83:151–163.
- McDonnell AM, Dang CH. Basic review of the cytochrome p450 system. J Adv Pract Oncol. 2013;4(4):263–268.
- Spector AA. Arachidonic acid cytochrome P450 epoxygenase pathway. J Lipid Res. 2009;50(Suppl):S52–6.
- Sowers MR, Wilson AL, Kardia SR, et al. Aromatase gene (CYP 19) polymorphisms and endogenous androgen concentrations in a multiracial/multiethnic, multisite study of women at midlife. Am J Med. 2006;119(9 Suppl 1):S23–30.
- Niwa T, Murayama N, Imagawa Y, et al. Regioselective hydroxylation of steroid hormones by human cytochromes P450. Drug Metab Rev. 2015;47(2):89–110.
- Zhou Y, Jiang L, Rui L. Identification of MUP1 as a regulator for glucose and lipid metabolism in mice. J Biol Chem. 2009;284(17):11152–11159.
- Gao H, Cao Y, Xia H, et al. CYP4A11 is involved in the development of nonalcoholic fatty liver disease via ROS‑induced lipid peroxidation and inflammation. Int J Mol Med. 2020;45(4):1121–1129.
- Abdelmegeed MA, Choi Y, Godlewski G, et al. Cytochrome P450-2E1 promotes fast food-mediated hepatic fibrosis. Sci Rep. 2017;7:39764.
- Grozdanov PN, Yovchev MI, Dabeva MD. The oncofetal protein glypican-3 is a novel marker of hepatic progenitor/oval cells. Lab Invest. 2006;86(12):1272–1284.
- Song Y, Liu C, Liu X, et al. H19 promotes cholestatic liver fibrosis by preventing ZEB1-mediated inhibition of epithelial cell adhesion molecule. Hepatology. 2017;66(4):1183–1196.
- Zhu J, Luo Z, Pan Y, et al. H19/miR-148a/USP4 axis facilitates liver fibrosis by enhancing TGF-β signaling in both hepatic stellate cells and hepatocytes. J Cell Physiol. 2019;234(6):9698–9710.
- Kuhlmann WD, Peschke P. Hepatic progenitor cells, stem cells, and AFP expression in models of liver injury. Int J Exp Pathol. 2006;87(5):343–359.
- Clinkenbeard EL, Turpin C, Jiang J, et al. Liver size and lipid content differences between BALB/c and BALB/cJ mice on a high-fat diet are due, in part, to Zhx2. Mamm Genome. 2019;30(7–8):226–236.
- Catlin JP, Marziali LN, Rein B, et al. Age-related neurodegeneration and cognitive impairments of NRMT1 knockout mice are preceded by misregulation of RB and abnormal neural stem cell development. Cell Death Dis. 2021;12(11):1014.
- Wu C, Qiu R, Wang J, et al. ZHX2 interacts with ephrin-B and regulates neural progenitor maintenance in the developing cerebral cortex. J Neurosci. 2009;29(23):7404–7412.
- Nail AN, Smith JJ, Peterson ML, et al. Evolutionary analysis of the zinc finger and homeoboxes family of proteins identifies multiple conserved domains and a common early chordate ancestor. Genome Biol Evol. 2020;12(3):174–184.