
Abstract
The CD8+ T-cell response comprises terminally differentiated effector cells and antigen-experienced memory T cells. The latter encompass central (TCM) and effector (TEM) memory cells. TCM cells are superior in their protection against viral and bacterial challenges and mediation of antitumor immunity due to their higher proliferative ability upon antigen re-encounter. Defining a mechanism to enhance TCM cells and delay terminal differentiation of CD8+ T cells is crucial for cancer immune therapy, as it can promote a better tumor immune response. The differentiation of CD8+ memory T cells is thought to be coordinated by the phosphoinositide 3-kinase (PI3K)/Akt pathway. We, therefore, investigated the role of Akt isoforms in the differentiation and proliferation of memory CD8+ T cells. We found that Akt1 and Akt2, but not Akt3, drive the terminal differentiation of CD8+ T cells, and their inhibition enhances the therapeutically superior TCM phenotype. Furthermore, the inhibition of Akt1 and Akt2, but not Akt 3, delays CD8+ T-cell exhaustion and preserves naïve and TCM CD8+ T cells, thus enhancing their proliferative ability and survival and prolonging their cytokine and Granzyme B production ability. Here, we define a mechanism in which proliferative potential, function, and survival of CD8+ T cells are enhanced by maintaining a reservoir of TCM and naïve cells using only Akt1 and Akt2 inhibition. Therefore, our findings strongly suggest the utility of using Akt1 and Akt2 inhibitors to modulate CD8+ T cells, both for adoptive cell transfer and vaccine-based cancer immune therapies.
Abbreviations:
- ACT, adoptive cell transfer
- CBA, cytometric bead array
- IFNγ, interferon gamma
- KLRG-1, killer cell lectin-like receptor subfamily G member 1
- KO, knockout
- SORP, special order research product
- TCM, central memory
- TEM, effector memory
- TCR, T-cell receptor
- TNF, tumor necrosis factor
- Tregs, regulatory T cells
- VCT, violet cell trace
- WT, wild type
Introduction
Upon antigen encounter and during the T-cell response, CD8+ T cells comprise effector and memory T cells.Citation1,2 Although effector CD8+ T cells become terminally differentiated and are eliminated by apoptosis following antigen clearance, approximately 10% of the remaining antigen-specific CD8+ T cells become memory T cells.Citation2 There are 2 types of CD8+ memory T cells: central (TCM) and effector (TEM) memory T cells.Citation2,3 Unlike TEM cells, TCM cells express a high level of CD62L and CCR7 and secrete high levels of interleukin (IL)-2 that correlates with their proliferative ability.Citation2-4 It is, therefore, no surprise that TCM cells are superior in their ability to protect against viral and bacterial challenges when compared to TEM cells.Citation4,5
The quality of tumor antigen-specific CD8+ T cells is crucial for an effective tumor immune response.Citation6 Adoptive cell transfer (ACT) of tumor-reactive CD8+ TCM cells has been shown to be a superior mediator of therapeutic antitumor immunity compared to TEM cells, because of their greater proliferative capacity upon antigen re-encounter.Citation4,5,7,8 Accordingly, understanding the regulation of CD8+ T cells into TEM or TCM cells is crucial, as defining a mechanism to enhance TCM cells and delay terminal differentiation of CD8+ T cells can promote a better tumor immune response.
The duration and intensity of antigenic stimulation control the magnitude of the CD8+ T-cell response as well as their differentiation into effector and memory CD8+ T cells. This differentiation is coordinated by the phosphoinositide 3-kinase (PI3K)/Akt pathway.Citation1 It has been established that Akt activation regulates the differentiation of CD8+ T cells into effector and memory T cells, where sustained Akt activation leads to terminal differentiation of effector CD8+ T cells, whereas inhibition of Akt in vivo increases the number of memory CD8+ T cells.Citation9
Here, we report that Akt1 and Akt2, but not Akt3, drive the terminal differentiation of CD8+ T cells, and their inhibition enhances the therapeutically superior central memory phenotype. Furthermore, the inhibition of Akt1 and Akt2, but not Akt 3, delays CD8+ T-cell exhaustion and preserves a reservoir of naïve and TCM CD8+ T cells, thus enhancing their proliferative ability and survival and prolonging their cytokine production ability.
Agents that slow down the terminal differentiation of CD8+ T cells without substantially impacting their proliferation are needed. Here, we define a mechanism in which proliferative potential, function, and survival are enhanced by maintaining a reservoir of TCM and naïve cells by inhibiting only Akt1 and Akt2. Therefore, our findings strongly suggest the utility of using Akt1 and Akt2 inhibitors to modulate CD8+ T cells as part of different cancer immune therapy regimens.
Results
Akt inhibition enhances the central memory phenotype of CD8+ T cells by diminishing their terminal differentiation and increasing their proliferative ability and survival
The TCM CD8+ T cells are superior mediators of therapeutic antitumor immunity due to their greater proliferative capacity upon antigen re-encounter.Citation4,5,7,8 Many T-cell functions are governed by PI3K/Akt signaling, including proliferation, survival, migration, and metabolism.Citation10,11 To test the role of Akt in the differentiation and proliferation of CD8+ T cells, we tested the effect of the pan Akt inhibitors MK-2206 and AZD5363 on stimulated CD8+ T cells. This was done using unfractionated splenocytes from pMel-1 mice activated with 1 µmol/L gp10025–33.
The phenotype of CD8+ T cells was assessed after 3 d of stimulation. We found that MK–2206-treated cells consisted mainly of TCM cells (CD62LhiCD44hi) and displayed a higher percentage of naïve cells (CD62LhiCD44lo) (). On the other hand, the majority of non–MK-2206-treated cells were TEM cells (CD62LloCD44hi). This was observed at all the concentrations used(), and plateaued at the 0.67 μmol/L concentration, which was, therefore, used as the optimal dose. The same pattern was also detected in AZD5363-treated cells (Fig. S1A), although the effect on CD8+ T cells was not as prominent as that with MK-2206 at the concentration used. This shows that Akt inhibition retards the terminal differentiation of CD8+ T cells and holds them in the central memory and earlier differentiation stages. The same effect was seen after the second and third stimulation with gp10025–33 on days 7, 14, and 21 in the presence of the Akt inhibitors (Fig. S2 and data not shown). It is worth mentioning that, although the percentage of TCM in inhibitor-treated cells decreased following the third and fourth stimulations, it was significantly higher than the percentage in the non-treated cells. This decrease could be attributed to the effect of consecutive stimulations with the antigen, which results in memory recall of TCM that eventually differentiate into TEM and effector cells. Taken together, these data show that Akt inhibition preserves a healthy reservoir of TCM cells even after several encounters with the antigen.
