
Abstract
We recently demonstrated that dual therapy combining AURKA and MDM2 antagonists is effective against melanoma in preclinical settings. Notably, besides inducing apoptosis, this regimen led to tumor senescence and stimulated the host's antitumor immune defenses. Treatments leveraging both cancer cell-intrinsic and extrinsic antitumor mechanisms can improve melanoma therapeutic outcomes.
Senescence is a biological process characterized by the loss of proliferative potential and the activation of a secretory response –known as senescence-associated secretory phenotype, or SASP– in affected cells. Normal cells within proliferative tissues initiate a senescence program when they reach the limits of their proliferative capacity, or prematurely in response to stress such as DNA damage or oncogene activation. Pro-inflammatory cytokines and chemokines (small secreted signaling proteins that induce directed chemotaxis in nearby responsive cells) produced by senescent tissues subsequently attract immune cells that, in turn, eliminate cells that pose a danger of oncogenic transformation. Therefore, senescence is considered to be among the critical mechanisms of early antitumor defense (reviewed in Citation1). However, it remains uncertain whether senescence can be leveraged to combat established cancerous lesions.
There are a number of drugs that induce a senescence-like state in cancer cells known as therapy-induced senescence (TIS). We have previously shown that inhibiting the mitotic kinase aurora kinase A (AURKA) in melanoma tumors leads to prominent senescence in vivo.Citation2 Such responses beg the question, is senescence a desirable outcome of cancer therapy? On the one hand, there are several obvious antitumor effects of senescence, such as reduced tumor growth due to cell cycle blockade and enhanced immune surveillance. In support, chemotherapy-induced senescence and p53 restoration has been shown to drive tumor regression in murine lymphoma, sarcoma and hepatocellular carcinoma models.Citation3-5 Notably, in these experiments myeloid cells of the host's immune system played a critical role in tumor clearance. On the other hand, paradoxical tumor-promoting effects of senescence have also been documented. For instance, some of the SASP components are known to promote aggressive cancer properties including proliferation, migration and invasion. SASP can also induce angiogenesis or lead to chronic inflammation, a condition that has been linked to tumorigenesis in many human tissues (summarized in Citation6). Furthermore, if the long-living senescent cells are not cleared by immune cells it is possible that such indolent malignant cells could eventually escape TIS leading to drug resistance.Citation7
In our prior preclinical study using an in vivo patient-derived xenograft model of melanoma a majority of the tested tumors (15 out of 19) responded to the senescence-inducing therapy with AURKA inhibitor with over 50% growth inhibition. However many of these tumors were still able to progress during the course of therapy or after the treatment ceased.Citation2 This finding suggests that melanoma tumors can indeed bypass senescence and, therefore, TIS alone may not be sufficient to control this aggressive disease.
In our resent study, we found that using TIS in a controlled manner by paring it with pro-apoptotic stimuli significantly improves therapeutic responses in murine and patient-derived melanoma models.Citation8 Specifically, we combined senescence-inducing AURKA inhibitor with an MDM2 antagonist that kills melanoma cells by stabilizing tumor suppressor p53 (). We showed that the lifespan of senescent melanoma cells with activated p53 was greatly limited as compared to control cells with low basal p53 activity. While control cells were able to survive indefinitely and some of them eventually escaped senescence, all of the cells with active p53 died within just a few days of the treatment. Notably p53 activation did not block the induction of either senescence or the SASP in our experimental conditions. In fact, we found that the secretion of the chemokines CCL1, CCL5 and CXCL9 was further increased in senescent tumors upon p53 induction. In addition to its critical role in senescence initiation, p53 has been also implicated in the maintenance of senescence, such that the loss of p53 has been shown to reverse senescence.Citation9 Our data demonstrate that upregulating p53 in cells with low basal p53 activity (such as melanoma cells) can subsequently reinforce the senescent phenotype.
Figure 1. The antitumor activity of combined therapy eliciting senescence and antitumor immunity. Anticancer responses to dual therapy co-targeting p53 with the MDM2 inhibitor (-)-Nutlin-3 and AURKA with the inhibitor MLN8237. AURKA – aurora kinase A; SA-βGal - senescence-associated β-galactosidase; SASP - senescence-associated secretory phenotype; Δψm – mitochondrial membrane potential; AIF – apoptosis-inducing factor; NK– NK cell; DC – Dendritic cell; Mφ - Macrophage.
