
Abstract
T–cell-based immunotherapy of melanoma becomes ineffective when β2m-deficient tumor cells of a human leukocyte antigen (HLA) class I-negative phenotype grow out. We demonstrated that an early-acquired chromosomal deletion and subsequent inactivating gene mutation lead to β2m deficiency, suggesting that melanoma cells can genetically evolve to avoid being recognized by CD8+ T cells.
Introduction
Specific killing of melanoma cells by autologous CD8+ T cells has been demonstrated in numerous in vitro studies over the last 2 decades and recent clinical trials have provided evidence of effective in vivo antitumor T-cell activity. In a subgroup of melanoma patients, objective and durable clinical responses have been achieved upon adoptive transfer of ex vivo expanded tumor-infiltrating T lymphocytes and treatment with immunomodulatory antibodies.Citation1,2 Thus, immunotherapy in melanoma holds great clinical promise, although mechanisms of intrinsic or acquired resistance to T–cell-based therapy are active in the majority of patients and need to be deciphered for further improvement of treatment regimens.
CD8+ T cells recognize peptide epitopes from tumor antigens in the context of human leukocyte antigen (HLA) class I molecules. In general, melanoma patients generate CD8+ T-cell responses toward multiple tumor antigens. Interestingly, the expressed somatic tumor mutations can function as neoantigens,Citation3 giving rise to new antigenic peptide epitopes that increase melanoma immunogenicity. On the other hand, a number of specific somatic mutations directly interfere with T-cell recognition of melanomas. As such, mutational inactivation of the B2M gene has been demonstrated to generate a T–cell-resistant HLA class I-negative melanoma phenotype.Citation4,5 The B2M gene encodes an essential component of all trimeric HLA class I antigen complexes, consisting of an antigenic peptide, a HLA heavy chain, and the constant β2m light chain.
Melanoma cells, particularly those from sun-exposed tumors, are characterized by a high mutation load.Citation6 Although a few mutations have been identified as shared “melanoma drivers,” the majority of the genetic alterations is “private.” This leads to marked tumor heterogeneity, both between and within patients, that further increases during disease progression. Due to the impact of genetic alterations on melanoma immunogenicity, it is conceivable that a tumor will give rise to different lineages of distinct immunogenicity. So far, the evolution of heterogeneity in melanoma immunogenicity, and in particular the development of T-cell resistance, is poorly understood.
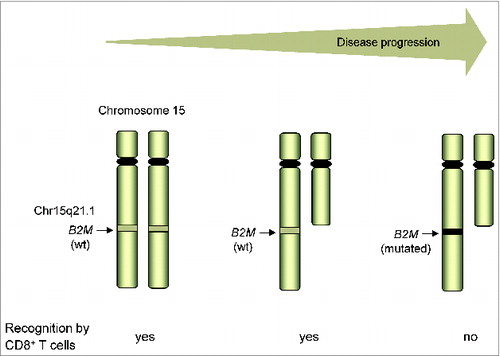
Recently, we studied the immunogenicity of tumor cells from 3 consecutive metastases of a melanoma patient (Ma-Mel-48) collected over a period of 1 year in the course of progressive stage IV disease.Citation7 From excised tumor tissues, we established melanoma cell lines that were studied, at first, for their immunophenotypes. Of the 3 cell lines, only 2, Ma-Mel-48a (established from a skin metastasis, July 2002) and Ma-Mel-48b (established from a lymph node lesion, January 2003) expressed HLA class I molecules whereas the Ma-Mel-48c cells (established from a lymph node metastasis, July 2003) were HLA class I-negative. These cellular phenotypes were also detected in the corresponding cryopreserved tumor tissues. Studying the capacity of the HLA class I-positive Ma-Mel-48a and Ma-Mel-48b cells to stimulate autologous CD8+ T cells, we found Ma-Mel-48a cells to be strong stimulators whereas the Ma-Mel-48b cells had only low T-cell-stimulatory capacity. The gradual decrease of melanoma immunogenicity culminated in the HLA class I-negative T–cell-resistant phenotype of Ma-Mel-48c cells that was caused by a lack of B2M expression. Molecular analyses found 2 genetic alterations to be responsible for the evolution of β2m deficiency in Ma-Mel-48c cells – a deletion on chromosome 15q where the B2M gene maps (Chr. 15q21.1) and an inactivating B2M gene mutation. Notably, the same deletion on chromosome 15q was already present in the Ma-Mel-48a and Ma-Mel-48b cells, indicating that the chromosomal aberration, associated with a B2M allele loss, occurred early in the course of disease.