Figure 1 (see previous page). Akt inhibition preserves the TCM phenotype and enhances the proliferative ability of CD8+ T cells. Non-fractionated splenocytes from pMel-1 mice were stained with VCT and activated with gp10025–33 peptide (1 µmol/L) in the presence or absence of MK-2206 (0.67, 2, and 6 µmol/L). The concentration of the inhibitors was maintained throughout the experiment. The cells were re-stimulated with gp10025–33 peptide on days 7, 14, and 21 and their phenotype and proliferation were assessed. The gated cells were viable (7AAD-) CD8+Vβ13+. The data are representative of at least 4 independent experiments. (A) In this representative example, CD8+ T cells from a naïve spleen (far left) are mainly (72%) naïve cells (CD62LhiCD44lo). Sixty-seven percent (67%) of non–MK-2206-treated CD8+ T cells (third graph from left) are TEM cells (CD62LloCD44hi) and less than 1% are naïve cells (CD62LhiCD44lo). This changes when cells are treated with MK-2206 (far right), as 65% of the cells possess the TCM phenotype (CD62LhiCD44hi) and 14% are naïve cells (CD62LhiCD44lo). (B) After 3 d of stimulation, the proliferation of CD8+ T cells was inhibited in a dose-dependent manner by MK-2206 (VCT dilution) (far left). CD8+ T cells treated with MK-2206 expand at a significantly high rate with further stimulations (middle graph; data normalized to the non-treated control [GP100]). MK–2206-treated CD8+ T cells secrete significantly higher levels of IL-2 following stimulations 2 and 3, which is consistent with their higher proliferative potential (far right). *, P < 0.05; **, P < 0.01; ****, P < 0.0001. (C) Akt inhibition by MK-2206 maintains a high level of CD62L expression in CD8+ T cells on Day 3, and on Day 7 after each stimulation with gp100. (D) Akt inhibition by MK-2206 maintains high levels of CD127 in CD8+ T cells on Day 3, and on Day 7 after each stimulation with gp100. (E) Akt inhibition by MK-2206 inhibits the upregulation of KLRG-1 in CD8+ T cells after the second and third stimulations with gp100.
![Figure 1 (see previous page). Akt inhibition preserves the TCM phenotype and enhances the proliferative ability of CD8+ T cells. Non-fractionated splenocytes from pMel-1 mice were stained with VCT and activated with gp10025–33 peptide (1 µmol/L) in the presence or absence of MK-2206 (0.67, 2, and 6 µmol/L). The concentration of the inhibitors was maintained throughout the experiment. The cells were re-stimulated with gp10025–33 peptide on days 7, 14, and 21 and their phenotype and proliferation were assessed. The gated cells were viable (7AAD-) CD8+Vβ13+. The data are representative of at least 4 independent experiments. (A) In this representative example, CD8+ T cells from a naïve spleen (far left) are mainly (72%) naïve cells (CD62LhiCD44lo). Sixty-seven percent (67%) of non–MK-2206-treated CD8+ T cells (third graph from left) are TEM cells (CD62LloCD44hi) and less than 1% are naïve cells (CD62LhiCD44lo). This changes when cells are treated with MK-2206 (far right), as 65% of the cells possess the TCM phenotype (CD62LhiCD44hi) and 14% are naïve cells (CD62LhiCD44lo). (B) After 3 d of stimulation, the proliferation of CD8+ T cells was inhibited in a dose-dependent manner by MK-2206 (VCT dilution) (far left). CD8+ T cells treated with MK-2206 expand at a significantly high rate with further stimulations (middle graph; data normalized to the non-treated control [GP100]). MK–2206-treated CD8+ T cells secrete significantly higher levels of IL-2 following stimulations 2 and 3, which is consistent with their higher proliferative potential (far right). *, P < 0.05; **, P < 0.01; ****, P < 0.0001. (C) Akt inhibition by MK-2206 maintains a high level of CD62L expression in CD8+ T cells on Day 3, and on Day 7 after each stimulation with gp100. (D) Akt inhibition by MK-2206 maintains high levels of CD127 in CD8+ T cells on Day 3, and on Day 7 after each stimulation with gp100. (E) Akt inhibition by MK-2206 inhibits the upregulation of KLRG-1 in CD8+ T cells after the second and third stimulations with gp100.](/cms/asset/565bf607-4b96-43c7-aa07-f9e3adce5655/koni_a_1005448_f0001_c.jpg)
Because TCM CD8+ T cells are known to possess a greater proliferative ability than TEM cells upon antigen re-encounter,Citation4,5,7,8 we then assessed the proliferation of CD8+ T cells. Following the first 3 d of stimulation (to differentiate antigen-specific CD8+ T cells), we found that the proliferation of CD8+ T cells treated with MK-2206 or AZD5363 was inhibited in a dose-dependent manner as measured by Violet cell trace (VCT) dilution, as expected ( and Fig. S1B). However, surprisingly, with further stimulation (on days 7, 14, and 21), the MK–2206-treated CD8+ T cells were found to expand at a significantly higher rate than the non-treated cells (). On the other hand, the non-treated cells lost the ability to expand following the third stimulation. This clearly shows that Akt inhibition leads to enhanced longevity and prolonged cell survival of the CD8+ T cells. The same effect was observed with AZD5363, although to a lesser extent than with MK-2206 at the concentration used (Fig. S1B).
Interestingly, we found that CD8+ T cells treated with MK-2206 maintained high expression levels of CD62L and CD127 (markers associated with high proliferative potential). This correlates with the enhanced proliferation ability of the TCM cells treated with the inhibitor. These high levels were observed on days 3, 7, 14, and 21 ().
The ability of CD8+ T cells to proliferate was further assessed by measuring the level of IL-2 secretion, which is diminished in terminally differentiated CD8+ T cells. We found that CD8+ T cells treated with MK-2206 maintained a significantly high level of IL-2 secretion when re-stimulated on days 7 and 14 ().