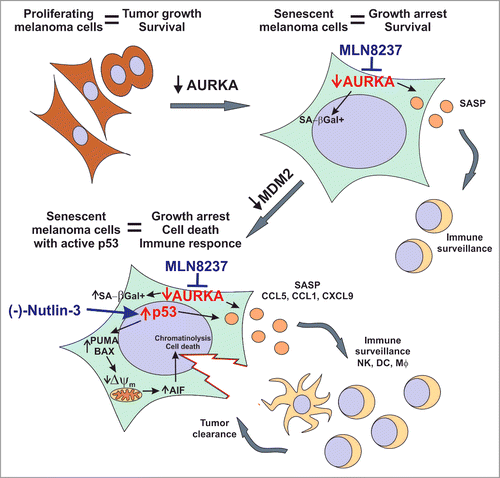
Interestingly, besides melanoma cells, non-malignant cells of the tumor microenvironment including tumor-associated fibroblasts, endothelial cells and tumor-infiltrating leukocytes, were also implicated in chemokine production in response to senescence-inducing therapy. The recruitment of immune cells and resultant tumor clearance was thus enhanced by combined MDM2 and AURKA antagonism in vivo.8 The recruited immune cells included antigen-presenting dendritic cells and macrophages, as well as natural killer (NK) and T cells. We further discovered that immune cells played a critical role in the response to the regimen because the therapeutic effect was compromised in severely immune-deficient NOD/SCID interleukin-2 receptor γ-chain null (NSG) mice that lack T, B, and NK cells, have functionally-defective macrophages and dendritic cells as well as defective cytokine signaling. In contrast, in immunocompetent animals the progression of aggressive B16F0 tumors was fully controlled by combined AURKA and MDM2 co-targeting. Of note, for melanoma tumors known for their immunogenicity, therapy-induced leukocyte infiltration may have an important implication for treatment through induction of the adaptive immune responses. Moreover, the presence of antitumor cells in the tumor immune infiltrate is a good prognostic factor in that it is associated with better survival, reduced prevalence of metastasis, and better response to immunotherapies (reviewed in Citation10).
In summary, our recent studies point out that combined therapies that induce senescence and stabilize p53 can target melanoma cells directly by inhibiting their growth and survival while also eliciting non-tumor cell autonomous antitumor responses through engagement of the host's immune system. These findings support the concept that rational combinatorial approaches to cancer therapy can improve patient outcomes.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank the members of Richmond laboratory for valuable discussion and research efforts.
Funding
The work described herein was supported by grants from NIH (CA116021 (AR), CA116021-S1 (AR), and GM084333 (JNJ)), the Department of Veterans Affairs (5101BX000196–04) (AR), and a Senior Research Career Scientist Award (AR). The support for this study was also provided by Vanderbilt Clinical Oncology Career Development program (AEV) funded through NIH K12 training grant (CA90625). Support for Core Facilities utilized in this study was provided by Vanderbilt Ingram Cancer Center (P30 CA68485).
References
- Pawlikowski JS, Adams PD, Nelson DM. Senescence at a glance. J Cell Sci 2013; 126(Pt 18): 4061–7; PMID:23970414; http://dx.doi.org/10.1242/jcs.109728
- Liu Y, Hawkins OE, Su Y, Vilgelm AE, Sobolik T, Thu YM, Kantrow S, Splittgerber RC, Short S, Amiri KI, Ecsedy JA, Sosman JA, Kelley MC, Richmond A. Targeting aurora kinases limits tumour growth through DNA damage-mediated senescence and blockade of NF-kappaB impairs this drug-induced senescence. EMBO Mol Med 2013; 5(1): 149–66; PMID:23180582; http://dx.doi.org/10.1002/emmm.201201378
- Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, Lowe SW. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002; 109(3): 335–46; PMID:12015983; http://dx.doi.org/10.1016/S0092-8674(02)00734-1
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007; 445(7128): 661–5; PMID:17251932; http://dx.doi.org/10.1038/nature05541
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445(7128): 656–60; PMID:17251933; http://dx.doi.org/10.1038/nature05529
- Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol 2011; 192(4): 547–56; PMID:21321098; http://dx.doi.org/10.1083/jcb.201009094
- Wang Q, Wu PC, Dong DZ, Ivanova I, Chu E, Zeliadt S, Vesselle H, Wu DY. Polyploidy road to therapy-induced cellular senescence and escape. Int J Cancer 2013; 132(7): 1505–15; PMID:22945332; http://dx.doi.org/10.1002/ijc.27810
- Vilgelm AE, Pawlikowski JS, Liu Y, Hawkins OE, Davis TA, Smith J, Weller KP, Horton LW, McClain CM, Ayers GD, et al. Mdm2 and aurora kinase a inhibitors synergize to block melanoma growth by driving apoptosis and immune clearance of tumor cells. Cancer Res 2015; 75(1): 181–93; PMID:25398437; http://dx.doi.org/10.1158/0008-5472.CAN-14-2405
- Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 2003; 22(16): 4212–22; PMID:12912919; http://dx.doi.org/10.1093/emboj/cdg417
- Oble DA, Loewe R, Yu P, Mihm MC, Jr. Focus on TILs: prognostic significance of tumor infiltrating lymphocytes in human melanoma. Cancer Immun 2009; 9: 3; PMID:19338264