This chronology of genetic alterations was also detected in tumor cells from consecutive metastases of the patient Ma-Mel-100.Citation7 The cell lines Ma-Mel-100a and Ma-Mel-100b, established from distinct lymph node lesions in April 2004 and May 2005, respectively, were both of a HLA class I-negative phenotype. In these 2 cell lines, we detected the same deletion on chromosome 15q, leading to an early loss of 1 B2M allele. Subsequently, the Ma-Mel-100a cells acquired a so-far-undefined post-transcriptional defect in B2M expression, whereas an inactivating B2M gene mutation occurred in Ma-Mel-100b cells.
In all our HLA class I-negative melanoma cell lines studied so far, we detected aberrations in chromosome 15q and B2M gene mutations.Citation4,8 The analyses in the 2 patient models now suggests that the B2M allele loss by deletions on chromosome 15q is an early-acquired genetic alteration that predisposes to the development of β2m deficiency in the course of progressive disease ().Citation7 Interestingly, Maleno et al. found deletions in the chromosome region 15q21 in 16% of the 70 metastatic melanoma samples analyzed,Citation9 indicating a substantial risk for the development of T-cell resistance.
Figure 1. Genetic evolution of T-cell resistance in melanoma in the course of disease progression. The HLA class I-negative phenotype of T–cell-resistant melanoma cells is caused by the lack of B2M expression that, in turn, originates from an early-acquired deletion on chromosome 15q, including 1 B2M allele, followed by mutational inactivation of the remaining B2M gene. wt, wild type.
Previously, the Ferrone group detected β2m loss in several recurrent melanoma metastases from patients receiving immunotherapy,Citation5 suggesting that the activity of tumor–antigen-specific T cells favored the outgrowth of HLA class I-negative melanoma lesions. Interestingly, a recent mouse model study by Matsushita et al. pointed to neo–antigen-specific T cells as important players in this so-called T–cell-mediated cancer immunoediting process.Citation10 It remains to be determined whether this can be translated to the human situation. Furthermore, it remains to be analyzed whether β2m-deficient metastases are present among the recurrent lesions from patients receiving adoptive T-cell therapy or treatment with immunomodulatory antibodies. In summary, we postulate that melanoma cells can genetically evolve to avoid being recognized by CD8+ T cells and propose that the screening of metastases for specific genetic alterations prior to and during immunotherapy could be relevant to predicting clinical responses.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 2011; 17:4550–7; PMID:21498393; http://dx.doi.org/10.1158/1078-0432.CCR-11-0116
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515:568–71; PMID:25428505; http://dx.doi.org/10.1038/nature13954
- Lennerz V, Fatho M, Gentilini C, Frye RA, Lifke A, Ferel D, Wölfel C, Huber C, Wölfel T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc Natl Acad Sci USA 2005; 102:16013–8.
- Paschen A, Méndez RM, Jimenez P, Sucker A, Ruiz-Cabello F, Song M, Garrido F, Schadendorf D. Complete loss of HLA class I antigen expression on melanoma cells: a result of successive mutational events. Int J Cancer 2003; 103:759–67; PMID:12516095; http://dx.doi.org/10.1002/ijc.10906
- Chang CC, Campoli M, Restifo NP, Wang X, Ferrone S. Immune selection of hot-spot beta 2-microglobulin gene mutations, HLA-A2 allospecificity loss, and antigen-processing machinery component down-regulation in melanoma cells derived from recurrent metastases following immunotherapy. J Immunol 2005; 174:1462–71; PMID:15661905; http://dx.doi.org/10.4049/jimmunol.174.3.1462
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature 2013 Aug 22; 500(7463):415–21; PMID:23945592; http://dx.doi.org/10.1038/nature12477
- Sucker A, Zhao F, Real B, Heeke C, Bielefeld N, Maβen S, Horn S, Moll I, Maltaner R, Horn PA, et al. Genetic evolution of T-cell resistance in the course of melanoma progression. Clin Cancer Res 2014; 20:6593–604; PMID:25294904; http://dx.doi.org/10.1158/1078-0432.CCR-14-0567
- Paschen A, Arens N, Sucker A, Greulich-Bode KM, Fonsatti E, Gloghini A, Striegel S, Schwinn N, Carbone A, Hildenbrand R, et al. The coincidence of chromosome 15 aberrations and beta2-microglobulin gene mutations is causative for the total loss of human leukocyte antigen class I expression in melanoma. Clin Cancer Res 2006; 12:3297–305; PMID:16740750; http://dx.doi.org/10.1158/1078-0432.CCR-05-2174
- Maleno I, Aptsiauri N, Cabrera T, Gallego A, Paschen A, López-Nevot MA, Garrido F. Frequent loss of heterozygosity in the β2-microglobulin region of chromosome 15 in primary human tumors. Immunogenetics 2011 Feb; 63(2):65–71; PMID:21086121; http://dx.doi.org/10.1007/s00251-010-0494-4
- Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, Arthur CD, White JM, Chen YS, Shea LK, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012; 482:400–4; PMID:22318521; http://dx.doi.org/10.1038/nature10755