The increase in longevity and survival observed in the MK–2206-treated CD8+ T cells prompted us to assess the expression level of KLRG-1, which is upregulated in terminally differentiated cells. We found that Akt inhibition maintained a low level of KLRG-1 after the second and third stimulations, whereas the non-treated CD8+ T cells expressed a significantly higher level of this marker (). The ability of MK-2206 to delay the exhaustion of CD8+ T cells corresponds to the cells' ability to survive and expand in response to more stimulations than the non-treated cells (). The same effect was observed when cells were treated with the Akt inhibitor AZD5363 (Fig. S1C-E), although to a lesser extent than with MK-2206 at the concentration used.
Taken together, these data show that Akt inhibition preserves TCM cells, thus enhancing their proliferative potential and survival while delaying the terminal differentiation and exhaustion of CD8+ T cells.
Akt inhibition rescues the ability of CD8+ T cells to produce cytotoxic cytokines and granzyme B after multiple stimulations
We have shown that targeting Akt using pan Akt inhibitors enhances proliferation, preserves the TCM phenotype, delays exhaustion, and conserves a larger pool of naïve cells. To assess the function of TCM cells, the secretion levels of interferon gamma (IFNγ) and tumor necrosis factor (TNF) were tested.
The CD8+ T cells were re-stimulated on days 7 and 14 with gp10025–33 and the level of IFNγ and TNF production after 24 h was assessed. After the second stimulation, MK–2206-treated and non-treated cells produced high and comparable levels of IFNγ and TNF in response to antigen re-encounter (). After the third stimulation, the secretion of IFNγ and TNF dropped significantly; however, Akt inhibition rescued the ability of CD8+ T cells to produce these cytokines, as their ability to secrete IFNγ and TNF was maintained at a significantly higher level (). This suggests that CD8+ T cells undergo terminal differentiation and reach exhaustion, thus losing their ability to secrete IFNγ and TNF upon several encounters with the antigen. Akt inhibition can clearly rescue the ability of CD8+ T cells to produce cytotoxic cytokines even with further stimulation.
Figure 2. Akt inhibition by MK-2206 maintains a high level of IFNγ and TNF secretion in CD8+ T cells. CD8+ T cells from pMel-1 mice were stimulated with gp10025–33 peptide (1 µmol/L) in the presence or absence of MK-2206 (0.67 µmol/L). On days 7 and 14, CD8+ T cells were re-stimulated with gp10025–33 peptide and the IFNγ and TNF levels in the supernatant were assessed after 24 h using CBA. Granzyme B expression was assessed on days 7 and 14. The data are representative of at least 2 independent experiments. (A) The ability of CD8+ T cells to produce IFNγ with subsequent stimulations is significantly diminished. CD8+ T cells treated with MK-2206 maintain their ability to secrete IFNγ with further stimulations. *, P < 0.05; ****, P < 0.0001. (B) CD8+ T cells treated with MK-2206 produce significantly higher levels of TNF and maintain this ability with further stimulations. *, P < 0.05. (C) Following the first stimulations, all the cells produce Granzyme B. A higher percentage of CD8+ T cells treated with MK-2206 or Akt-1/2 inhibitor produce a high level of Granzyme B following the second stimulation.
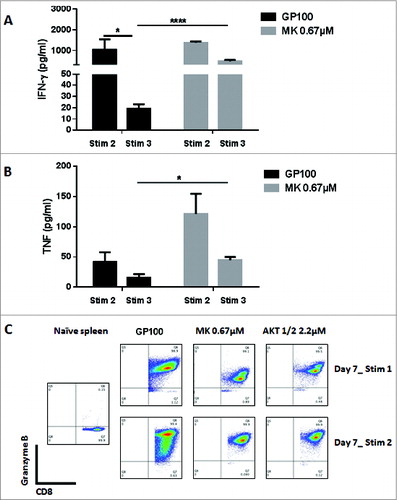
To further test the cytotoxic ability of the CD8+ T cells, we assessed the level of Granzyme B production by intracellular staining. Following the first stimulation, all of the cells produced Granzyme B (in comparison to naïve cells); however, MK–2206-treated cells produced a lower level in comparison to the non-treated cells. Remarkably, following the second stimulation, a higher percentage of MK–2206-treated cells produced higher levels of Granzyme B (). Interestingly, a small population of MK–2206-treated cells produced a higher level of Granzyme B than the rest of the cells, and these corresponded to the TCM population with the highest CD62L expression level (data not shown).
The maintained levels of IFNγ and TNF secretion suggest that CD8+ T cells do not lose their cytotoxic functionality as a result of Akt inhibition. In fact, Akt inhibition enhances their proliferative ability and survival by delaying their terminal differentiation and prolongs their ability to produce cytotoxic cytokines.
Akt 1 and Akt 2 are the two isoforms responsible for terminal differentiation of CD8 T cells
We have shown that Akt inhibition in CD8+ T cells delays their terminal differentiation, preserves TCM cells, enhances their proliferative ability and cytokine secretion, and prolongs their survival. The role of specific Akt isoforms (Akt1, -2, and -3) in the development, proliferation and function of CD8+ T cells is only known during thymic development, where Akt1 and Akt2 are the main isoforms contributing to the transition toward the double-positive (CD4+CD8+) stage and are involved in the differentiation of single-positive T cells.Citation12,13 To further dissect the role of specific Akt isoforms in the differentiation and proliferation of CD8+ T cells, we tested the effect of Akt1 and Akt2 inhibition using an Akt-1/2 inhibitor on stimulated CD8+ T cells. This was done using unfractionated splenocytes from pMel-1 mice activated with 1 µmol/L gp10025–33.
The phenotype of the cells was assessed after 3 d of stimulation. Similar to what we observed with the pan Akt inhibitors, CD8+ T cells treated with the Akt-1/2 inhibitor consisted mainly of TCM cells and displayed a higher percentage of naïve cells (). On the other hand, the majority of the non-treated CD8+ T cells were TEM cells. This effect was persistent even after the second and third stimulations (). These findings suggest that Akt1 and -2 are the isoforms responsible for terminal differentiation of CD8+ T cells and that their inhibition maintains CD8 T cells in earlier stages of differentiation (naïve and TCM).
Figure 3 (see previous page). The Inhibition of Akt1 and Akt2 preserves TCM cells and enhances the proliferative ability of CD8+ T cells. Non-fractionated splenocytes from pMel-1 mice were stained with VCT and activated with gp10025–33 peptide (1 µmol/L) in the presence or absence of Akt-1/2 inhibitor (2.2, 6.7, and 20.1 µmol/L). The cells were re-stimulated with gp10025–33 on days 7, 14, and 21. The gated cells were viable (7AAD-) CD8+Vβ13+. (A) Akt1 and Akt2 inhibition preserves the TCM phenotype. In this representative example, 76% of non-treated CD8+ T cells are TEM cells (CD62LloCD44hi) and less than 1% are naïve cells (CD62LhiCD44lo), whereas CD8+ T cells treated with an Akt-1/2 inhibitor consist of 76% TCM cells (CD62LhiCD44hi) and 22% naïve cells (CD62LhiCD44lo). (B) The proliferation of CD8+ T cells is inhibited by the Akt-1/2 inhibitor in a dose-dependent manner (Day 3). The expansion of CD8+ T cells treated with the inhibitor is significantly enhanced with further stimulations. Data are normalized to the non-treated control (GP100). *, P < 0.05. (C) Akt1 and Akt2 inhibition maintains a high level of CD62L on Day 3, and on Day 7 after each stimulation. (D) Akt1 and Akt2 inhibition maintains a high level of CD127 on Day 3, and on Day 7 after each stimulation. (E) Akt1 and Akt2 inhibition mitigates the upregulation of KLRG-1 in CD8+ T cells after the second and third stimulations. Following the third stimulation, the KLRG-1 level was dramatically increased and, therefore, the biexponential scale of the graph had to be adjusted for presentation purposes.
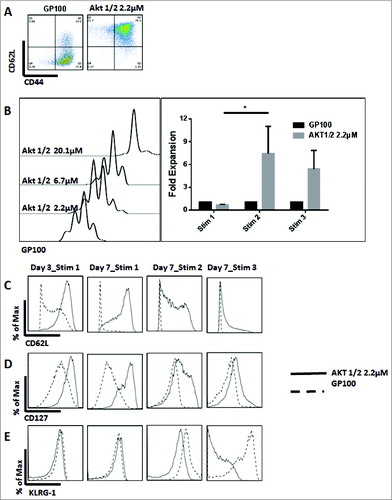
To test if the TCM phenotype generated by the inhibition of Akt1 and -2 possesses an enhanced proliferative ability, the proliferation of CD8+ T cells was assessed after 3 d of stimulation. We found that the proliferation of CD8 T cells treated with Akt-1/2 inhibitor was inhibited in a dose-dependent manner (). Additionally, the inhibition of Akt1 and Akt2 significantly enhanced the proliferative ability of CD8+ T cells with further stimulations (days 7 and 14) (). Treatment of CD8+ T cells with an Akt-1/2 inhibitor also maintained high expression levels of CD62L and CD127 () and high secretion levels of IL-2 (data not shown), consistent with the enhanced proliferative ability of TCM cells.
Similar to what we observed with pan Akt inhibitors, treatment of CD8 T cells with an Akt-1/2 inhibitor prolonged their survival and delayed their exhaustion, as evidenced by the maintained proliferative ability and the low levels of KLRG-1 following multiple stimulations ().
Treating the cells with an Akt-1/2 inhibitor also preserved high levels of TNF and IFNγ secretion (data not shown), suggesting an enhanced cytotoxic functionality of CD8+ T cells. Remarkably, the inhibition of Akt1 and Akt 2 did not significantly affect the production of Granzyme B following the first stimulation, as the cells produced a comparable level to the non-treated cells (). Following the second stimulation, a higher percentage of Akt–1/2-treated cells produced higher levels of Granzyme B (). Similar to treatment with pan inhibitors, a small population of Akt–1/2-treated cells produced a higher level of Granzyme B than the rest of the cells, and these corresponded to the TCM population with highest CD62L expression level (data not shown).
To rule out any influence of nonspecific inhibition of different isoforms and kinases using Akt inhibitors, the phenotype of stimulated CD8+ T cells from Akt1, -2, and -3 knockout (KO) and wild-type (WT) mice was assessed. After 7 d of stimulation, the highest percentage of TCM cells was observed from Akt1 KO mice, followed by Akt2 KO CD8+ T cells. The CD8+ T cells from both WT and Akt3 KO mice had comparable levels of TCM cells, which were significantly lower than the Akt1 and -2 KO CD8+ T cells ().
Figure 4. The absence of Akt1 and Akt2 isoforms, but not Akt3, preserves the TCM phenotype. Enriched CD8+ T cells from Akt1, -2, and -3 KO and WT mice were stimulated with anti-CD3 (1 μg/mL) and co-stimulated with anti-CD28 (2.5 μg/mL) antibodies. The phenotype of the cells was assessed on Day 7. The gated cells were viable (7AAD-) CD8+. (A) In this representative example, WT CD8+ T cells consisted of 83% TEM cells (CD62LloCD44hi). Akt1 KO CD8+ T cells consisted of 55% TCM cells (CD62LhiCD44hi), with 43% of the cells being TEM cells. Akt2 KO CD8 T cells consist of 36% TCM and 63% TEM cells. Akt3 KO CD8+ T cells consisted of 86% TEM cells, whereas only 12% were TCM cells. (B) Akt1 KO cells express a higher level of CD62L than Akt2 KO cells, which in turn express higher levels of these markers than WT and Akt3 KO cells that express similar levels. (C) Akt1 KO cells express a higher level of CD127 than Akt2 KO cells, which in turn express higher levels of these markers than WT and Akt3 KO cells that express similar levels.
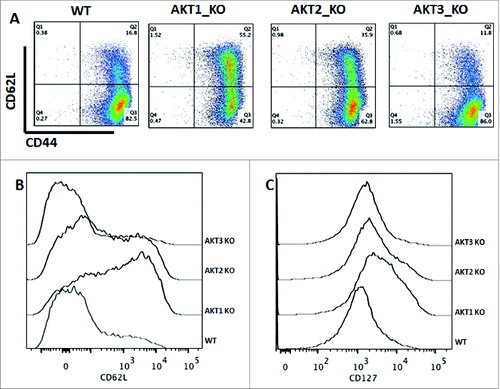
The Akt1 KO cells expressed a higher level of CD62L and CD127 than Akt2 KO cells, which in turn expressed higher levels of these markers than WT and Akt3 KO cells (). However, CD8+ T cells from WT and from different Akt KO mice did not show any differences in proliferation after several stimulations (data not shown). This could be explained by compensation from the different isoforms once the CD8+ T cells are stimulated in vitro.
To assess the contribution of each Akt isoform to the differentiation of TCM cells, stimulated CD8+ T cells from WT and Akt KO mice were treated with the pan Akt and Akt-1/2 inhibitors. Interestingly, cells from all the Akt KO mice and from WT mice behaved similarly, where the inhibition of Akt resulted in the enhancement of the TCM phenotype (). In Akt1 and -2 KO cells, the inhibitors target the existing isoform, thus enhancing the effect of the absent isoform. Therefore, TCM cells from Akt1 and Akt2 KO mice are maintained at comparable levels to WT treated with the same inhibitors. On the other hand, the absence of Akt3 in the KO mice did not affect the differentiation of CD8+ T cells, and the inhibition of the existing isoforms (Akt1 and Akt2) resulted in a similar outcome to Akt inhibition in WT mice. This finding suggests that, unlike Akt1 and -2, Akt3 does not play a significant role in the development of CD62LhiCD44hiCD8+ T cells.
Figure 5. Akt inhibition preserves the TCM phenotype in WT and Akt KO mice. Enriched CD8+ T cells from Akt1, -2, and -3 KO and WT mice were stimulated anti-CD3 (1 μg/mL) and co-stimulated with anti-CD28 (2.5 μg/mL) antibodies in the presence or absence of MK-2206 (0.67 µmol/L) or an Akt-1/2 inhibitor (2.2 μmol/L). The phenotype of the cells was assessed on Day 7. The gated cells were viable (7AAD-) CD8+. (A) Untreated CD8+ T cells from WT mice consist mainly of TEM cells, whereas those treated with MK-2206 or Akt-1/2 inhibitors consist mainly of TCM cells. (B) CD8+ T cells from Akt1 KO mice possess significantly more TCM cells than WT without any inhibitors. Once treated with MK-2206 or Akt-1/2 inhibitors, more TCMcells (with a higher CD62L expression) are maintained comparable to treated WT cells. (C) CD8+ T cells from Akt2 KO mice possess significantly more TCM cells than WT without any treatments, although less than that observed from Akt1 KO mice. Treatment with MK-2206 or Akt-1/2 inhibitors maintains a significantly higher percentage of TCM cells comparable to WT and Akt1 KO treated cells. (D) Similar to WT, CD8+ T cells from Akt3 KO mice consist mainly of TEM cells. Treatment with MK-2206 or Akt-1/2 inhibitors maintains a significantly higher percentage of TCM cells comparable to WT and Akt1 and -2 KO treated cells.
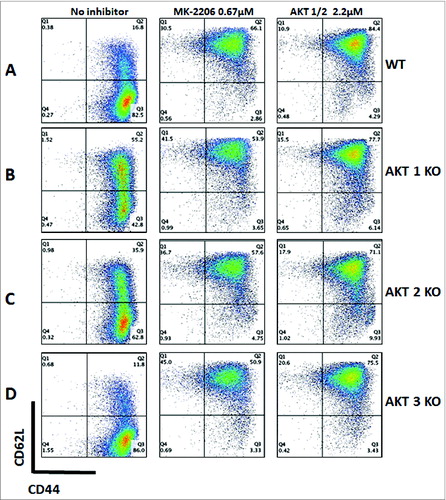
Taken together, our data demonstrate that the inhibition of Akt1 and -2, but not Akt 3, mitigates the terminal differentiation of CD8+ T cells and preserves TCM cells, thus enhancing their proliferative potential, longevity, and survival and rescues their ability to produce cytokines.
Discussion
During the T-cell response, CD8+ T cells comprise effector and memory T cells.Citation1,2 CD8+ memory T cells can be classified into TCM and TEM cells.Citation2,3 The TCM cells are superior in their ability to protect against viral and bacterial challenges Citation4,5 and mediation of therapeutic antitumor immunity when compared to TEM cells, due to their greater proliferative capacity upon antigen re-encounter.Citation4,5,7,8
Multiple T-cell functions are governed by PI3K/Akt signaling, including proliferation, survival, migration, and metabolism.Citation10,11 In fact, the differentiation of CD8+ cells into memory T cells is thought to be coordinated, at least in part, by the PI3K/Akt pathway.Citation1,14,15 Sustained Akt activation leads to terminal differentiation of effector CD8+ T cells, whereas inhibition of Akt in vivo increases the number of memory CD8+ T cells,Citation9 and the downstream inhibition of mTOR augments the functional quality of cytotoxic CD8+ T-cell responses by prompting a CD8 memory phenotype.Citation14-16 However, mechanisms that enhance the TCM phenotype and delay the terminal differentiation of CD8+ T cells without significantly impacting their proliferation and function are still needed.
Here, we show, or the first time, that the Akt1 and -2 isoforms drive the terminal differentiation of antigen-specific CD8+ T cells. We further report that the inhibition of Akt1 and -2 delays the exhaustion of CD8+ T cells, prolongs their survival, preserves a remarkably high percentage of TCM cells, and significantly increases their proliferative potential upon re-encountering the antigen.
Our data show that Akt1 and -2 inhibition enhances the proliferative potential of CD8 cells and maintains a high expression of CD62L and CD127, while mitigating their exhaustion.
Interestingly, inhibition of mTOR by rapamycin has been shown to enhance the memory phenotype of CD8+ T cells as a result of modulating the functional quality of CD8+ T cells rather than their proliferation.Citation14-16 In fact, in the tumor microenvironment, mTOR inhibition leads to a decrease in the proliferation of CD8+ T cells by direct inhibition coupled with a significant increase in the suppressive regulatory T-cell (Treg) population.Citation17 Rapamycin has been shown to support the proliferation and survival of Treg cells Citation18-21 due to a feedback loop where mTOR inhibition results in PI3K-dependent Akt activation, which sustains signaling through mTOR.Citation22 Accordingly, antibody-based depletion of Treg cells was proposed recently as a strategy to counteract this inhibition of CD8+ T cells when rapamycin is used to augment memory T cells.Citation17 Although this suggested strategy can minimize the suppression of CD8+ T cells by Tregs, it does not overcome the direct proliferation inhibition exerted on CD8+ cells by rapamycin. Here, we demonstrate that inhibition of the PI3K/Akt pathway at the level of Akt significantly enhances the proliferation of memory CD8+ T cells. Furthermore, we have recently shown that PI3K/Akt pathway inhibitors selectively target Tregs with resultant significant enhancement of antitumor immune response, including a significant decrease in Treg cells and increase in CD8+ T cells within the tumor microenvironment.Citation23 Therefore, using Akt inhibitors can simultaneously enhance the effector arm by augmenting the memory CD8+ T cells and diminish the suppressive arm by selectively inhibiting Treg cells.
The CD8+ T cells progressively lose IL-2 production as a function of differentiation from naïve to effector cells.Citation3 Here, we show that Akt inhibition in CD8+ T cells maintains a significantly higher level of IL-2 secretion. This is consistent with their maintained ability to proliferate and their phenotype as TCM cells. Remarkably, the enhanced proliferative ability of CD8+ T cells exerted by Akt inhibition is isoform-specific, suggesting the possibility of precise targeting of Akt1 and -2 to modulate the CD8+ T-cell response with minimal effects on other cellular functions.
Additionally, we show that Akt1 and -2 inhibition can rescue the ability of CD8+ T cells to secrete high levels of TNF and IFNγ and produce Granzyme B, even following multiple stimulations, thus suggesting a prolonged and potent antitumor cytotoxic ability. This suggests the use of Akt isoform-specific inhibitors to produce a sustained and powerful antitumor T-cell response when combined with different cancer immune therapies.
Our work shows that Akt1 and Akt2 inhibition leads to the preservation of a percentage of naïve CD8+ T cells. It has been shown that naïve CD8+ T cells have greater proliferative potential Citation24,25 and display elevated levels of IL-2 and IFNγ secretion following secondary stimulations.Citation24 Additionally, effector T cells derived from naïve T cells were found to promote more potent in vivo antitumor activity.Citation24,25 Therefore, our data show that Akt1 and Akt2 inhibition maintains a higher percentage of both naïve and TCM cells, which are both superior mediators of antitumor activity in comparison to effector and TEM CD8+ T cells.Citation4,5,7,8,24,25
To rule out any influence of undesired inhibition of different isoforms and different kinases using Akt inhibitors, the phenotype of CD8+ T cells from different Akt isoform KO mice was assessed. Our data confirm that the complete absence of Akt1, and to a lesser extent Akt2, display an increased proportion of TCM developing upon stimulation, when compared to WT CD8+ T cells. Furthermore, the absence of Akt3 has no effect on the percentage of TCM cells and is comparable to cells from WT mice.
This suggests that, in CD8+ T cells, Akt1 and -2 push toward the more powerful and acute TEM response. Although this is the desired situation in cases of acute infections, a continuous exposure to the antigen in cases of chronic infections and tumors leads to depletion of the TEM cells and the small reservoir of TCM CD8+ T cells. The inhibition of Akt1 and Akt2 isoforms seems to reverse this effect and favors a more sustainable response without affecting the number or function of available CD8+ T cells.
Here, we report that Akt1 and Akt2, but not Akt3, drive the terminal differentiation of CD8+ T cells and that their inhibition enhances the TCM phenotype, improves CD8+ T-cell survival, prolongs their cytokine and Granzyme B production ability, and enhances their proliferative potential.
To date, mechanisms that slow down terminal differentiation of CD8+ T cells without substantially impacting proliferation after T-cell receptor (TCR) stimulation are still lacking. Here, we define a mechanism in which proliferative potential, function, and survival are enhanced by maintaining a reservoir of TCM and naïve cells using only Akt1 and -2 inhibition. These findings strongly suggest the utility of using Akt isoform inhibitors to modulate the immune response as part of cancer immune therapy.
Materials and Methods
Mice and reagents
In vitro experiments used pMel-1 mice [B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J] that carry a rearranged TCR transgene (Vβ13) specific for the mouse homolog (pmel-17) of human (gp100).Citation26 C57BL/6(H-2b) WT, Akt1 KO, Akt2 KO, and Akt3 KO mice were also used. Akt3 KO mice were a generous gift from Dr Morris Birnbaum (University of Pennsylvania, PA) and Dr Phillip Dennis (NCI, NIH, MD) and were extensively backcrossed onto WT C57BL/6(H-2b) mice. All other mouse strains were purchased from the Jackson Laboratory. The animals were housed under pathogen-free conditions.
MK-2206 was purchased from Selleckchem. It is a highly selective inhibitor of all Akt isoforms with an IC50 of 8 nmol/L for Akt1, 12 nmol/L for Akt2, and 65 nmol/L for Akt3. The inhibitor was used in vitro at an optimized concentration of 0.67 µmol/L. AZD5363 was purchased from Selleckchem. It is a highly specific Akt inhibitor and has an IC50 of 3 nmol/L for Akt1, 8 nmol/L for Akt2, and 8 nmol/L for Akt3. The inhibitor was used in vitro at the optimized concentration of 2.4 μmol/L. The Akt kinase 1/2 inhibitor [1,3-Dihydro-1-(1-((4-(6-phenyl-1H-imidazo[4,5-g]quinoxalin-7-yl)phenyl)methyl)-4-piperidinyl)-2H-benzimidazol-2-one trifluoroacetate salt hydrate (Sigma)] has an IC50 of 58 nmol/L for Akt1 and 210 nmol/L for Akt2 and only inhibits Akt3 at a concentration of 2.12 mmol/L. The inhibitor was used in vitro at the optimal concentration of 2.2 µmol/L, which is 103-fold lower than the IC50 for Akt3 and, therefore, ensures specificity for Akt1 and Akt2. The inhibitors were titrated based on the IC50 of specific isoforms, and the doses used showed optimal inhibition with minimal effect on viability. Pan Akt inhibitors were used at doses ensuring the inhibition of all 3 isoforms.
The gp10025–33 9-mer peptide (KVPRNQDWL) (ANASPEC) was used for in vitro activation of pMel-1 splenocytes at a 1 µmol/L concentration. Briefly, GP100 is an enzyme involved in pigment synthesis that is expressed by different melanoma cell lines (including B16) and normal melanocytes.
CD8+ enrichment kits (Miltenyi) were used according to the manufacturer's instructions. Fluorochrome-labeled antibodies used for flow cytometry were purchased from BD.
In Vitro activation of CD8+ T cells
Tumor antigen-specific CD8+ T cells
Unfractionated splenocytes from pMel-1 mice were homogenized and stimulated in vitro by gp10025–33 peptide at a 1 µmol/L concentration (Day 0). Cells were cultured in RPMI-1640 (Lonza) supplemented with 10% fetal bovine serum (FBS), penicillin (100 U/mL), streptomycin (100 mg/mL), 0.1% β-mercaptoethanol (Life Technologies, Invitrogen), and IL-2 (100 U/mL) (Peprotech) at 37°C with 5% CO2. The cells were cultured with or without MK-2206 (0.67, 2, or 6 µmol/L), AZD5363 (0.27, 0.81, or 2.4 µmol/L), or Akt-1/2 inhibitor (2.2, 6.7, or 20.1 µmol/L). The concentration of the inhibitors was maintained throughout the culture by changing the media every 48–72 h.
On days 7, 14, and 21, cells were re-stimulated with gp10025–33 peptide at a 1 µmol/L concentration using feeder cells (irradiated WT splenocytes, 4,000 Rads) at 1:1 ratio using the same culture conditions.
TCR stimulation and co-stimulation
The CD8+ T cells from WT, Akt1 KO, Akt2 KO, and Akt3 KO mice were enriched using CD8+ enrichment kits (Miltenyi) according to the manufacturer's instructions (purity was on average 91%) or CD8+ cells were sorted using FACS ARIA II (BD Biosciences; purity > 99%).
The cells were then stimulated using feeder cells (irradiated WT splenocytes, 4000 Rads) at 1:1 ratio (Day 0) using TCR stimulation (anti-CD3 antibody 1 µg/mL, BD Biosciences) and co-stimulation (anti-CD28 antibody 2.5 µg/mL BD Biosciences) in the presence of 100 U/mL IL-2 (Peprotech). The cells were cultured with or without the optimized doses of the inhibitors MK-2206 at a 0.67 µmol/L concentration or Akt-1/2 inhibitor at a concentration of 2.2 µmol/L, and the concentration was maintained throughout the culture by changing the media every 48–72 h. Cells were cultured in the same conditions described earlier.
Proliferation assay and phenotyping of CD8+ T cells
Prior to their stimulation (Day 0), cells were labeled with 5 µmol/L VCT proliferation dye (Life Technologies, Invitrogen) according to the manufacturers' instructions. Samples were then evaluated for CD8+ T-cell expansion via VCT dye dilution (Day 3) using an LSRII SORP with HTS Flow Cytometer (BD Biosciences). The data were analyzed using FlowJo 9 or 10 (Tree Star).
To assess the phenotype of the T cells, the cultured cells were harvested and analyzed on days 3, 7, 14, and 21. The cells were stained with APC-Cy7-labeled anti-CD8, fluorescein isothiocyanate (FITC)-labeled anti-Vβ13, phycoerythrin (PE) labeled anti-CD62L, APC-labeled anti-CD44, PE-CF594-labeled anti-CD127, and APC-labeled anti-KLRG-1 in addition to the viability stain 7AAD (BD Biosciences). The same APC-Cy7-labeled anti-CD8 was used for sorting the cells using the FACS ARIA II (BD Biosciences).
For proliferation and phenotyping, the analyses were conducted on CD8+ T cells specific for the gp100 antigen and were gated on viable (7AAD-), Vβ13+CD8+ T cells. For WT and KO cells, the cells were gated on viable (7AAD-), CD8+ T cells.
For intracellular staining, cells were stained with the fixable near infrared Live/Dead viability stain (Life Technologies, Invitrogen), and fixed, permeabilized, and stained with APC-labeled anti-CD8, V450-labeled anti-Vβ13, PE-labeled anti-CD62L, and PE-CF594-labeled anti-CD44 (BD Biosciences), and FITC-labeled Granzyme B (Biolegend). The analyses were conducted on CD8+ T cells specific for the gp100 antigen and were gated on viable (Live/Dead negative), Vβ13+CD8+ T cells.
Cytometric bead array
Following the same activation protocol mentioned earlier, the pMel-1 gp100-specific CD8+ T cells were harvested on Day 7 after the first and second stimulation. The viable cells (trypan blue negative) were then counted and co-incubated (at 1:1 ratio) with 1 µmol/L gp10025–33 pulsated irradiated splenocytes (4000 Rads) for 24 h using the same culture conditions. The supernatant was collected and the levels of IL-2, TNF, and IFNγ were assessed using the mouse Th1/Th2/Th17 Cytokine Kit BD™ Cytometric Bead Array (CBA) kit. The data were collected using an LSRII SORP with HTS flow cytometer (BD Biosciences), and analyzed using the FCAP Array Software v3.0 (BD Biosciences).
Statistical analysis
All statistical parameters (average values, SD, significant differences between groups) were calculated using GraphPad Prism Software. Statistical significance between groups was determined by paired t-test or one-way ANOVA with post hoc Tukey's multiple comparison test (P < 0.05 was considered statistically significant).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website
1005448_Supplementary_Materials.zip
Download Zip (564 KB)Acknowledgments
The authors thank Dr Esteban Celis and Dr Rhea-Beth Markowitz for reviewing the manuscript and for their valuable suggestions and also thank Dr Lei Huang for his suggestions.
Funding
This work was supported by the Georgia Regents University Cancer Center (GRUCC) and a Fellowship Grant from King Hussein Institute for Biotechnology and Cancer (KHIBC, Jordan to RAE).
References
- Kim EH, Suresh M. Role of PI3K/Akt signaling in memory CD8 T cell differentiation. Front Immunol 2013; 4:20; PMID:23378844
- Klebanoff CA, Gattinoni L, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev 2006; 211:214–24; PMID:16824130; http://dx.doi.org/10.1111/j.0105-2896.2006.00391.x
- Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999; 401:708–12; PMID:10537110; http://dx.doi.org/10.1038/44385
- Klebanoff CA, Gattinoni L, Torabi-Parizi P, Kerstann K, Cardones AR, Finkelstein SE, Palmer DC, Antony PA, Hwang ST, Rosenberg SA, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A 2005; 102:9571–6; PMID:15980149; http://dx.doi.org/10.1073/pnas.0503726102
- Wherry EJ, Teichgraber V, Becker TC, Masopust D, Kaech SM, Antia R, , von Andrian UH, Ahmed R. Lineage relationship and protective immunity of memory CD8 T cell subsets. Nat Immunol 2003; 4:225–34; PMID:12563257; http://dx.doi.org/10.1038/ni889
- Rosenberg SA, Sherry RM, Morton KE, Scharfman WJ, Yang JC, Topalian SL, Royal RE, Kammula U, Restifo NP, Hughes MS, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol 2005; 175:6169–76; PMID:16237114; http://dx.doi.org/10.4049/jimmunol.175.9.6169
- Roberts AD, Ely KH, Woodland DL. Differential contributions of central and effector memory T cells to recall responses. J Exp Med 2005; 202:123–33; PMID:15983064; http://dx.doi.org/10.1084/jem.20050137
- Wu F, Zhang W, Shao H, Bo H, Shen H, Li J, et al. Human effector T cells derived from central memory cells rather than CD8T cells modified by tumor-specific TCR gene transfer possess superior traits for adoptive immunotherapy. Cancer Lett 2013; 339(2):195–207.
- Kim EH, Sullivan JA, Plisch EH, Tejera MM, Jatzek A, Choi KY, Suresh M. Signal integration by Akt regulates CD8 T cell effector and memory differentiation. J Immunol 2012; 188:4305–14; PMID:22467649; http://dx.doi.org/10.4049/jimmunol.1103568
- Finlay D, Cantrell D. Phosphoinositide 3-kinase and the mammalian target of rapamycin pathways control T cell migration. Ann N Y Acad Sci 2010; 1183:149–57; PMID:20146713; http://dx.doi.org/10.1111/j.1749-6632.2009.05134.x
- Kane LP, Weiss A. The PI-3 kinase/Akt pathway and T cell activation: pleiotropic pathways downstream of PtdIns(3,4,5)P3. Immunol Rev 2003; 192:7–20; PMID:12670391; http://dx.doi.org/10.1034/j.1600-065X.2003.00008.x
- Mao C, Tili EG, Dose M, Haks MC, Bear SE, Maroulakou I, Horie K, Gaitanaris GA, Fidanza V, Ludwig T, et al. Unequal contribution of Akt isoforms in the double-negative to double-positive thymocyte transition. J Immunol 2007; 178:5443–53; PMID:17442925; http://dx.doi.org/10.4049/jimmunol.178.9.5443
- Juntilla MM, Wofford JA, Birnbaum MJ, Rathmell JC, Koretzky GA. Akt1 and Akt2 are required for alphabeta thymocyte survival and differentiation. Proc Natl Acad Sci U S A 2007; 104:12105–10; PMID:17609365; http://dx.doi.org/10.1073/pnas.0705285104
- Li Q, Rao R, Vazzana J, Goedegebuure P, Odunsi K, Gillanders W, Shrikant PA. Regulating mammalian target of rapamycin to tune vaccination-induced CD8(+) T cell responses for tumor immunity. J Immunol 2012; 188:3080–7; PMID:22379028; http://dx.doi.org/10.4049/jimmunol.1103365
- Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009; 460:108–12; PMID:19543266; http://dx.doi.org/10.1038/nature08155
- Mineharu Y, Kamran N, Lowenstein PR, Castro MG. Blockade of mTOR signaling via rapamycin combined with immunotherapy augments anti-glioma cytotoxic and memory t cells' functions. Mol Cancer Ther 2014; 13(12):3024–36; PMID:25256739
- Kim HL. Antibody-based depletion of Foxp3+ T cells potentiates antitumor immune memory stimulated by mTOR inhibition. Oncoimmunology 2014; 3:e29081; PMID:25083329; http://dx.doi.org/10.4161/onci.29081
- Long SA, Buckner JH. Combination of rapamycin and IL-2 increases de novo induction of human CD4(+)CD25(+)FOXP3(+) T cells. J Autoimmun 2008; 30:293–302; PMID:18313267; http://dx.doi.org/10.1016/j.jaut.2007.12.012
- Strauss L, Whiteside TL, Knights A, Bergmann C, Knuth A, Zippelius A. Selective survival of naturally occurring human CD4+CD25+Foxp3+ regulatory T cells cultured with rapamycin. J Immunol 2007; 178:320–9; PMID:17182569; http://dx.doi.org/10.4049/jimmunol.178.1.320
- Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood 2005; 105:4743–8; PMID:15746082; http://dx.doi.org/10.1182/blood-2004-10-3932
- Basu S, Golovina T, Mikheeva T, June CH, Riley JL. Cutting edge: Foxp3-mediated induction of pim 2 allows human T regulatory cells to preferentially expand in rapamycin. J Immunol 2008; 180:5794–8; PMID:18424697; http://dx.doi.org/10.4049/jimmunol.180.9.5794
- Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, Khuri FR. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res 2005; 65:7052–8; PMID:16103051; http://dx.doi.org/10.1158/0008-5472.CAN-05-0917
- Abu-Eid R, Samara RN, Ozbun L, Abdalla MY, Berzofsky JA, Friedman KM, Mkrtichyan M, Khleif SN. Selective inhibition of regulatory T cells by targeting the PI3K-Akt pathway. Cancer Immunol Res 2014; 2:1080–9; PMID:25080445; http://dx.doi.org/10.1158/2326-6066.CIR-14-0095
- Wen M, Xu W, Ren L, Gao F, Cui N, Wen J, Li X, Lin L, Ma Z, Chen B, et al. Effector cells derived from naive T cells used in tumor immunotherapy of mice bearing B16 melanoma. Chin Med J (Engl) 2014; 127:1328–33; PMID:24709189
- Hinrichs CS, Borman ZA, Gattinoni L, Yu Z, Burns WR, Huang J, Klebanoff CA, Johnson LA, Kerkar SP, Yang S, et al. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood 2011; 117:808–14; PMID:20971955; http://dx.doi.org/10.1182/blood-2010-05-286286
- B6.Cg-Thy1a/Cy Tg(TcraTcrb)8Rest/J. The Jackson Laboratory; 2004. Available at: http://jaxmice.jax.org/strain/005023.html